

Abstract
The purpose of this study was to determine optimal lipid concentration range for lyophilization of sterically stabilized phospholipid nanomicelles (SSM) and the freeze drying feasibility of self-associated therapeutic peptide-SSM assemblies. SSM at 5 to 20 mM 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-methoxy-poly(ethylene glycol 2000 (DSPE-PEG2000) were analyzed for particle size and viscosity before and after freeze drying which showed no significant changes (p>0.05). However, a steep increase in viscosity was seen for SSM above 15 mM phospholipid implying micelle-micelle interaction. Greater shrinkage of lyophilized cakes was observed below 10 mM phospholipid while they were more fibrous above 15 mM. Therefore, 10 - 15 mM DSPE-PEG2000 was chosen as the optimal phospholipid concentration for lyophilized SSM. When vasoactive intestinal peptide (VIP), glucagon-like peptide 1 (GLP-1) or gastric inhibitory peptide (GIP) (each, 67 μM) was added to SSM (10 mM), formulations showed no significant change in particle size, peptide fluorescence and peptide α-helicity before and after lyophilization. In conclusion, we found that peptide drug-SSM interactions are conserved during lyophilization.
Keywords: Nanomedicine, Nanobiotechnology, Drug delivery, Nanocarrier, DSPE-PEG2000, VIP, GLP-1, GIP
1. Introduction
Clinical applications of peptide drugs emanating from proteomics research is hampered by their short half-life (minutes) and non-targeted biodistribution in vivo (Lu et al., 2006). Despite recent advances in medical biotechnology (Ye et al., 2006, Menei et al., 2005; Liang et al., 2006, Ishihara et al., 2006, Onyuksel et al., 2006), delivery of peptide drugs still represents an unmet scientific need within the pharmaceutics community.
To this end, we showed that self-association of amphipathic peptide drug candidates, such as vasoactive intestinal peptide (VIP), with long-circulating, biocompatible and biodegradable sterically stabilized phospholipid nanomicelles (SSM) increases peptide stability in vitro and prolongs and amplifies its bioactivity in vivo (Önyüksel et al., 1999; Tseushita et al., 2002; Krishnadas et al., 2003). Importantly, given their distinct physico-chemical characteristics, these nanosized constructs are preferentially targeted to inflamed and injured tissues through local ‘leaky’ microcirculation (Sethi et al., 2003a).
Unfortunately, aqueous formulations of self-associated peptide drugs with SSM are stable for only seven days at 25 °C after preparation thereby precluding their storage before clinical use (Sethi et al., 2003b). To address this problem, recently our laboratory has successfully lyophilized SSM in the absence of cryo- and lyo-protectants and showed that physico-chemical properties of these nanomicelles are preserved upon reconstitution in aqueous media (Koo et al., 2005). Conceivably, shelf life of SSM in dried state could be increased due to absence of water and dissolved oxygen thereby circumventing phospholipid hydrolysis and oxidation. Whether self-associated peptide drug-SSM formulations can be freeze dried without compromising the integrity of peptide-nanomicelle interactions is uncertain.
Accordingly, the purpose of this study was to begin to address this issue by determining optimal phospholipid concentration for SSM lyophilization that mitigates micelle-micelle interaction, and by determining freeze drying feasibility of self-associated peptide drug-SSM constructs. For the latter, we chose vasoactive intestinal peptide (VIP), glucagon-like peptide (7-36) (GLP-1) and gastric inhibitory peptide (GIP) that have been tested in humans (Nauck et al., 1997; Gautier et al., 2005; Drucker et al., 2006; Groneberg et al., 2006).
2. Materials and Methods
2.1. Materials
1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-methoxy-poly(ethylene glycol 2000) (DSPE-PEG2000) were purchased from LIPOID GmbH (Ludwigshafen, Germany). Synthetic human VIP was synthesized by the Protein Research Laboratory, University of Illinois at Chicago (Chicago, IL). Synthetic human GLP-1 and GIP were ordered from American Peptides (Sunnyvale, CA). Saline (0.9% NaCl injection USP) and sterile water for irrigation (USP) were purchased from Baxter Healthcare Corporation (Deerfield, IL). All peptide and lipid samples were high performance liquid chromatography purified and the peptide purity was always greater than 90% as ascertained by RP-HPLC.
2.2. Preparation of SSM Dispersion and Peptide Stock Solutions
SSM were prepared as previously described in our laboratory (Gandhi et al., 2002). Briefly, SSM was prepared by dissolving DSPE-PEG2000 in saline, vortexing (Thermolyne Maxi Mix II) until complete dissolution before undergoing 1 h incubation at 25 °C (VWR SHEL LAB Incubator) in the dark. Stock solutions of individual peptides (VIP, GLP-1 or GIP) were prepared by dissolution of peptides in saline just before use. Depending on the final peptide concentration required, measured aliquots were added to saline or SSM.
2.3. Lyophilization of SSM and SSM containing model peptides
To determine optimal lipid concentration of SSM for lyophilization, 5, 10, 15 and 20 mM of SSM were prepared. For each lipid concentration, 1 ml of the equilibrated SSM sample was transferred into a 2-ml vial (n=3 for each concentration). The samples were frozen overnight at -20 °C, followed by freezing in liquid nitrogen for 3 min before overnight lyophilization using Labconco FreeZone® 6 Litre Freeze Dry System (Labconco, Kansas, MO).
For samples of SSM with peptides, 10 mM of SSM was first prepared. After 1 h incubation at 25 °C, peptides were added (VIP, GLP-1 or GIP) (each, 67 μM) to achieve lipid: peptide molar ratio of 150:1. After an additional 2 h incubation at 25 °C, the samples were transferred as 0.5 ml aliquots into separate Target DP™ vials (n=3 for each peptide). The samples were then subjected to the same lyophilization protocol as for SSM.
To reconstitute, 1 ml of sterile water was added into each lyophilized sample of SSM. For SSM containing peptides, 0.5 ml of sterile water was added during reconstitution. All samples were mechanically swirled in circular motion till complete dissolution, followed by 2 h incubation at 25 °C to ensure equilibration of the micelles and peptide-SSM interaction. Two hours was chosen since this was the duration previously found to be required for optimal bioactivity of amphipathic peptide molecules when added to SSM (Onyuksel et al., 1999). It is possible that shorter incubation time may be sufficient post reconstitution since peptide molecules are already associated with SSM in the lyophilized cake but further studies will be needed to draw a definite conclusion.
2.4. Characterization of SSM
Before freezing the SSM dispersions, the samples were analyzed for particle size by quasi-elastic light scattering (Nicomp® 380 particle size analyzer; Santa Barbara, CA) and viscosity (Brookfield® DVII + Pro viscometer; Middleboro, MA). After lyophilization, the lyophilized cakes of SSM were inspected visually for appearance and cake height. The reconstituted SSM were analyzed for particle size and viscosity again.
2.5. Characterization of SSM containing peptides
Samples of SSM containing different peptides were analyzed for their particle size, fluorescence emission spectra (fluorescence spectroscopy) and peptide secondary structures (CD) before and after lyophilization. The lyophilized cakes of peptide-SSM were subjected to the same visual inspection as with SSM.
2.5.1. Fluorescence emission spectroscopy
The fluorescence emission spectra of SSM containing peptides were measured using SLM Aminco 8000 Spectrofluorimeter at room temperature [Exλ (nm)/ Emλ (nm): VIP, 275/305; GLP-1, 275/340; GIP, 278/348] before and after freeze drying. The spectra of peptides (in the presence of SSM) (I) were normalized against spectra of saline (Ins) and presented as I/Ins.
2.5.2. Circular dichroism spectroscopy (CD)
The secondary structure of peptides was determined using Jasco J-710 Spectropolarimeter (Jasco, Easton, MD). Spectra were scanned at room temperature in a 0.1 cm path length fused quartz under the following conditions: 190 to 260 nm at 1 nm bandwidth and 2 s response time averaged over 3 runs. Spectra were corrected for empty nanomicelle scans and smoothed using manufacturer's Savitzky Golay algorithm. Deconvolution of Spectra was done by fitting the data into simulations using SELCON® to calculate the percentage of α-helical structures (Sreerama et al., 1994).
2.6. Data and statistical analyses
Data of the above experiments were expressed as mean ± standard deviation (S.D.). Statistical analysis was performed by Student's paired t test. P <0.05 was considered statistically significant.
3. Results and Discussion
For progression of a drug formulation into the clinical phase, an acceptable in vitro shelf life is very critical for mass production, bulk handling and storage. For these reasons, although the delivery of therapeutic peptides in SSM showed improved and prolonged in vivo bioactivities, widespread clinical application of peptide-SSM in the aqueous formulation would have been hindered by its shelf storage instability, due to susceptibility of the phospholipid content of SSM to oxidation and hydrolysis. To overcome these stability problems, our approach was to test the peptide formulations for feasibility of lyophilization.
3.1 Optimal phospholipid concentration range of SSM for lyophilization
To determine optimal phospholipid concentration range for freeze drying of SSM, 5 to 20 mM of SSM was tested in our study. Sample viscosity was measured to determine the maximum lipid concentration that can be used without adversely affecting the flow properties of SSM dispersion. At lipid concentration above 15 mM, our data showed that the rheological property of SSM changed and sample viscosity increased steeply compared to lower concentrations. However, the lyophilization process did not alter the viscosity of SSM, compared to freshly-prepared samples, for each lipid concentration investigated (Figure 1). Likewise, particle size of reconstituted SSM was not significantly different from its pre-lyophilized sample at all lipid concentrations studied (Table 1).
Figure 1.
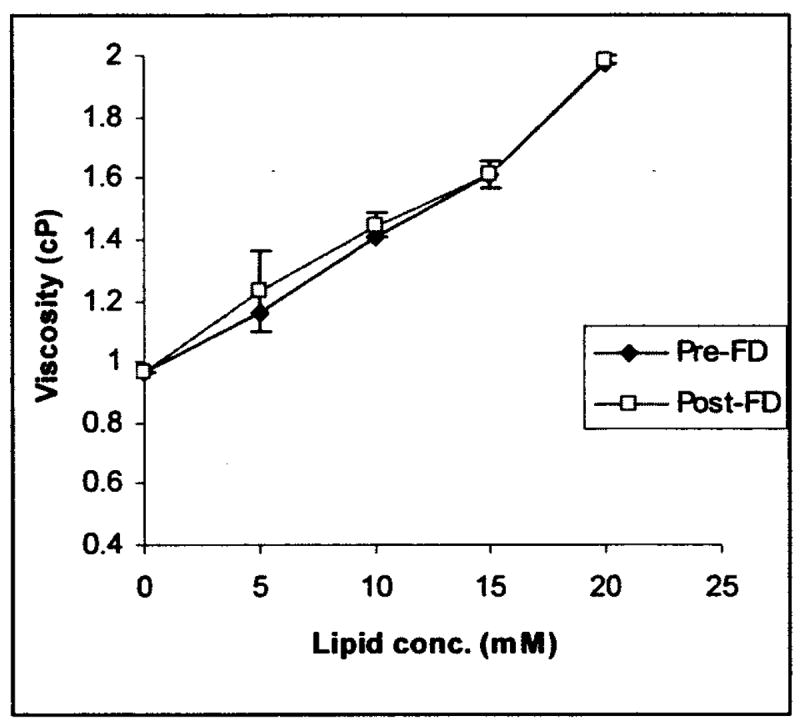
Viscosity of SSM at lipid concentrations of 5, 10, 15 and 20 mM before and after freeze drying (FD).
Table 1.
SSM particle size before and after freeze drying.
| Particle size (nm)* | ||
|---|---|---|
| Lipid concentration (mM) | before FDa | after FDa |
| 5 | 13.1±2.6 | 14.1±2.4 |
| 10 | 12.9±2.8 | 12.9±3.1 |
| 15 | 12.1±2.0 | 12.3±2.7 |
| 20 | 10.9±2.3 | 11.8±2.0 |
For all lipid concentrations, comparison before and after FD were not statistically significant with p > 0.05.
FD, freeze drying
Appearance wise, the freeze dried samples below 20 mM looked fluffy while those at 20 mM appeared more fibrous (Figure 2). All the lyophilized samples shrunk slightly from the side walls of container, with the most apparent shrinkage observed at 5 mM. Furthermore, cake heights of samples were also lower (0.6 cm) at 5 mM compared to higher lipid concentrations (10, 15 and 20 mM; 0.8 to 0.9 cm). Despite these differences, all the freeze dried samples dissolved upon reconstitution with sterile water to form clear, colorless solutions.
Figure 2.

Freeze dried cakes of SSM at (a) 5 mM, (b) 10 mM, (c) 15 mM and (d) 20 mM.
In addition to these observations, there was also a progressive trend for decreasing particle size of SSM with increasing lipid concentrations (Table 1). Therefore, it is speculated that micelle-micelle interaction occurred at 20 mM due to close proximity of neighboring micelles, resulting in folding of the PEG polymers to form the constrained “mushroom” conformation and measuring smaller particle size. At lower lipid concentrations, greater distances between SSM would allow the PEG palisade to form the relaxed ‘brush’ conformation and hence larger particle sizes were measured. Consequently, to avoid micelle-micelle interaction and minimize shrinkage of lyophilized cakes, 10 to 15 mM was found to be the optimal lipid concentration range for lyophilization of SSM. At these lipid concentrations, SSM is robust to the lyophilization conditions with elegant appearance of the resulting lyophilized cakes. For maximum loading of peptide drug per unit volume of SSM dispersion, 15 mM of DSPE-PEG2000 would be the maximum lipid concentration for the nanocarrier. However, for our study purpose, 10 mM of lipid was used for our subsequent experiments to minimize the amount of lipid and peptide drugs required. For the same reason, sample size was also reduced to 0.5 ml using Target DP™ vial (volume capacity, 1.5 ml). To ensure that the change in sample volume and size of vial would not affect the quality of lyophilized cake, lyophilization process was repeated and the resulting lyophilized products appeared similar to those as previously obtained (data not shown).
3.2 Lyophilization of SSM containing model peptides
The lyophilized cakes of SSM containing VIP, GLP-1 or GIP (each, n=3) looked similar to that of empty SSM (Figure 3), with the same time required for complete dissolution upon reconstitution (∼2 min). Moreover, the particle sizes of peptide associated SSM were comparable to that of empty SSM (10 mM) (Table 1) and did not change significantly pre and post lyophilization (Table 2). Therefore, the addition of peptides to SSM did not affect the freeze drying ability of the micellar formulation.
Figure 3.

Freeze dried cakes of peptide (67μM) associated SSM (10 mM) for (a) VIP, (b) GLP-1 and (d) GIP.
Table 2.
Particle size of SSM containing VIP, GLP-1 and GIP before and after freeze drying.
| Particle size (nm)* | ||
|---|---|---|
| Peptideb | before FDa | after FDa |
| VIP | 12.6±2.9 | 12.8±2.8 |
| GLP-1 | 12.8±2.7 | 13.2±2.4 |
| GIP | 12.5±3.6 | 12.8±2.8 |
For all peptides, comparison before and after FD were not statistically significant with p > 0.05.
FD, freeze drying
peptide (67 μM) - SSM at lipid concentration of 10 mM
For the associated peptides, their fluorescence spectra showed similar magnitude of Emmax and the corresponding peak wavelengths before and after freeze drying (Figure 4). The intrinsic fluorescence intensity of peptides increases in the presence of SSM compared to peptides in saline. This phenomenon occurred due to changes in the environment of the peptide fluorophores from a hydrophilic environment in saline to a relatively more hydrophobic surrounding in the nanomicelle palisade, demonstrating the presence of spontaneous association of peptides to nanomicelles. Since there were no significant changes in the fluorescence Emmax and the peak wavelengths of the associated peptides after lyophilization, it would indicate the persistency of peptide interaction at the same location of SSM even after relatively harsh treatment of freeze drying. Consequently, it was concluded that the extent of peptide-micelle interaction was not significantly affected by the lyophilization process. It should be noted that the Emmax of each model peptide differs due to the differences in the type and number of fluorophores interacting with SSM and is hence not comparable between each other. VIP fluorophore is tyrosine which has lower quantum yield and peak wavelength than tryptophan, the dominant fluorophores in GIP and GLP-1. Although GIP molecule has two tryptophan residues compared to one in GLP-1, it had lower Emmax at the same peptide concentration due to fewer molecules associating with each nanomicelle (Lim et al., 2007).
Figure 4.
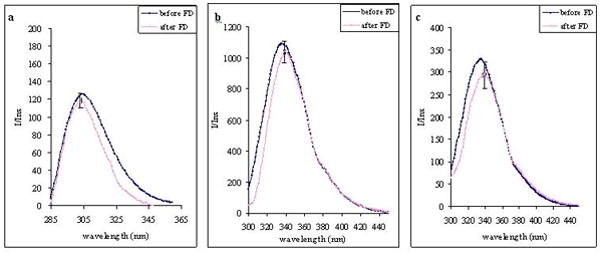
Fluorescence spectra of (a) VIP, (b) GLP-1 and (c) GIP (each, 67 uM) in association with SSM (10 mM) before and after freeze drying.
With respect to the peptide conformation, the percent α-helicity of VIP (before FD, 38.0±6.2; after FD, 37.5±2.9), GLP-1 (before FD, 32.6±7.4; after FD, 31.8±8.0) and GIP (before FD, 25.5±6.8; after FD, 16.8±7.4) in association with SSM did not differ significantly pre and post lyophilization (Figure 5). Representative CD spectra of each peptide (in the presence of SSM) before and after freeze drying are shown in Figure 6. We had previously shown that for amphipathic peptides that have the propensity to form helical structures in a hydrophobic environment, their α-helicity increased when associated with SSM compared to their respective random conformations in saline (Krishnadas et al., 2003). Since our lyophilization process did not induce significant changes to the secondary structure of the model peptides in the presence of SSM, these data confirm our earlier deduction that peptides remained associated with SSM after being lyophilized and reconstituted.
Figure 5.
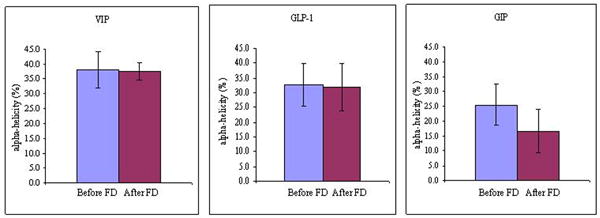
Percent α-helix of VIP, GLP-1 and GIP (each, 67 uM) in association with SSM (10 mM) before and after freeze drying (FD).
Figure 6.
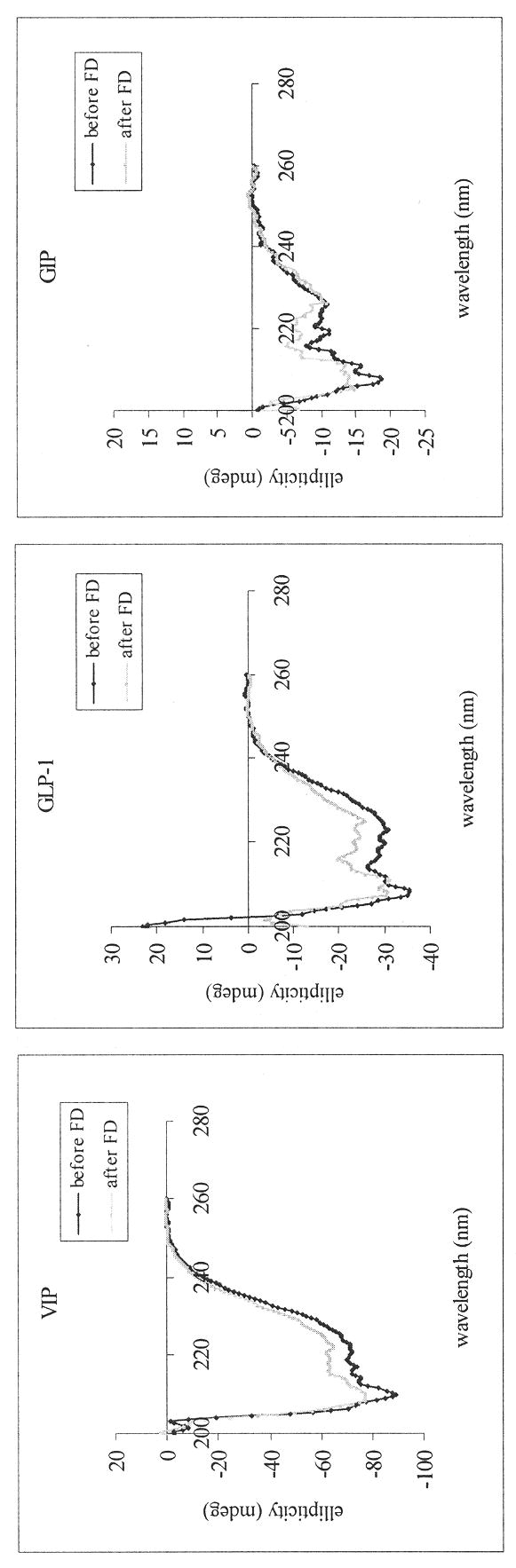
Representative CD spectra of VIP, GLP-1 and GIP (each, 67 uM) in association with SSM (10 mM) before and after freeze drying (FD).
Given that there were no significant differences in the properties of peptide self-associated with SSM before and after lyophilization, the freeze drying process did not adversely affect the integrity of the formulations. Accordingly, we propose this process could be utilized to prepare self-associated peptide drugs-SSM formulations.
4. Conclusions
We found that optimal DSPE-PEG2000 concentration for SSM lyophilization required to obtain reproducible freeze dried cake with minimal shrinkage and avoid potential micelle-micelle interaction ranged from 10 to 15 mM. In addition, freeze drying did not elicit significant changes in SSM particle size and in secondary conformation and extent of self-associated peptide drug-SSM interaction of VIP, GLP-1, and GIP. We propose that self-associated peptide drug-SSM constructs could be successfully lyophilized to potentially increase the shelf life of these products due to likely decrease in drug and lipid degradation in the dried state.
Acknowledgments
We thank Dr. Megaridis and Ms. Tiwari for assistance with the Brookfield DVII + Pro viscometer. This study was supported, in part, by Parenteral Drug Association Research Grant, NIH grants RO1 AG024026 and R01 CA121797, and VA Merit Review. The investigation was conducted in a facility constructed with support from Research Facilities Improvement Program Grant Number C06 RR15482 from the National Center for Research Resources, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ashok B, Rubinstein I, Tsueshita T, Onyuksel H. Effects of peptide molecular mass and PEG chain length on the vasoreactivity of VIP and PACAP(1-38) in pegylated phospholipid micelles. Peptides. 2004;25(8):1253–8. doi: 10.1016/j.peptides.2004.05.013. [DOI] [PubMed] [Google Scholar]
- Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368(9548):1696–705. doi: 10.1016/S0140-6736(06)69705-5. [DOI] [PubMed] [Google Scholar]
- Gandhi S, Tsueshita T, Onyuksel H, Chandiwala R, Rubinstein I. Interactions of human secretin with sterically stabilized phospholipid micelles amplify peptide-induced vasodilation in vivo. Peptides. 2002;23(8):1433–9. doi: 10.1016/s0196-9781(02)00092-x. [DOI] [PubMed] [Google Scholar]
- Gautier JF, Fetita S, Sobngwi E, Salaun-Martin C. Biological actions of the incretins GIP and GLP-1(7-36) and therapeutic perspectives in patients with type 2 diabetes. Diabetes Metab. 2005;31(3 Pt 1):233–42. doi: 10.1016/s1262-3636(07)70190-8. [DOI] [PubMed] [Google Scholar]
- Groneberg DA, Rabe KF, Fischer A. Novel concepts of neuropeptide-based drug therapy: vasoactive intestinal polypeptide and its receptors. Eur J Pharmacol. 2006;533(13):182–94. doi: 10.1016/j.ejphar.2005.12.055. [DOI] [PubMed] [Google Scholar]
- Ishihara M, Fujita M, Obara K, Hattori H, Nakamura S, Nambu M, Kiyosawa T, Kanatani Y, Takase B, Kikuchi M, Maehara T. Controlled releases of FGF-2 and paclitaxel from chitosan hydrogels and their subsequent effects on wound repair, angiogenesis, and tumor growth. Curr Drug Deliv. 2006;3(4):351–8. doi: 10.2174/156720106778559047. [DOI] [PubMed] [Google Scholar]
- Koo OM, Rubinstein I, Onyuksel H. Camptothecin in sterically stabilized phospholipid micelles: a novel nanomedicine. Nanomedicine. 2005;1(1):77–84. doi: 10.1016/j.nano.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Krishnadas A, Onyuksel H, Rubinstein I. Interactions of VIP, secretin and PACAP(1-38) with phospholipids: a biological paradox revisited. Curr Pharm Des. 2003;9(12):1005–12. doi: 10.2174/1381612033455206. [DOI] [PubMed] [Google Scholar]
- Liang MT, Davies NM, Blanchfield JT, Toth I. Particulate systems as adjuvants and carriers for peptide and protein antigens. Curr Drug Deliv. 2006;3(4):379–88. doi: 10.2174/156720106778559029. [DOI] [PubMed] [Google Scholar]
- Lim S, Rubinstein I, Onyuksel H. Study of sterically stabilized phospholipid simple and mixed micelles as nanocarriers for peptide drugs. SFB 2007 Annual Meeting; Chicago, IL. 2007. [Google Scholar]
- Lu Y, Yang J, Sega E. Issues related to targeted delivery of proteins and peptides. AAPS J. 2006;8(3):E466–78. doi: 10.1208/aapsj080355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onyuksel H, Ikezaki H, Patel M, Gao XP, Rubinstein I. A novel formulation of VIP in sterically stabilized micelles amplifies vasodilation in vivo. Pharm Res. 1999;16:155–160. doi: 10.1023/a:1018847501985. [DOI] [PubMed] [Google Scholar]
- Onyuksel H, Sejourne F, Suzuki H, Rubinstein I. Human VIP-alpha: a long-acting, biocompatible and biodegradable peptide nanomedicine for essential hypertension. Peptides. 2006;27(9):2271–5. doi: 10.1016/j.peptides.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Menei P, Montero-Menei C, Venier MC, Benoit JP. Drug delivery into the brain using poly(lactide-co-glycolide) microspheres. Expert Opin Drug Deliv. 2005;2(2):363–76. doi: 10.1517/17425247.2.2.363. [DOI] [PubMed] [Google Scholar]
- Nauck MA, Niedereichholz U, Ettler R, Holst JJ, Orskov C, Ritzel R, Schmiegel WH. Glucagon-like peptide 1 inhibition of gastric emptying outweighs its insulinotropic effects in healthy humans. Am J Physiol. 1997;273(5 Pt 1):E981–8. doi: 10.1152/ajpendo.1997.273.5.E981. [DOI] [PubMed] [Google Scholar]
- Sethi V, Onyuksel H, Rubinstein I. Enhanced circulation half-life and reduced clearance of vasoactive intestinal peptide (VIP) loaded in sterically stabilized micelles (SSM) in mice with collage-induced arthritis (CIA) AAPS PharmaSci. 2003a;5:M1045. [Google Scholar]
- Sethi V, Onyuksel H, Rubinstein I. A novel therapy for rheumatoid arthritis using α-helix VIP. FASED 2003 conference proceedings; San Diego, CA. 2003b. p. 660. [Google Scholar]
- Sreerama N, Woody R. Poly (pro)II helices in globular proteins: identification and circular dichroic analysis. Biochemistry. 1994;33:10022–10025. doi: 10.1021/bi00199a028. [DOI] [PubMed] [Google Scholar]
- Tseushita T, Gandhi S, Onyuksel H, Rubinstein I. Phospholipids modulate the biophysical properties and vasoactivity of PACAP-(1–38) J Appl Physiol. 2002;93:1377–1383. doi: 10.1152/japplphysiol.00277.2002. [DOI] [PubMed] [Google Scholar]
- Ye M, Bhat G, Johnston KA, Tan H, Garnick M. Proprietary Rel-Ease drug delivery technology: opportunity for sustained delivery of peptides, proteins and small molecules. Expert Opin Drug Deliv. 2006;3(5):663–75. doi: 10.1517/17425247.3.5.663. [DOI] [PubMed] [Google Scholar]