

Abstract
Molecular dynamics simulations of three related helical antimicrobial peptides have been carried out in zwitterionic diphosphocholine (DPC) micelles and anionic sodiumdodecylsulfate (SDS) micelles. These systems can be considered as model mammalian and bacterial membrane interfaces, respectively. The goal of this study is to dissect the differences in peptide composition which make the mutant peptides (novispirin-G10 and novispirin-T7) less toxic than the parent peptide ovispirin (OVIS), although all three peptides have highly antibacterial properties. Compared to G10 and T7, OVIS inserts deepest into the DPC micelle. This correlates well with the lesser toxicity of G10 and T7. There is strong evidence which suggests that synergistic binding of hydrophobic residues drives binding of OVIS to the micelle. The helical content of G10 and T7 is reduced in the presence of DPC, and this leads to less amphipathic peptide structures, which bind weakly to the micelle. Simulations in SDS were carried out to compare the influence of membrane electrostatics on peptide structure. All three peptides bound strongly to SDS, and retained helical form. This corresponds well with their equally potent antibacterial properties. Based on the simulations, we argue that secondary structure stability often leads to toxic properties. We also propose that G10 and T7 operate by the carpet mechanism of cell lysis. Toxicity of peptides operating by the carpet mechanism can be attenuated by reducing the peptide helical content. The simulations successfully capture experimental binding states, and the different depths of binding of the three peptides to the two micelles correlate with their antibacterial and toxic properties.
Keywords: Ovispirin-1, Molecular dynamics simulations, DPC micelle, Peptide-membrane interactions, SDS micelle, Secondary structure induction, AMP activity, AMP toxicity, AMP
1. Introduction
All animals possess evolutionarily ancient mechanisms for recognizing and resisting attack by microorganisms. Much of the immune resistance comes without the delay associated with the development of a complete specific immune response. Gene encoded antimicrobial peptides (AMPs) are now well known to be a pervasive component of the immune defense system all across the plant and animal kingdom [14]. Hundreds of such peptides have been discovered from a plethora of living organisms, and many have been synthesized in laboratories with the aim of designing active molecules to combat antibiotic-resistant bacterial strains [2]. Indeed, our knowledge and comprehension of how AMPs function to kill microbial cells by non-specific disruption of the plasma membrane lipid bilayer has improved significantly in recent years.
The ultimate goal of research in this area is to engineer peptides with low toxicity and high antimicrobial activity. We now have a rudimentary comprehension of how helical AMPs function to selectively kill microbial cells. The plasma membrane of bacterial cells contains a significant amount of anionic components, while mammalian membranes are composed of mainly zwitterionic (phosphatidylcholine) lipids. Most AMPs are highly cationic, so they bind selectively to anionic bacterial membranes and permeabilize them [15]. It is believed that the electrostatic differences in the composition of bacterial and mammalian plasma membranes are responsible for selective toxicity of AMPs [10]. However, besides attracting cationic AMPs, the anionic surface of bacterial membranes might influence other processes which affect selectivity. It is known that most AMPs are unstructured in solution, and achieve their helical form after binding to the membrane. The extent of helicity induced in an AMP in presence of a membrane interface can influence its hemolytic and antibacterial properties [34]. Generally, no comprehensive attempt has been made to correlate peptide toxicity with the differences in the abilities of anionic and zwitterionic membranes to induce secondary structure in AMPs. Only few studies have compared the extent of AMP helix induction in anionic and zwitterionic membranes. For example, the peptide maculatin 1.1 and its analog P15A were found to be more helical in presence of anionic DMPG lipid vesicles than in DMPC lipids [6]. Both peptides inserted preferentially in DMPG vesicles. Mutations in pardaxin-1 which lead to lower helical content caused reduced antibacterial and hemolytic properties. All the above three peptides had only +1 cationic charge [36]. On the other hand, reduction of helicity in a pardaxin analog with +5 charge retained its antibacterial activity while reducing the hemolytic activity [35]. Variants of magainin (charge ≥ +5) which retained helical structure in presence of POPC membranes were found to be highly hemolytic, while those with reduced helical structure were less hemolytic [9]. There are various other instances where the helicity of peptides and membrane protein fragments is conserved in anionic lipids, but not in zwitterionic lipids [3,18,20,29,32]. Often these differences affect the ultimate cellular functions and structural roles of the proteins. The same should apply to the action of antimicrobial peptides. AMPs are believed to disrupt membranes either by an orderly pore-forming mechanism (barrel-stave or toroidal pore models), or by disordered binding of peptides to the membrane surface eventually causing membrane destabilization (carpet models). In general, a highly helical structure is required for membrane lysis mediated by the barrel-stave mechanism [36]. Although the same does not directly apply to the carpet or the toroidal pore models of cell lysis, a more amphipathic structure has long been correlated with increased antibacterial properties. Amphipathicity is usually promoted by helix formation. The observations made above paint a complicated picture of the influence of peptide structure on antibacterial activity and hemolysis. In effect, it seems that peptides which retain high helical content in presence of zwitterionic lipids tend to be more hemolytic. The correlation with antibacterial properties is not as distinct, although it appears that higher activity is observed for more helical cationic peptides, especially when the barrel mechanism of action is in effect. What is clear is that the secondary structure content of AMPs near membranes has a significant influence of peptide activity and selectivity. If different types of membranes (anionic versus neutral) do induce different peptide structures, then further investigation into these differences is warranted.
To explicitly reveal the influence of secondary structure stability on the molecular details of peptide–membrane interactions, we have carried out molecular dynamics (MD) simulations of three related helical peptides in the presence of zwitterionic dodecylphosphocholine (DPC) micelles. Ovispirin-1 (OVIS) is an 18-amino acid (KNLRR IIRKI IHIIK KYG) helical peptide, and has significant antimicrobial activity [22]. It is unsuitable for therapeutic use owing to its hemolytic properties [22]. Novispirin-G10 (KNLRR IIRKG IHIIK KYG) and novispirin-T7 (KNLRR ITRKI IHIIK KYG) are single residue mutants of OVIS which retain the antibacterial activity, but are less toxic. The three-dimensional structures of all three peptides were evaluated in presence of tri-fluoro-ethanol (TFE). The point mutations led to significant structural changes in the peptides, inducing a helix kink in G10 and N-terminal flexibility in T7 [33]. The structural and functional properties of these three peptides make them very well suited to carry out investigations to answer some of the questions raised above regarding the interplay between membrane-influenced structure and peptide–membrane interactions. All three peptides are highly cationic (+7), and based on the aforementioned discussion, we attempt to answer two questions: firstly, how is the helical content of the three peptides affected by the type of interface present? Secondly, how does this influence the affinity of the three peptides to the two types of micelles? Specifically, the simulations should capture the differences in the peptides’ affinity to DPC micelles because G10 and T7 are less toxic than OVIS.
In the reported work, we do not explicitly advocate the use of micelles as accurate models for membranes. However, micelles are excellent model systems to study the influence of interfacial membrane electrostatics on the structure and properties of small peptides. The primary advantage of using micelles as opposed to lipid bilayers is the faster time scales of motion of DPC and SDS lipids. It has been shown both experimentally [13] and by simulations [31,38–40] that the slowest relaxation times of lipids in micellar solutions are of the order of 500–2000 ps. The interaction of the peptide with the lipid macromolecular assembly induces a much faster response in micelles as opposed to bilayers. The micelle contains about half the number of atoms of a typical 128-lipid peptide–water bilayer simulation cell. This allows much longer simulations and permits monitoring of biological phenomena of longer time scales. In our recent work, we have shown that the micelle model is successful in capturing experimentally observed binding states of small peptides [23,24]. DPC lipids carry a choline head group like zwitterionic phospholipids (DMPC, DPPC, etc.), and the micelle is thus well suited as a model to study peptide–membrane interactions on a zwitterionic interface. For structure comparison purposes, simulations have also been carried out in anionic sodiumdodecylsulfate (SDS) micelles. We have previously reported simulations in SDS micelles [23,26]. In those simulations, the peptide was initially placed along a micelle diameter. In the current simulations, the peptide starts out in the aqueous phase.
2. Methods
The starting coordinates of the DPC micelle–water complex were obtained from simulations carried out by Wymore and Wong [39]. This structure was obtained after extensive minimization and dynamics of about 1 ns in a cubic simulation cell. We placed the micelle consisting of 60 DPC molecules and 4377 waters in a cubic simulation box of cell size 56.15 Å. The cell dimensions were setup so as to obtain the equilibrium bulk water density (0.033 Å−3) far away from the interface. Water was modeled using the TIP3P potential [21]. Seven chloride counterions and five Na+ and Cl− ions as 0.15 mM electrolyte were randomly distributed in the aqueous phase. In principle, to observe the folding of the peptide on the membrane interface, the simulation should start with a random coil peptide structure. However, the very long time scales of peptide folding on membrane interfaces precludes such simulatious. The experimental peptide structures in TFE were used to start the simulations. The initial structure of the peptides were obtained from the PDB databank (pdb ids: OVIS:1HU5, G10:1HU6, T7:1HU7) [33].
Solid-state NMR experiments of OVIS in lipid bilayers [43] suggest that the majority of the OVIS helix is oriented parallel to the interface, with the hydrophobic face embedded in the lipid core. For the simulations, the peptide was placed 5 Å away from the surface of the micelle, with the peptide helical axis oriented parallel to the micelle surface. The hydrophobic residues of the peptide faced the micelle. The fast dynamical time scales of DPC molecules ensured that the repositioning of the peptide to its final conformation would be computationally tractable.
To remove initial bad contacts between the peptide and the DPC core, and prevent penetration of water, the peptide and bulk water were kept under weak harmonic constraints with spring constants of 10 and 5 kcal/(mol Å), respectively, during equilibration. The constraints were gradually removed in 20,000 steps of minimization (steepest descent method). The entire system was then minimized for 20,000 more steps without any constraints. Thereafter, the system consisting of about ~16,000 atoms was gradually heated to 303 K. The entire assembly was subjected to NPT dynamics at pressure P = 1 atm and temperature T = 303.15 K for 39 ns. The constant pressure–temperature module of CHARMM [5] was used for the simulation with a leap-frog integrator. A time step of 2 fs was used. The temperature was set at 303.15 K using Hoover temperature control [19]. For the extended system pressure algorithm employed, all the components of the piston mass array were set to 500 amu [12]. The electrostatic interactions were simulated using the particle mesh Ewald (PME) summation [8,11] without truncation and a real space Gaussian width of 0.25 Å−1, a β-spline order of 4, and a FFT grid of about one point per Å. The non-bonded van der Waals interactions were smoothly switched off over a distance of 3.0 Å, between 9 and 12 Å. SHAKE was used to eliminate the fastest degrees of freedom involving bonds with hydrogen atoms. The simulation was carried out using CHARMM Version c30b2 with the all atom param22 parameter set. We have shown earlier that unnatural π-helices are not formed with the param22 parameter sets in peptide–micelle simulation setups [23]. Because the equilibrium steady state was reached after 25 ns (see later), ensemble average properties were calculated for the last 14 ns of the simulation. For calculation of most dynamical properties, trajectories were sampled every 4 ps.
The simulations in SDS micelles were carried out as described in our previous work [23]. The only difference being that in the current work, the peptide was initially placed in the aqueous phase. Standard protocols of non-bonded cutoffs, PME and constraint-based equilibration were the same as for the DPC simulations. Simulations in SDS were run for 40 ns each.
3. Results
3.1. Peptide location
In the first 25 ns of simulation, OVIS and G10 diffuse to the DPC interface. T7, on the other hand, quickly diffuses away from the interface in the first 3 ns of simulation. In the next 22 ns, it diffuses to the surface of the micelle. All three peptides are bound to the micelle thereafter (Fig. 1); hence, all ensemble averages have been calculated after 25 ns. For calculation of most properties, trajectories were sampled every 4 ps.
Fig. 1.
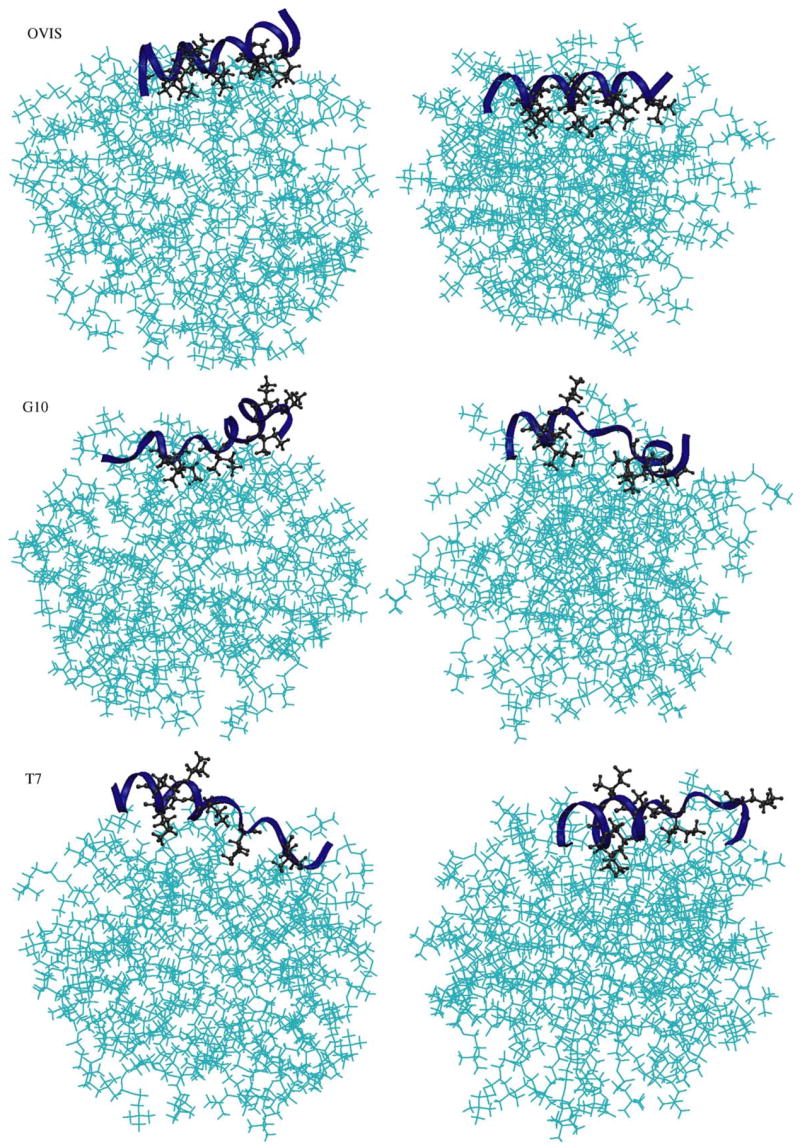
The starting structures (left) and the final conformations (right) of the three simulations. The “final” snapshots have been taken at t = 32 ns.
OVIS continues to remain deeply inserted in the interface for the remaining 14 ns of simulation, with its hydrophobic face buried in the micellar core. The peptide helix lies parallel to the interface. We have recently reported simulations of the OVIS peptide in DPC with a different starting conformation where the peptide was placed along a micellar diameter [24]. The final binding conformation of OVIS obtained in that simulation is the same as that obtained in the current simulation. The peptide starts in the aqueous phase in the current simulation. Such an initial setup is more appropriate for capture the influence of the interface on peptide structure. Solid-state NMR experiments conducted in lipid bilayers suggest that the bulk of the OVIS helix lies parallel to the membrane interface, with the hydrophobic residues buried in the bilayer core [42]. The final binding conformation of OVIS is thus in excellent agreement with solid-state NMR experiments.
In the simulations, the final binding conformations of G10 and T7 are similar to those of OVIS. However, the depth of insertion of both mutants is less than that of OVIS (Fig. 2). The ensemble-averaged electron density data in Fig. 2 indicate that OVIS is embedded deepest in the micelle, while the position of T7 w.r.t. to the micelle fluctuates most. No experimental binding conformation data is available for G10 and T7. However, both peptides are less toxic than OVIS, and are thus expected to insert less into zwitterionic interfaces compared to OVIS. The simulations neatly reproduce this correlation.
Fig. 2.
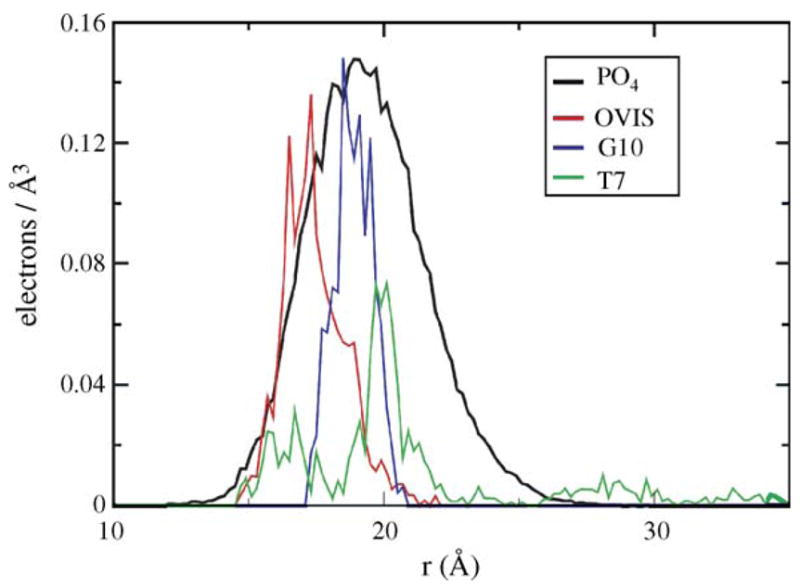
The electron densities of the three peptides shown w.r.t. the phosphates. The phosphates in all three simulations had nearly the same profile; hence, only one profile has been shown for clarity.
In SDS micelles, all three peptides remain tightly bound to the interface. The comparable depth of binding correlates well with the high antimicrobial activity of the three peptides.
3.2. Peptide structure
The helical structure of OVIS is further stabilized as the peptide diffuses to the interface. All the (i, i + 4) backbone–backbone H-bonds are satisfied, leading to a compact interface-bound helix which lies parallel to the interface (Figs. 3 and 4). On the other hand, both G10 and T7 become less helical in DPC micelles as the simulation progresses. The experimental TFE-induced structure of G10 has a bend as positions 12–13 induced by inclusion of a glycine at position 10 [33]. Fig. 3 shows the ensemble averaged dihedral angles of the G10 peptide from the simulation. The bend in the G10 helix now extends from position 7 to 11. This results in partial unfolding of the second N-terminal helix turn. Effectively, G10 is reduced to less than three helical turns during the simulation (Fig. 4). The C-terminal helical turn is retained in the ensemble structures. The experimental structure of T7 in TFE is flexible at the N-terminus. Residues 1–5 have dihedral angles that do not confirm to the native helical (ϕ, ψ) pair value of (−50, −60). The flexibility is enhanced in presence of the DPC micelle. Residue 6 (Ile) also acquires non-native dihedral angles.
Fig. 3.
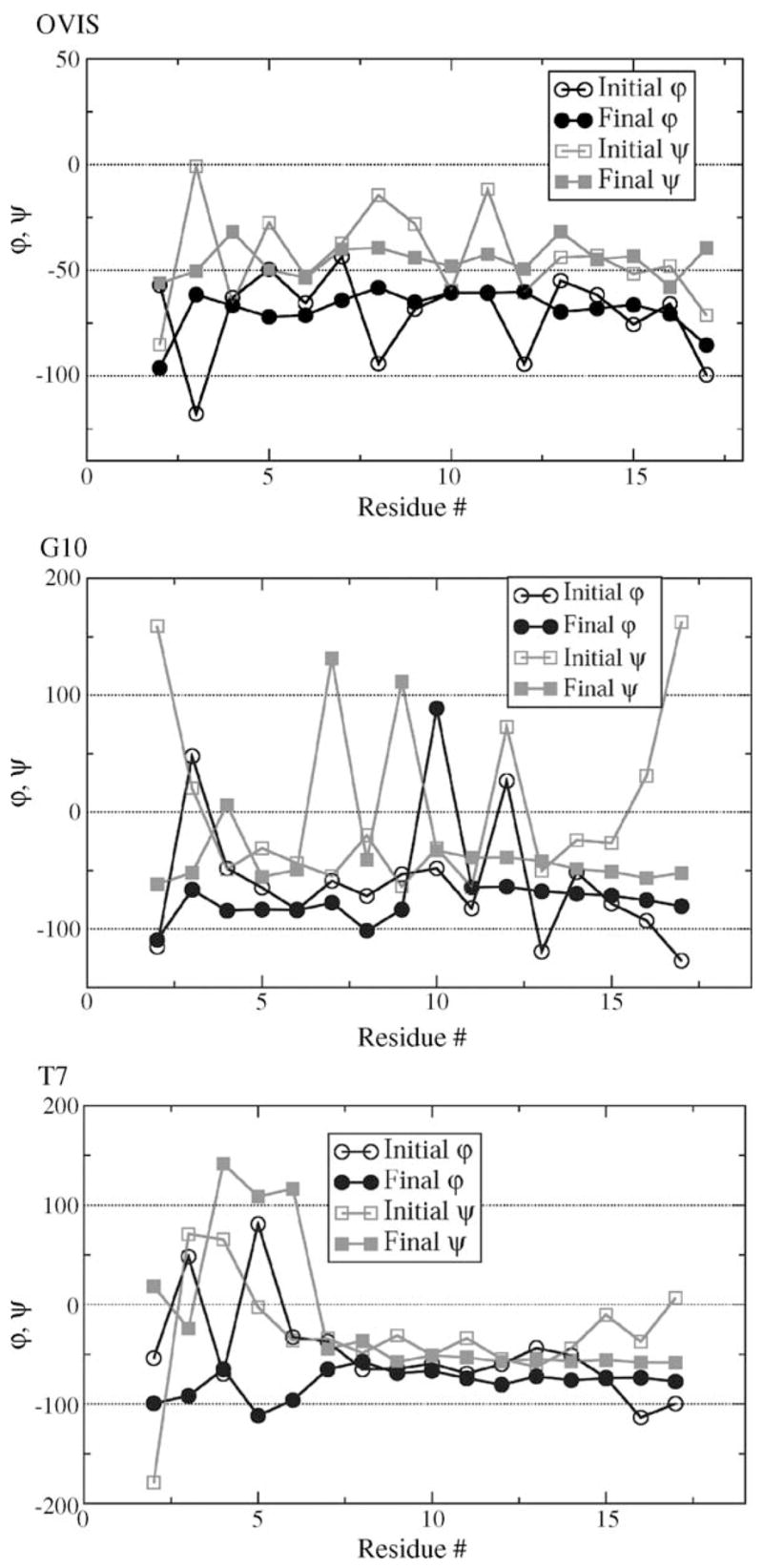
The initial and final dihedral angles of the peptides. The initial values were calculated from the pdb-deposited structures of the peptides. The final values were calculated by averaging over the last 14 ns of simulation.
Fig. 4.
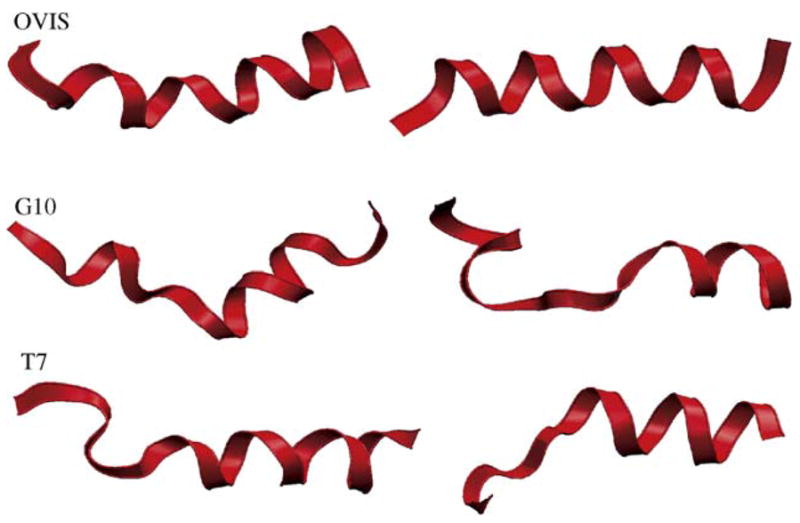
The initial and final structures of the peptides. The “final” snapshots were taken at t = 32 ns.
We counted the number of hydrogen bonds being formed between the peptide backbone atoms and the lipids and water molecules. A H-bond was counted if the bond distance was less than 3.0 Å, and the donor–hydrogen–acceptor angle was less than 120°. The total number of H-bonds formed was averaged for the last 15 ns of simulation. OVIS forms 1.67 ± 0.71 H-bonds with DPC and 10.94 ± 3.3 H-bonds with water. For G10 and T7, these numbers are 5.77 ± 1.24 and 25.03 ± 3.55, and 2.91 ± 0.67 and 18.82 ± 5, respectively. The lower helicity of G10 and T7 correlates well with these H-bonding patterns. The backbone atoms of OVIS are least available for intermolecular H-bond formation with the rest of the lipids and water. G10 and T7 are less helical, and a greater number of backbone amide and carbonyl groups are free to form hydrogen bonds. The greater number of H-bonds with water in G10 and T7 also indicate that the backbone is more hydrated then that of OVIS, which is embedded deeper into the micelle. Although the structural changes in G10 and T7 induced by DPC may seem insignificant, they have a drastic influence on peptide–DPC interactions and the final binding conformations (see later).
The structures of the three peptides are as helical in SDS micelle simulations as they are in the experimentally obtained structures in TFE. The SDS lipids do not induce any unfolding of the helices. In fact, the helices in SDS are tighter and conform better to the native helical (ϕ, ψ) pair value of (−50, −60).
3.3. Peptide–DPC interactions
Radial distribution functions (RDFs) give an accurate and detailed picture of specific microscopic interactions between the peptide and the lipids. All the RDFs were constructed for the last 15 ns of simulations, and the trajectories were sampled every 20 ps. All RDFs were normalized by a bulk density of 0.01. The phosphorus atoms on DPC were used for RDFs drawn with the lipid head groups.
Interestingly, there are no pronounced differences in the three peptides when it comes to interactions of cationic peptide residues with the DPC head groups (RDF data not shown). Additionally, the number of hydrogen bonds formed between the polar side chains of the peptide and the DPC head groups are nearly the same in all three peptides. However, the hydrophobic interactions of the peptide with the micellar core differ significantly in OVIS and the other two peptides (Fig. 5). Specifically, all isoleucine residues on OVIS are well inserted into the micellar core, compared to their position in G10 or T7. The hydrophobic interactions of T7 are the weakest, partly owing to the mutation of Ile-7 to Thr-7. Residues Thr-7, Ile-10 and Ile-11 evince no preference to the micellar core. G10 also lacks an isoleucine at position 10, but the isoleucine peaks at positions 6, 7, 11 and 14 are also all lower than those of OVIS. The peaks of OVIS side chains, on the other hand, are high at all hydrophobic residue positions.
Fig. 5.
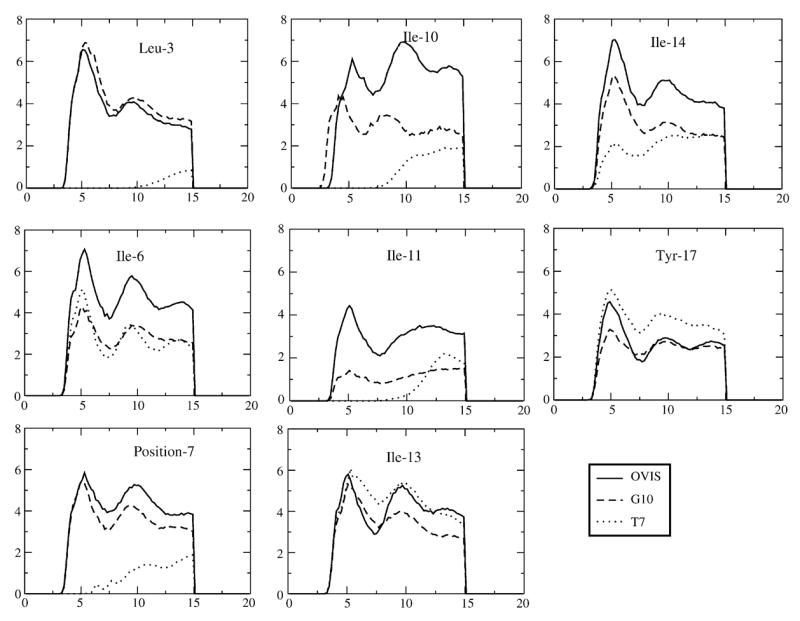
Radial distribution functions drawn between the hydrophobic peptide side chains and the DPC hydrocarbon atoms.
The differences in the hydrophobic interactions of the peptides with the micelle appear to be responsible for their different binding depths in the micelle. Although all three peptides were shown to be amphipathic in TFE [33], OVIS which is the most helical peptide, retains the maximum spatial differentiation of hydrophobic and polar regions. This allows the hydrophobic residues of OVIS to cooperatively bind the micellar core, with all the side chains facing the micelle interior. The deeper binding of these residues to the micellar core reduces the flexibility of the peptide backbone (Fig. 6) and presumably is the key factor that keeps OVIS helical. G10 and T7, on the other hand, are not as amphipathic to begin with, and the amphipathicity is further reduced by their loss of helicity in presence of DPC. All hydrophobic residues on G10 and T7 are not isolated into the same spatial region (Fig. 1b and c), and as a result, the hydrophobic insertion into the zwitterionic micelle is weaker.
Fig. 6.
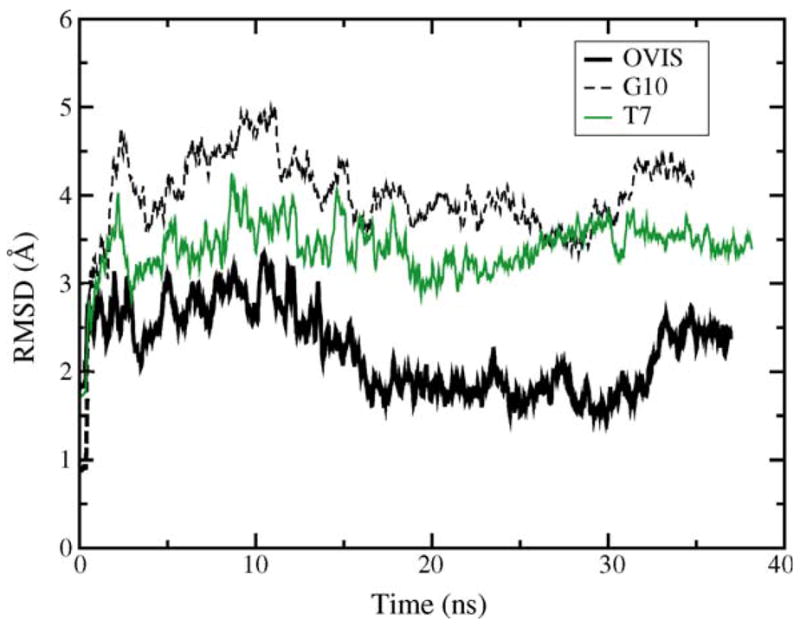
Temporal profile of the backbone RMSD of the three peptides.
4. Discussion
We have carried out MD simulations of an active and toxic helical AMP and its two active, but non-toxic analogs near zwitterionic interfaces. Simulations were also carried out in SDS micelles to compare the influence of membrane electrostatics on peptide secondary structure and toxicity. The goal of the study was to dissect the differences in the peptides that make the analogs (G10 and T7) less toxic compared to the native peptide (OVIS). We demonstrate that the simulations have converged to their final equilibrium state w.r.t. the position of the peptide relative to the micelle. However, it is uncertain how much more sampling may be required to guarantee that conformational equilibrium w.r.t. peptide structure has also been achieved. The results in this work should be interpreted in this light.
There are no significant differences in the three peptides with respect to interaction of polar residues with the DPC micelle. The simulations explicitly demonstrate that hydrophobic interactions drive the strong association of the OVIS peptide with the DPC lipids. This observation has been widely documented in literature for various helical AMPs. More importantly, the simulations indicate that the most toxic peptide, OVIS, remains helical, while the less toxic G10 and T7 lose helicity in presence of DPC. The stabilization of the helical structure of OVIS in presence of DPC lipids allows the hydrophobic residues of the peptide to bind cooperatively to the micelle. The reduced helicity induced in G10 and T7 by DPC leads to lower amphipathicity resulting in lesser binding depth into the micelle. For this reason, G10 and T7 are suggested to be less toxic than OVIS. Both G10 and T7 have one less hydrophobic residue than OVIS, and indeed, this also contributes to the lower toxicity, but ramifications of the point mutations in G10 and T7 extend well beyond the reduction of total hydrophobic content. The mutations induce critical changes in peptide secondary structure that prevents the cooperative isolation of hydrophobic residues into the membrane core. The situation is different in SDS micelles, where all three peptide retain their secondary structure, and remain equidistant from the micellar center of mass. The equal depth of binding of all three peptides to SDS, and the lesser binding depth of G10 and T7 in DPC correlate perfectly with the high antimicrobial activity of all three peptides, and the reduced toxicity of G10 and T7.
Given its interface-parallel orientation in lipid bilayers [43], OVIS has been putatively thought to operate by the carpet mechanism. As discussed in Section 1, a highly helical structure is not a requirement for membrane lysis mediated by the carpet mechanism [36]. Both G10 and T7 are less helical than OVIS in presence of DPC, and are still highly antibacterial. Additionally, they are oriented parallel to the interface in SDS micelles. It is thus highly probable that G10 and T7 also lyse cells by the carpet mechanism.
Based on our simulations and the above discussion, we suggest that mutations which marginally lower peptide helical content should attenuate toxic properties by lowering overall peptide amphipathicity. This deduction should hold consistent for peptides which operate by the carpet mechanism of action. Interestingly, we found at least two such examples in literature. Pleurocidin (GWGSFFKAAHVGKHVGKAALTHYL) is a 25-residue amphipathic helical AMP isolated from the Winter Flounder [7]. The sequence is 68% similar to dermaseptin, indicating that it might operate by the carpet mechanism. Mutations of glycines to alanines at positions 13 and 17 increased both overall the helical content and hemolytic activity against erythrocytes [28]. Similar observations were made about the scorpion-derived AMP IsCT and its analogs, although there is no direct evidence of a carpet-based mechanism [27]. The major families of helical peptides which operate by the carpet mechanism include the dermaseptins [37] and cecropins [17]. OVIS falls in this category, and is an analog of the human AMP LL-37 [30], which is thought to operate by the carpet mechanism as well.
Although simulations of peptides in DPC [16,23,38] and SDS [1,4,24,25,40,41] micelles have been carried out in the past, the current study focuses on the larger context of secondary structure induction near membrane interfaces, and the remarkable influence this has on the toxicity of antimicrobial peptides. It is also one of the most computationally intensive studies that we are aware of, with more than 200 ns of combined molecular dynamics simulations. The simulations show that zwitterionic interfaces discourage helix formation, and that this correlates neatly with the reduced toxicity of peptides which is reflected in their shallower binding to the DPC micelle. The results from the simulations can be generalized to other non-AMP membrane-active peptide fragments as well. The simulations confirm various scattered experimental observations that peptide secondary structure is preferentially induced in the presence of anionic membranes [3,18,20,29,32].
Acknowledgments
This work was supported by grants from NIH (GM 070989) and NSF (EEC-0234112). Computational support from the Minnesota Supercomputing Institute (MSI) is gratefully acknowledged. This work was also partially supported by National Computational Science Alliance under MCA04T033 and utilized the marvel cluster at the Pittsburgh Supercomputing Center. We thank Prof. Alan Waring for useful discussions.
References
- 1.Bond PJ, Cuthbertson JM, Deol SS, Sansom MS. MD simulations of spontaneous membrane protein/detergent micelle formation. J Am Chem Soc. 2004;126:15948–9. doi: 10.1021/ja044819e. [DOI] [PubMed] [Google Scholar]
- 2.Brahmachary M, Krishnan SP, Koh JL, Khan A, Seah SH, Tan TW, et al. ANTIMIC: a database of antimicrobial sequences. Nucleic Acids Res. 2004;32(database issue):D586–9. doi: 10.1093/nar/gkh032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brandenburg K, Harris F, Dennison S, Seydel U, Phoenix D. Domain V of m-calpain shows the potential to form an oblique-orientated alpha-helix, which may modulate the enzyme’s activity via interactions with anionic lipid. Eur J Biochem. 2002;269:5414–22. doi: 10.1046/j.1432-1033.2002.03225.x. [DOI] [PubMed] [Google Scholar]
- 4.Braun R, Engelman DM, Schulten K. Molecular dynamics simulations of micelle formation around dimeric glycophorin A transmembrane helices. Biophys J. 2004;87:754–63. doi: 10.1529/biophysj.104.040279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brooks BR, Bruccoleri RE, Olfson BD, States DJ, Swaminathan S, Karplus K. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comp Chem. 1983;4:187–217. [Google Scholar]
- 6.Chia CS, Torres J, Cooper MA, Arkin IT, Bowie JH. The orientation of the antibiotic peptide maculatin 1.1 in DMPG and DMPC lipid bilayers. Support for a pore-forming mechanism. FEBS Lett. 2002;512:47–51. doi: 10.1016/s0014-5793(01)03313-0. [DOI] [PubMed] [Google Scholar]
- 7.Cole AM, Weis P, Diamond G. Isolation and characterization of pleurocidin, an antimicrobial peptide in the skin secretions of winter flounder. J Biol Chem. 1997;272:12008–13. doi: 10.1074/jbc.272.18.12008. [DOI] [PubMed] [Google Scholar]
- 8.Darden T, Pedersen L, Toukmaji A, Crowley M, Cheatham T. Particle-Mesh based methods for fast Ewald summation in molecular dynamics simulations. 1998 http://www.ee.duke.edu/~wrankin/SciComp/Papers/SIAM97_darden.html.
- 9.Dathe M, Nikolenko H, Beyermann M, Bienert M. Optimization of the antimicrobial activity of magainin peptides by modification of charge. FEBS Lett. 2001;501:146–50. doi: 10.1016/s0014-5793(01)02648-5. [DOI] [PubMed] [Google Scholar]
- 10.Epand RM, Vogel HJ. Diversity of antimicrobial peptides and their mechanisms of action. Biochim Biophys Acta. 1999;1462:11–28. doi: 10.1016/s0005-2736(99)00198-4. [DOI] [PubMed] [Google Scholar]
- 11.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577–93. [Google Scholar]
- 12.Feller SE, Zhang Y, Pastor RW, Brooks BR. Constant pressure molecular dynamics simulation: the Langevin piston method. J Chem Phys. 1995;103:4613–21. [Google Scholar]
- 13.Fernandez P, Schroedle S, Buchner R, Kunz W. Micelle and solvent relaxation in aqueous sodium dodecylsulfate solutions. Chem Phys Chem. 2003;4:1065–72. doi: 10.1002/cphc.200300725. [DOI] [PubMed] [Google Scholar]
- 14.Ganz T. The role of antimicrobial peptides in innate immunity. Integrative Comp Biol. 2003;43:300–4. doi: 10.1093/icb/43.2.300. [DOI] [PubMed] [Google Scholar]
- 15.Ganz T, Lehrer RI. Antimicrobial peptides. Innate Immun. 2003:287–303. [Google Scholar]
- 16.Gao X, Wong TC. Molecular dynamics simulation of adrenocorticotropin (1–10) peptide in a solvated dodecylphosphocholine micelle. Biopolymers. 2001;58:643–59. doi: 10.1002/1097-0282(200106)58:7<643::AID-BIP1037>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 17.Gazit E, Miller IR, Biggin PC, Sansom MSP. Structure and orientation of the mammalian antibacterial peptide cecropin P1 within phospholipid membranes. J Mol Biol. 1996;258:860–70. doi: 10.1006/jmbi.1996.0293. [DOI] [PubMed] [Google Scholar]
- 18.Halsall A, Dempsey CE. Intrinsic helical propensities and stable secondary structure in a membrane-bound fragment (S4) of the shaker potassium channel. J Mol Biol. 1999;293:901–15. doi: 10.1006/jmbi.1999.3194. [DOI] [PubMed] [Google Scholar]
- 19.Hoover WG. Canonical dynamics: equilibrium phase-space distributions. Phys Rev A. 1985;31:1695–7. doi: 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- 20.Johnson JE, Cornell RB. Membrane-binding amphipathic alpha-helical peptide derived from CTP:phosphocholine cytidylyltransferase. Biochemistry. 1994;33:4327–35. doi: 10.1021/bi00180a029. [DOI] [PubMed] [Google Scholar]
- 21.Jorgensen WL, Chandrasekhar J, Medura JD, Impey RW, Klein ML. Comparison of simple potential function for simulating liquid water. J Chem Phys. 1983;79:926–35. [Google Scholar]
- 22.Kalfa VC, Jia HVP, Kunkle RA, McCray PB, Tack BF, Brogden KA. Congeners of SMAP29 kill ovine pathogens and induce ultrastructural damage in bacterial cells. Antimicrob Agents Chemother. 2001;45:3256–61. doi: 10.1128/AAC.45.11.3256-3261.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khandelia H, Kaznessis YN. Molecular dynamics simulations of helical antimicrobial peptides: what do point mutations achieve? Peptides. 2005;26:2037–49. doi: 10.1016/j.peptides.2005.03.058. [DOI] [PubMed] [Google Scholar]
- 24.Khandelia H, Kaznessis YN. Molecular dynamics simulations of the helical antimicrobial peptide ovispirin-1 in zwitterionic dodecylphoshocholine micelles. J Phys Chem B. 2005;109:12990–6. doi: 10.1021/jp050162n. [DOI] [PubMed] [Google Scholar]
- 25.Langham A, Kaznessis YN. Simulation of the N-terminus of HIV-I glycoprotein 41000 fusion peptide in micelles. J Pept Sci. 2005;11:215–24. doi: 10.1002/psc.623. [DOI] [PubMed] [Google Scholar]
- 26.Langham A, Khandelia H, Kaznessis YN. How can a β-sheet peptide be both a potent antimicrobial and harmfully toxic? Molecular dynamics simulations of protegrin-1 in micelles. J Pept Sci. 2005;11:215–24. doi: 10.1002/bip.20397. [DOI] [PubMed] [Google Scholar]
- 27.Lee K, Shin S, Kim K, Lim SS, Hahm KS, Kim Y. Antibiotic activity and structural analysis of the scorpion-derived antimicrobial peptide IsCT and its analogs. Biochem Biophys Res Commun. 2004;323:712–9. doi: 10.1016/j.bbrc.2004.08.144. [DOI] [PubMed] [Google Scholar]
- 28.Lim SS, Song YM, Jang MH, Kim YY, Hahm KS, Shin SY. Effects of two glycine residues in positions 13 and 17 of pleurocidin on structure and bacterial cell selectivity. Protein Pept Lett. 2004;11:35–40. doi: 10.2174/0929866043478383. [DOI] [PubMed] [Google Scholar]
- 29.Liu LP, Deber CM. Anionic phospholipids modulate peptide insertion into membranes. Biochemistry. 1997;36:5476–82. doi: 10.1021/bi970030n. [DOI] [PubMed] [Google Scholar]
- 30.Oren Z, Lerman JC, Gudmundsson GH, Agerberth B, Shai Y. Structure and organization of the human antimicrobial peptide LL-37 in phospholipid membranes: relevance to the molecular basis for its non-cell-selective activity. Biochem J. 1999;341:501–13. [PMC free article] [PubMed] [Google Scholar]
- 31.Rakitin AR, Pack GR. Molecular dynamics simulations of ionic interactions with dodecyl sulfate micelles. J Phys Chem B. 2004;108:2712–6. [Google Scholar]
- 32.Rourke AM, Cha Y, Collins D. Stabilization of granulocyte colony-stimulating factor and structurally analogous growth factors by anionic phospholipids. Biochemistry. 1996;35:11913–7. doi: 10.1021/bi960491t. [DOI] [PubMed] [Google Scholar]
- 33.Sawai MV, Waring AJ, Kearney WR, McCray PB, Forsyth WR, Jr, Lehrer RI, et al. Impact of single-residue mutations on the structure and function of ovispirin/novispirin antimicrobial peptides. Protein Eng. 2002;15:225–32. doi: 10.1093/protein/15.3.225. [DOI] [PubMed] [Google Scholar]
- 34.Shai Y, Oren Z. Diastereoisomers of cytolysins, a novel class of potent antibacterial peptides. J Biol Chem. 1996;271:7305–8. doi: 10.1074/jbc.271.13.7305. [DOI] [PubMed] [Google Scholar]
- 35.Shai Y, Oren Z. From “carpet” mechanism to de-novo designed diatereomeric cell-selective antimicrobial peptides. Peptides. 2001;22:1629–41. doi: 10.1016/s0196-9781(01)00498-3. [DOI] [PubMed] [Google Scholar]
- 36.Strahilevitz J, Mor A, Nicolas P, Shai Y. Spectrum of antimicrobial activity and assembly of dermaseptin-b and its precursor form in phospholipid membranes. Biochemistry. 1994;33:10951–60. doi: 10.1021/bi00202a014. [DOI] [PubMed] [Google Scholar]
- 37.Wong TC, Kamath S. Molecular dynamics simulations of adrenocorticotropin (1-24) peptide in a solvated dodecylphosphocholine (DPC) micelle and in a dimyistoylphosphatidylcholine (DMPC) bilayer. J Biomol Struct Dyn. 2002;20:39–57. doi: 10.1080/07391102.2002.10506821. [DOI] [PubMed] [Google Scholar]
- 38.Wymore T, Gao XF, Wong TC. Molecular dynamics simulation of the structure and dynamics of a dodecylphosphocholine micelle in aqueous solution. J Mol Struct. 1999;485–486:195–210. [Google Scholar]
- 39.Wymore T, Wong TC. Molecular dynamics study of substance P peptides partitioned in a sodium dodecylsulfate micelle. Biophys J. 1999;76:1213–27. doi: 10.1016/S0006-3495(99)77285-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wymore T, Wong TC. The structure and dynamics of ACTH (1-10) on the surface of a sodium dodecylsulfate (SDS) micelle: a molecular dynamics simulation study. J Biomol Struct Dyn. 2000;18:461–76. doi: 10.1080/07391102.2000.10506681. [DOI] [PubMed] [Google Scholar]
- 41.Yamaguchi S, Hong M. Determination of membrane peptide orientation by 1H-detected 2H NMR spectroscopy. J Mag Reson. 1997;155:244–50. doi: 10.1006/jmre.2002.2517. [DOI] [PubMed] [Google Scholar]
- 42.Yamaguchi S, Huster D, Waring AJ, Lehrer RI, Kearney W, Tack BF, et al. Dynamics of an antimicrobial peptide in the lipid bilayer by solid-state NMR spectroscopy. Biophys J. 2001;81:2203–14. doi: 10.1016/S0006-3495(01)75868-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415:389–95. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]