

Abstract
Androgens exert significant organizational and activational effects on the nervous system and behavior. Despite the fact that female mammals generally produce low levels of androgens, relative to the male of the same species, increasing evidence suggests that androgens can exert profound effects on the normal physiology and behavior of females during fetal, neonatal, and adult stages of life. This review examines the effects of exposure to androgens at three stages of development – as an adult, during early postnatal life and as a fetus, on reproductive hormone secretions in female rats. We examine the effects of androgen exposure both as a model of neuroendocrine sexual differentiation and with respect to the role androgens play in the normal female. We then discuss the hypothesis that androgens may cause epigenetic modification of estrogen target genes in the brain. Finally we consider the clinical consequences of excess androgen exposure in women.
Introduction
Female mammals generally produce low levels of androgens, relative to the male of the same species, and therefore relatively little attention is usually given to the study of androgen effects on the female reproductive axis. There are, however, compelling reasons to study the impact of androgen actions on female reproductive hormone secretions – their effects may have physiological significance, clinical importance, and relevance to the study of sex differences in neuroendocrine function.
Several studies have implicated circulating androgens as physiological regulators of the neuroendocrine mechanisms governing ovulatory cyclicity. In female rats, serum testosterone (T) and dihydrotestosterone (DHT) levels vary across the estrous cycle (Dunlap and Sridaran, 1988; Rush and Blake, 1982). These variations appear to be important in the regulation of reproductive hormone gene expression (Burger et al., 2007; Haisenleder et al., 1997; Yasin et al., 1996). A physiological role for androgens in the female reproductive axis has also been suggested by the finding that androgen receptor (AR) -deficient mice exhibit a reproductive phenotype, including premature ovarian failure (Shiina et al., 2006).
The pathophysiological actions of androgens in females are also of major importance, particularly as they relate to hyperandrogenic disorders in women. Hyperandrogenemia is a core feature of common reproductive and metabolic disorders such as polycystic ovarian syndrome (PCOS). This disorder is often accompanied by anovulation, oligo- or amenorrhea, polycystic ovaries, and hirsuitism, as well as metabolic traits such as obesity, hyperinsulinemia, and insulin resistance (Dunaif and Thomas, 2001; Rosenfield, 1997). Evidence from both clinical and animal studies suggests that PCOS has a developmental origin, in which androgen excess during fetal or prepubertal life can reprogram multiple tissues to manifest the syndrome in adolescence and adulthood (Abbott et al., 2005; Xita and Tsatsoulis, 2006). Recent evidence also suggests that hyperandrogenemia and AR activation in adult PCOS patients may sustain the reproductive features of the syndrome during the adult reproductive lifespan (Eagleson et al., 2000). Despite the obvious clinical importance of hyperandrogenism in women, the molecular and cellular mechanisms that mediate the pathophysiological effects of androgens remain distressingly unclear.
Androgen actions in the female nervous system have also been examined as a means to study the origins of sex differences in brain structure and function. Although recent genetic studies have revealed a direct role for sex chromosomes in sexual differentiation of the brain prior to the expression of sex steroids (Carruth et al., 2002), classical studies demonstrate that the exposure to testosterone in utero and in the early postpartum period is an essential component of the sexual differentiation process. In the male fetus, the nascent testes release a surge of T that functions to masculinize, as well as defeminize developing reproductive tissues, including brain areas such as the preoptic area and hypothalamus (Becu-Villalobos et al., 1997; Gorski et al., 1978; Simerly, 2002). Masculinization refers to the enhancement of the anatomical, behavioral, and endocrine characteristics that are exhibited either exclusively or to a greater extent by males than females. These characteristics are dependent on the presence of androgens during development and the associated behaviors (e.g., courtship and copulatory behavior) require steroids for their activation in adults (Becu-Villalobos et al., 1997; Kudwa et al., 2006). A separate process, defeminization, reduces the ability of males to exhibit female-typical anatomical, behavioral, or endocrine characteristics (Becu-Villalobos et al., 1997; Kudwa et al., 2006). In rats, one example of defeminization resulting from early exposure to androgens is the loss of the positive feedback response to estrogen by the hypothalamus and pituitary necessary to induce the LH-surge required to stimulate ovulation. In the female fetus, the absence of prenatal androgen exposure permits feminization of target tissues, including the expression of the positive feedback response.
Classical and recent studies have employed a variety of experimental paradigms to study sexual differentiation of the brain, including the prenatal or neonatal exposure of female rats to T (Becker et al., 2005). These studies are typically performed to characterize the consequences of T actions that would presumably occur in the male in response to endogenous T secretion. Additionally, the studies inform our understanding of the pathophysiology of androgen excess in females.
The actions of androgens are thus important in understanding normal physiology, pathophysiology, and development of female reproductive systems. This review focuses on the effects of androgens on reproductive hormone secretions in female rats, and considers their implications in all three of these domains. More specifically, we address the effects of androgens on the “master regulator” of the reproductive axis, the neurohormone gonadotropin-releasing hormone (GnRH), and gonadotropin secretions that are in turn governed by GnRH release. The physiological role of endogenous androgens and the pathophysiological effects of androgen excess at different stages of development – adult, neonatal, and fetal – are examined with respect to their impact on GnRH and LH secretion, and fertility, in the female rat. We pay particular attention to the mechanisms by which androgens may “program” permanent disruptions of gonadotropin secretion. In so doing, we begin to explore the hypothesis that androgens render animals acyclic by epigenetically programming refractoriness of the brain to the effects of estrogen, specifically conferring resistance to the ability of estrogen to induce the expression of an important downstream mediator of its actions – the nuclear progesterone receptors (PRs), PRA and PRB.
Cellular Actions of Androgens in Females
There is little evidence to suggest that the basic signaling pathways that mediate androgen actions are qualitatively different in males and females. In both the male and female brain, the actions of the major gonadal androgen, T, are exerted through three principle routes: (a) activation by T of the intracellular AR, (b) conversion of T to DHT by the enzyme 5α-reductase and subsequent activation of the AR by DHT, and (c) conversion of T to estrogen by the enzyme aromatase and subsequent activation of the intracellular estrogen receptor (ER). The latter action may be mediated by either or both of two different ER isoforms, ERα and ERβ. Both ARs and ERs primarily function as ligand-induced transcription factors. When bound by ligand, they are capable of binding hormone response elements in the promoter regions of target genes, where they recruit co-activators and co-repressor proteins to a DNA-binding complex that regulates the transcription of those genes. Numerous “non-classical” signaling mechanisms have also been characterized, in which activated receptors exert their genomic actions indirectly, through protein-protein interactions with other transcription factors that in turn bind DNA response elements (Glidewell-Kenney et al., 2007; Jakacka et al., 2002). Receptors localized to the plasma membrane have also been shown to convey signals to the interior of the cell, culminating in both genomic and non-genomic effects (Ronnekleiv and Kelly, 2005).
In a given target cell, nevertheless, the relative degree of signaling through each of the three pathways may vary greatly between males and females, usually as a function of the concentration of androgen present. On several days during development male and female rats have similar whole-body concentrations of androgens (Baum et al., 1991), however, there are distinct periods when young males experience acute surges of androgens which are critical to the organizational role of steroids on the masculinization and defeminization of the neuroanatomy (Gorski, 1985; Gorski et al., 1978), behavior (Hart, 1968; Larsson, 1966), and endocrinology of male rats (Barraclough, 1961; Barraclough and Haller, 1970). The first of these surges occurs prenatally between gestational days 16–19 (Baum et al., 1991; Weisz and Ward, 1980). The second occurs 1–3h postpartum (Baum et al., 1988; Slob et al., 1980). Testosterone produced by the testes of the male fetuses can enter the uterine circulation or diffuse into the uterine fluid to influence the development of neighboring female fetuses as can exogenous androgens (Ryan and Vandenbergh, 2002). Following birth testosterone levels of female rats are low, equivalent to those of adult females, while those of males are elevated, but highly variable, from day 1 to day 19 before subsiding to the low levels expected of a prepubertal animal (Dohler and Wuttke, 1975). Following puberty, testosterone levels remain 2–10 fold higher in the male than female throughout the remainder of life (Dohler and Wuttke, 1975), exerting activational androgenic effects on neuroendocrine systems. In both male and female rats, the majority of T production occurs in the gonads, with an additional fraction of T secretion by the adrenal cortex.
The actions of androgens may also differ according to the level of AR or ER expression, the expression level or bioactivity of 5α-reductase and aromatase, the compliment of co-activators and co-repressors present in a given cell type, and the pattern of interacting and convergent signaling by other regulators in target cells. Any of these variables may be sexually differentiated, and thereby yield different cellular responses to androgens in the female versus the male neuron, glial cell, gonadotrope, or other relevant cell type. For example, expression levels of AR mRNA in the medial preoptic area (mPOA) and bed nucleus of the stria terminalis increase with age in both sexes. However, they are significantly greater in the male than in the respective female brain nuclei (McAbee and DonCarlos, 1998). The expression of 5α-reductase type 2 mRNA is also higher in males than in females, particularly in the neonate (Colciago et al., 2005). Aromatase expression exhibits two male-specific peaks, just before and after birth (Colciago et al., 2005). The steroid receptor co-activators SRC-1 and cAMP-response element binding protein (CBP) are similarly expressed at higher levels in the male neonate (Auger et al., 2002; Bousios et al., 2001). These differences in receptor, metabolizing enzyme, and transcription factor expression may occur both as a consequence of the perinatal androgen surges, and as a mechanism that mediates amplification of androgen signaling in the male hypothalamus during these developmental periods.
The potency and magnitude of androgen signaling is thus greater in the male than in the female hypothalamus, owing to differences in concentrations of ligand as well as metabolizing enzymes, receptors and transcriptional co-activators. These molecules are nevertheless present, albeit at lower levels, in female tissues throughout prenatal, neonatal, and adult life, and may mediate some physiological actions of androgens in females. Furthermore, it is very likely that they are among the mediators of the pathophysiological consequences of androgen excess.
Pathophysiological Actions of Androgens in Females
Exogenous androgen treatments have long been known to exert profound effects on fertility in female mammals, depending upon the dose, route of administration, and the timing of the androgen exposure. There are undoubtedly androgenic actions at every level of the female reproductive axis, making the job of identifying direct and indirect effects on specific tissues a challenging one. For example, androgens may alter GnRH release, and thereby effect gonadotropin and steroid hormone secretion; they may alter pituitary responsiveness to GnRH, and thereby alter gonadotropin secretions and ovarian physiology; they may disrupt ovarian function directly, producing changes in ovarian cyclicity and steroid output; additionally, androgens may change the responsiveness of the female hypothalamus and pituitary gland to the feedback actions of ovarian steroids, leading to deregulated GnRH and LH secretions. A given exposure of a female animal to androgen may evoke one, some, or all of these types of these actions, making it difficult to identify cause and effect in androgenic disruption of the hypothalamic-pituitary-ovarian axis.
Androgens can also exert both organizational and activational effects on the female reproductive neuroendocrine system, depending upon the duration and developmental stage at which the exposure occurs. The organizational actions of androgens are those that alter the development of the GnRH and LH secretory control systems, thereby programming the operating characteristics that they will exhibit in adulthood. Androgens are known to manifest organizational effects on neuroendocrine systems during fetal life in primates and other precocial species, and during late prenatal and early postnatal life in altricial animals, such as rats and mice. Thus, androgen treatments may evoke changes in GnRH, LH, and steroid secretions that differ in magnitude, direction, and duration, depending upon whether they are given during the prenatal, perinatal, prepubertal, or adult periods. We consider the effects of androgen excess during each of these life stages on the two major features of GnRH and gonadotropin secretions: basal pulsatile release, and preovulatory GnRH and gonadotropin surges. The neuroendocrine processes underlying these two different modes of secretion are first described below.
Androgen Excess in the Adult: Effects on Pulsatile GnRH Release
Endogenous or exogenous androgens may impact the adult female reproductive axis by altering the basal production and pulsatile release of GnRH. The GnRH decapeptide is synthesized in preoptic and periventricular neurons, transported intraneuronally to neurovascular junctions in the median eminence of the hypothalamus, and released into the hypothalamic-hypophysial portal vasculature (Chappell et al., 1997). Following transport to the anterior pituitary, GnRH binds to its G-protein coupled receptors in the plasma membranes of gonadotropes, activates associated signal-transduction pathways and thereby stimulates synthesis and secretion of LH and FSH. Neurosecretion of GnRH is almost invariably intermittent, consisting of regular pulses occurring at intervals as short as 20 min, or as long as 3h, depending upon species and physiological circumstance. The pulsatile release pattern, moreover, is obligatory for sustaining normal gonadotropin secretion and synthesis, and is thus considered a critical feature of the cascade of hormone secretions that constitute the reproductive axis. Neurons and processes that function to release GnRH in this rhythmic manner are collectively referred to as the “GnRH pulse generator” (Knobil, 1981). The hypothalamic GnRH pulse generator thus sustains the operation of the reproductive axis: pulsatile GnRH release stimulates pulsatile LH and FSH secretion, which in turn stimulate secretion of the ovarian steroid hormones, estrogen and progesterone, and of protein hormones such as inhibin. The ovarian hormones, in turn, exert feedback regulatory actions on GnRH release, and on the responsiveness of the pituitary gland to GnRH stimulation.
There is ample evidence that endogenous androgens exert homeostatic negative feedback actions on GnRH release in male animals. Thus, removal of the testes results in an acceleration of GnRH pulsatility (Caraty and Locatelli, 1988) and T treatments prevent or reverse this effect (Levine and Duffy, 1988; Meredith and Levine, 1992). The effects of T on GnRH pulsatility have not been similarly assessed in females. Nevertheless, there is circumstantial evidence to suggest that androgens may exert similar effects in the female hypothalamus. For instance, ARs and high-affinity binding sites are present in the female hypothalamus (McAbee and DonCarlos, 1998; Roselli et al., 1989), and in vivo treatments of female rats with T can inhibit LH secretion to an extent that may not be fully accounted for by alterations in pituitary sensitivity to GnRH (Hassani et al., 1978; Roos et al., 1980; Turgeon and Waring, 1999).
If indeed androgens can act to modulate basal GnRH release in the female, this could conceivably occur within the GnRH neuron itself, in neurons or glial cells that constitute the microcircuitries that control GnRH release, or via both mechanisms. There remains considerable disagreement about the capacity of GnRH neurons in either sex to express any of the gonadal steroid receptors, and thus the ability of estrogens, progestins, and androgens to exert direct effects on GnRH neurons has likewise remained controversial. Although early studies found GnRH neurons to be largely devoid of gonadal steroid receptor expression, more recent analyses have suggested that subsets of GnRH neurons express ERβ during development (Temple et al., 2004) and in adulthood (Herbison, 1995; Hrabovszky et al., 2000; Skynner et al., 1999). Moreover, some immortalized GnRH-producing cell lines have been shown to express both ER isoforms and to possess specific high-affinity ER binding sites (Navarro et al., 2003; Poletti et al., 1994; Roy et al., 1999). Expression of ARs has also been demonstrated in GT1–7 cells (Belsham et al., 1998) where they can mediate both genomic and non-genomic effects on GnRH release and gene expression (Shakil et al., 2002). Interestingly, both T and DHT were found to exert inhibitory effects on forskolin-stimulated cAMP accumulation, intracellular Ca++ mobilization, and GnRH release, while both androgens also suppressed GnRH gene expression. The complex physiological roles played by steroid receptors in GnRH neurons may be resolved in the near future using conditional gene targeting methods to specifically eliminate the genes encoding ER, PR, and ARs in GnRH neurons (Gaveriaux-Ruff and Kieffer, 2007; Yeh et al., 2002). At the present time, however, it remains unclear whether androgens, or estrogens derived from the aromatization of androgens, exert any physiological or pathophysiological actions in males or females by activating ARs and/or ERs expressed in GnRH neurons.
There are several afferent neuronal groups that may function as androgen sensitive circuitries communicating androgenic stimuli to GnRH neurons. Subsets of GABAergic, glutaminergic, opiatergic, noradrenergic, dopaminergic, neuropeptide Y (NPY)-ergic and kisspeptin-producing neurons have been shown to express ARs and/or ERs, and to release neurotransmitters that exert neuromodulatory actions on GnRH neurosecretion (Brann et al., 1992; Petersen et al., 2003; Smith et al., 2005b; Smith et al., 2006; Smith and Jennes, 2001; Sullivan and Moenter, 2004). In large part, the capacity to express ARs and ERs in most of these cell groups is overlapping (Herbison, 1995; Simerly et al., 1990), although the magnitude of expression of either receptor can differ markedly between the sexes and according to developmental and physiological circumstances. It remains virtually unknown, nevertheless, if any of these afferent neuronal groups mediate the inhibitory or stimulatory effects of androgens on basal GnRH neurosecretion in adult female animals.
Androgen Excess in the Adult: Effects on Basal Gonadotropin Secretions
There is far more evidence that androgens can modulate gonadotropin synthesis and secretion by direct actions within the gonadotrope. The preponderance of studies have focused on the effects of androgens on LH secretion in the male, specifically as they relate to physiological negative feedback actions of T within the hypothalamic-pituitary-gonadal axis. Both T and DHT have thus been shown to suppress GnRH-induced LH secretion in vivo (Jackson et al., 1991) and in vitro (Winters et al., 1992), most likely via ARs and possibly ERs expressed within the gonadotrope (Clay et al., 1993; McGinnis et al., 1983; Pelletier et al., 2000). The suppression of GnRH-induced LH and FSH secretion is associated with T-induced reductions in glycoprotein subunit α, LHβ subunit, and FSHβ subunit gene expression. Androgens may additionally inhibit GnRH-induced gonadotropin secretion by modulating GnRH receptor number (Giguere et al., 1981), arachidonic acid release (Kamel and Kubajak, 1988), Ca++ mobilization (Kamel and Krey, 1983), PKC activation (Kamel and Kubajak, 1988), and other GnRH-dependent signaling events. Paradoxically, T also appears to exert stimulatory effects on FSHβ gene expression and FSH secretion that are independent of GnRH stimulation (Burger et al., 2007; Winters et al., 1992), however, the physiological significance of these actions is not clear.
Androgen treatments in female rats, specifically, treatments that sustain male-typical serum androgen levels, suppress gonadotropin secretion similar to effects observed in the male, suggesting that similar mechanisms mediate androgen effects in the two sexes. Thus, androgens readily suppress LH secretion in adult females in vivo (Foecking and Levine, 2005) and in vitro (Turgeon and Waring, 1999), while they are either without effect (Wierman et al., 1988; Wierman et al., 1990) or stimulatory on FSH secretion (Drouin and Labrie, 1976; Kamel and Kubajak, 1987; Winters et al., 1992). We recently confirmed that T can exert these effects in vivo, while also comparing the relative abilities of T and DHT to modulate LH and FSH secretion (Foecking and Levine, 2005). Treatments consisted of implanting subcutaneous silastic capsules containing vehicle, T or DHT for four days. These treatments have previously been shown to sustain circulating androgens at levels equivalent to those present in adult male rats (Urban et al., 1996). We found that compared to controls, both steroids suppressed LH secretion (Figure 1) regardless of the presumptive estrous cycle stage at which the animals were sampled (Foecking, unpublished). In addition, both androgens dramatically suppressed LH secretion in ovariectomized animals (Figure 2) (Foecking, unpublished). These data confirm previous reports which suggest that AR is involved in mediating the suppression of LH secretion by androgens in females (Foecking and Levine, 2005; Turgeon and Waring, 1999).
Figure 1. Serum LH levels in female rats treated with androgens in adulthood.
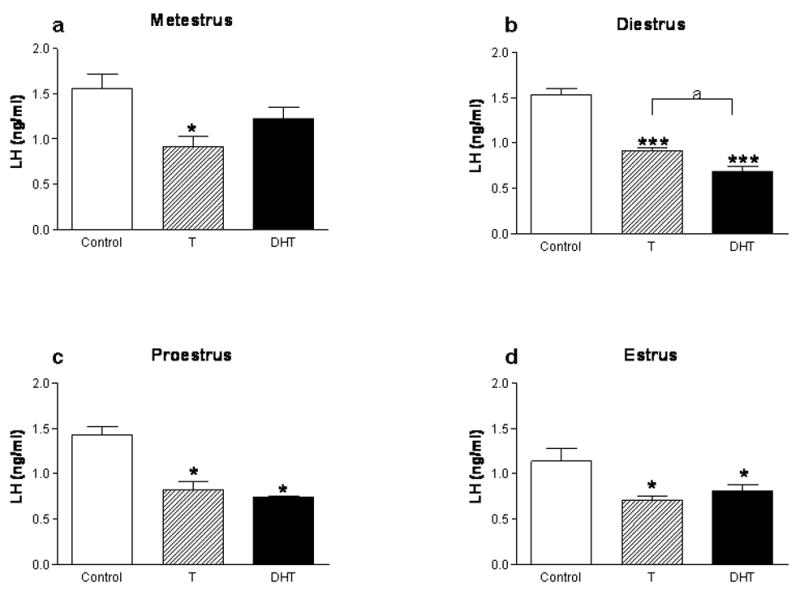
Cycling female rats were implanted with either empty Silastic capsules or capsules filled with either crystalline T, or DHT on metestrus, diestrus, proestrus or estrus. After four consecutive days of treatment, animals were sacrificed on presumptive metestrus (a), diestrus (b), proestrus (c), or estrus (d), respectively (n=5 for all groups). Trunk blood was collected and serum LH levels were determined by RIA (*p<0.05, ***p<0.001, as compared to the Control group). The data are represented as mean ± SEM.
Figure 2. Androgen treatment in adulthood suppresses serum LH levels in ovariectomized rats.
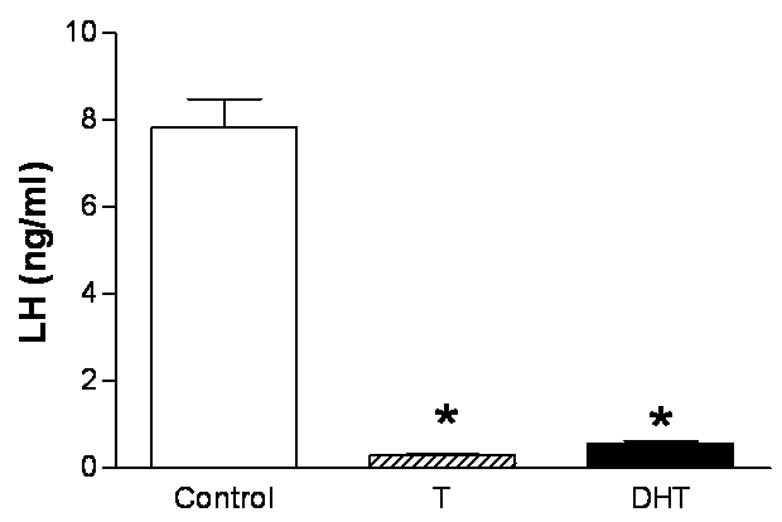
Ovariectomized female rats were implanted with either empty Silastic capsules or capsules filled with either crystalline T, or DHT (n=5 for all groups). Seven days later, trunk blood was collected and serum LH levels were determined by RIA. Animals treated with T and DHT showed a significant reduction (p<0.001) serum LH levels compared to oil-treated controls.
In agreement with previous studies (Drouin and Labrie, 1976; Kamel and Kubajak, 1987; Winters et al., 1992), we also found that neither androgen treatment produced a concomitant suppression of FSH secretion, but instead both augmented FSH secretion on the presumptive day of proestrous (Figure 3). The observation that both T and DHT can stimulate FSH secretion is consistent with recent findings that T, but not estrogen, increased expression of the primary transcript of FSHβ and increased phosphorylation of SMAD2, a signaling protein that mediates the stimulatory effects of activin on FSHβ expression (Burger et al., 2007).
Figure 3. Serum FSH levels in female rats treated with androgens in adulthood.
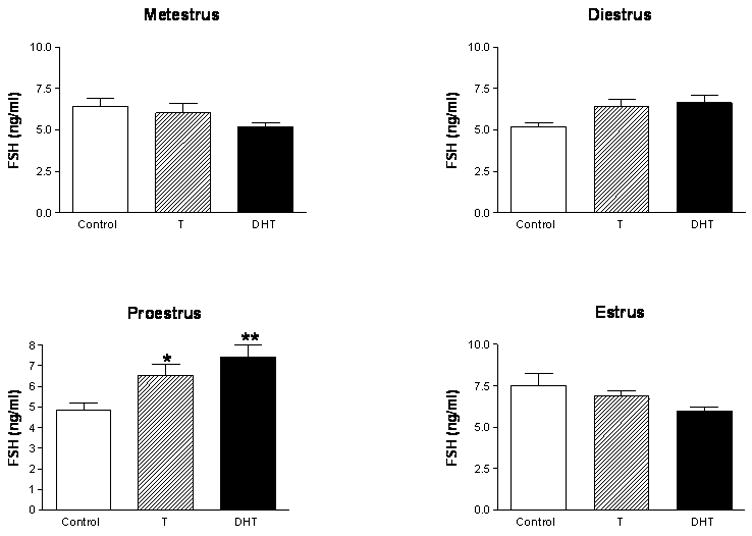
Cycling female rats were implanted with either empty Silastic capsules or capsules filled with either crystalline T, or DHT on metestrus, diestrus, proestrus or estrus. After four consecutive days of treatment, animals were sacrificed on presumptive metestrus (a), diestrus (b), proestrus (c), or estrus (d), respectively (n=5 for all groups). Trunk blood was collected and serum FSH levels were determined by RIA (*p<0.05, **p<0.01, as compared to the Control group). The data are represented as mean ± SEM.
Endogenous Androgens: Sculpting the Proestrous LH Surge
It is thus clear that exogenous androgen treatments can exert robust suppressive effects on basal LH secretion, while stimulating FSH secretion, by direct actions on the female pituitary gland. The actions of endogenous androgens in females, however, may be qualitatively different from those produced by exogenous T and DHT, especially when the latter produce male-typical levels of the androgens. In female rats, endogenous serum T and DHT concentrations are generally less than 30% of those in males (Dohler and Wuttke, 1975; Pang et al., 1979; Rhoda et al., 1984) and vary across the estrous cycle, reaching peak levels on the afternoon of proestrus and a nadir on the morning of estrous (Dunlap and Sridaran, 1988; Rush and Blake, 1982). As already noted, when exogenous T and DHT are administered at doses that elevate androgens to high physiological or supraphysiological (i.e. male-typical) levels, both androgens suppress GnRH-induced LH secretion; however, maintenance of T at levels typical for the female (0.42 ng/ml), results in an enhancement of GnRH-induced LHβ subunit gene expression (Turgeon and Waring, 1999). It has been suggested that the ability of androgens to facilitate LH release at lower concentrations, together with their inhibitory actions at higher concentrations, may function to shape the preovulatory gonadotropin surge that occurs on proestrus (Turgeon and Waring, 1999). True androgen excess, however, may produce only suppressive effects on release of preovulatory LH surges, as well as the preovulatory GnRH surges that stimulate them, as addressed in the next section.
Androgen Excess in the Adult: Effects on Preovulatory Gonadotropin Surges
The frequencies and amplitudes of pulsatile GnRH, LH and FSH secretions during the ovulatory cycle of the rat are maintained at basal levels on estrus, metestrus, diestrus, and the morning of proestrus, primarily through the negative feedback actions of the ovarian steroids, estrogen and progesterone, and of protein hormones, such as inhibin. Superimposed upon this basal secretory pattern is the release of preovulatory gonadotropin surges on the afternoon of proestrus. Numerous studies have examined the acute effects of androgens on basal gonadotropin secretion, yet few have specifically assessed their effects on release of preovulatory GnRH and gonadotropin surges. We recently revisited the effects of short-term androgen exposure on the release of gonadotropin surges in female rats (Foecking and Levine, 2005), and found that treatment with androgen for four days completely blocked their release. Both T and DHT were effective in preventing release of LH surges in ovary-intact and ovariectomized, estrogen-treated animals (Figure 4) (Foecking and Levine, 2005). Thus, inhibitory actions of T appear to be at least partially mediated by androgen receptors expressed in the hypothalamic neurons responsible for the stimulation of GnRH surges and/or pituitary gonadotropes.
Figure 4. Androgen treatment in adulthood blocks the EB-induced LH surge.
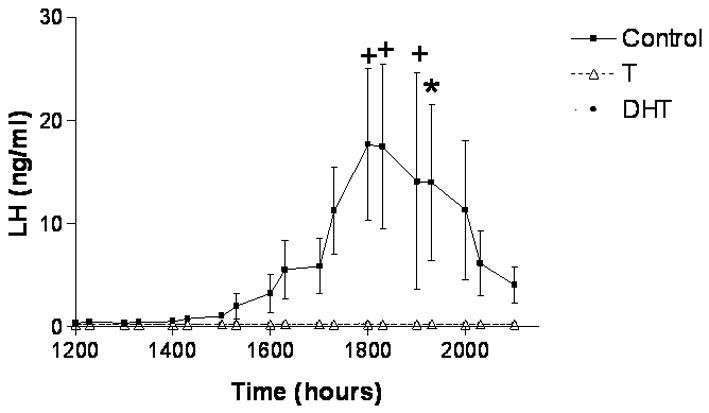
Adult female rats were implanted with either empty Silastic capsules or capsules filled with either crystalline T, or DHT. Three days later on diestrus, at 0900h, the female rats were ovariectomized (OVX) and treated with estradiol benzoate (EB; 30 μg sc). At the same time, the rats were fitted with jugular catheters. The following day, blood samples were collected every 30 minutes from 1200h to 2100h to assess the release of the LH.. Statistically significant LH surges were detected in control females but not in T- or DHT-treated females (n=4 per group). The data are represented as mean ± SEM. (From Foecking and Levine, 2005).
The preovulatory gonadotropin surges are evoked by the positive feedback actions of estrogen. Under the influence of a rising tide of estrogen secreted by the ripening ovarian follicle(s), an abrupt surge of GnRH occurs and both gonadotropins (FSH and LH) are released on the afternoon of proestrus; this in turn, triggers ovulation on the following morning of estrus. A prolonged, secondary phase of FSH secretion continues throughout the morning of estrus and most likely functions to recruit ovarian follicles for the subsequent cycle. Ovarian progesterone released just before or during the surge greatly amplifies the surge and prevents their recurrence during the same period on the next day. It is generally held that there are two critically important, steroid-dependent processes that together mediate the generation of the gonadotropin surge: 1) hypothalamic stimulation of the coordinated preovulatory release of GnRH and 2) the simultaneous increase in pituitary responsiveness to the release of GnRH.
The preovulatory release of GnRH consists of a 2–4h increase in the overall amount of GnRH secreted, occurring between 1600–2000h on the afternoon of proestrus (Levine and Ramirez, 1982). Classical neuroendocrine studies demonstrated that the neuroendocrine mechanisms that mediate the release of a GnRH surge require the integration of two obligatory signals – the preovulatory estrogen surge, and a daily neural signal that is synaptically conveyed from the circadian clock resident in the suprachiasmatic nucleus (Levine, 1997). The major action of estrogen is to “couple” the daily neural signal to the neuronal circuitries that mediate release of GnRH surges (Legan et al., 1975; Legan and Karsch, 1975). In the absence of a sufficient elevation of estrogen, the daily signal is not communicated to neurons controlling the release of GnRH, and no surge takes place; with exposure of the hypothalamus to a sufficient estrogen stimulus, the pathways that convey the daily neural signal for the surge are rendered patent, and the GnRH surge is released into the hypophysial portal vasculature. At the same time, estrogen greatly enhances the responsiveness of the gonadotropes to this GnRH surge (Bauer-Dantoin et al., 1995; Drouva et al., 1983; Shupnik, 1996; Taga et al., 1982). Both of these processes, along with the ability of GnRH to “self-prime” the estrogen-exposed pituitary to its own actions (Fink, 1995; Kamel et al., 1987) culminate in the release of a massive, but transient surge on the afternoon of proestrus, which in turn triggers ovulation on the following morning of estrous.
How does estrogen couple the daily neuronal signal to the neurons responsible for the release of GnRH surges? One major locus of this integrative activity is the anteroventral periventricular (AVPV) nucleus of the hypothalamus, where estrogen appears to activate ERα in neurons that receive afferents from the SCN and project to GnRH neurons (Tsukahara, 2006; Van der Beek et al., 1997; Watson et al., 1995). We have examined the cellular actions of estrogen that may mediate these effects, focusing on the roles that PRs may play. Estrogen induces the expression of both isoforms of PR, PRB and the N-terminally truncated PRA, in the AVPV, as well as other hypothalamic and preoptic nuclei. We have determined that induction of PRs is obligatory for the successful release of GnRH surges; thus, GnRH and LH surges are absent in ovarian intact and estrogen-treated PR gene knockout (PRKO) mice (Chappell et al., 1997; Chappell et al., 1999), and in rats treated with a progesterone receptor antagonist (Figure 5) or intracerebroventricular PR antisense oligonucleotides (Chappell and Levine, 2000). We have proposed a model for the release of GnRH surges that includes (Levine, 1997) 1) the induction of PRs by estrogen in AVPV neurons, 2) delivery of the daily neural signal from the SCN to AVPV neurons by neurotransmitter circuitries, e.g. vasopressin or VIP-producing projections, 3) the neurotransmitter-mediated activation of intracellular second messenger production that in turn transactivates the PRs in a ligand-independent manner. It should be noted that PRs can be activated either by progesterone (ligand-dependent activation) or by neurotransmitters (e.g. dopamine) (ligand-independent activation) (Mani, 2006). Thereafter, downstream signals evoke the neurosecretion of the GnRH surge, which is further amplified by ovarian progesterone release in response to the LH surge. The net result of all of these integrated physiological events is the release of a robust LH surge that is timed to trigger ovulation in concert with behavioral heat and maximal wakefulness, thereby maximizing the chances for successful fertilization.
Figure 5. GnRH and LH surges are absent in ovariectomized female rats treated with a PR antagonist.
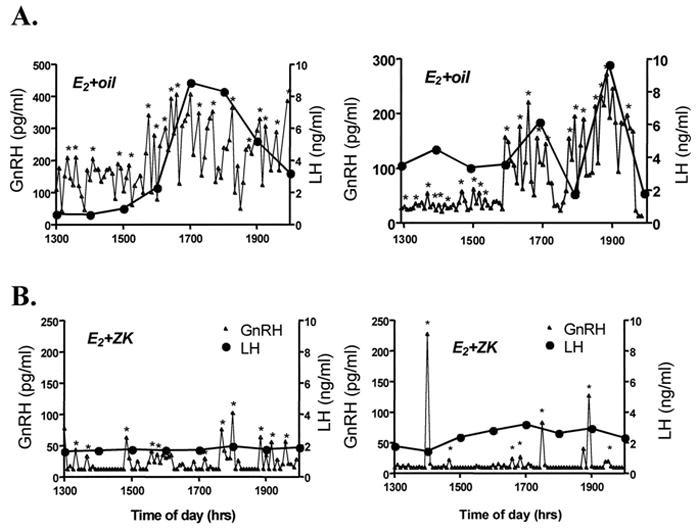
Representative GnRH and LH release profiles of OVX, E2-primed rats (A) and OVX, E2-primed rats treated with ZK98299 (B). A, E2-priming stimulated significant increases in GnRH pulse amplitude commencing at approximately 1600 h, accompanied by a concomitant 3- to 5-fold increase in plasma LH. B, Treatment of E2-primed, OVX rats with ZK98299 prevented this E2-stimulated rise in both GnRH and LH release without affecting pulse frequency. Asterisks represent significant pulses as determined by the ULTRA pulse analysis program. (From Chappell et.al., 2000).
Given the central role of estrogen-induced PRs in the integrated processes leading to release of the GnRH surge, we tested the hypothesis that the ability of androgens to suppress the release of GnRH surges results from the interference of androgens with the induction of PR expression. Consistent with this hypothesis, treatment with androgens for 4 days was found to produce a near total suppression of PR expression in adult female rats during presumptive proestrous (Figure 6) (Foecking, unpublished) and a total blockade of PR expression in preoptic-hypothalamic tissues (Figure 7) (Foecking and Levine, 2005). Similar to the blockade of the LH surge by androgens, both T and the non-aromatizable DHT were effective in blocking induction of PR, implicating the androgen receptor in these suppressive actions. Moreover, we consider it unlikely that activation of ERs could play a major role in the actions of androgens, because additional activation of ERs by estrogen derived from T would have enhanced, not suppressed the induction of PRs by the exogenous estrogen stimulus.
Figure 6. Androgen treatment in adulthood suppresses progesterone receptor mRNA expression in the hypothalamus.
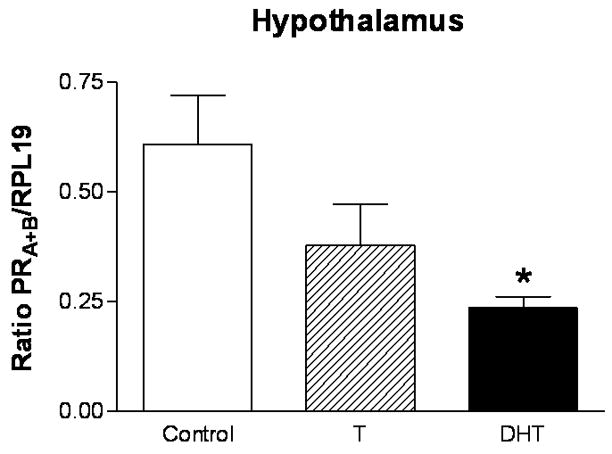
Cycling female rats were implanted with either empty Silastic capsules or capsules filled with either crystalline T, or DHT on proestrus. After four consecutive days of treatment, animals were sacrificed on presumptive proestrus. The hypothalamus was dissected out of each brain. RNA was isolated from the hypothalamus, reverse transcribed, and processed for PRA+B mRNA expression via semi-quantitative PCR. PRA+B mRNA expression is significantly decreased (*p<0.05) in the hypothalamus of DHT-treated rats compared to controls (n=5 for all groups). The semi-quantitative RT-PCR results for PRA+B are normalized to the housekeeping gene RPL19 and are represented as mean ± SEM.
Figure 7. Androgen treatment in adulthood suppresses EB-induced progesterone receptor mRNA expression in the POA-hypothalamus.
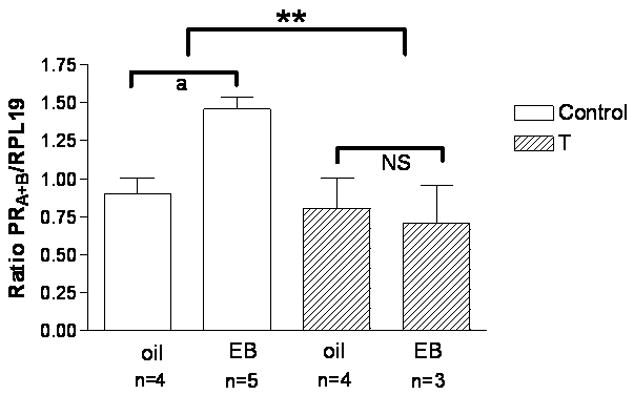
Adult female rats were either empty Silastic capsules or capsules filled with either crystalline T, or DHT for four days, ovariectomized (OVX) and treated with estradiol benzoate (EB; 30 µg sc) or sesame oil vehicle (oil). The POA-hypothalamus was dissected out of each brain. RNA was isolated from the POA-hypothalamus, reverse transcribed, and processed for PRA+B mRNA expression via semi-quantitative PCR. PRA+B mRNA expression is significantly increased in the POA-hypothalamus of EB-treated, ovariectomized control rats (n=5) compared to their oil-treated (n=4) counterparts (p=0.0026). PRA+B mRNA expression is not induced by EB in testosterone-treated females (OVX, n=4, OVX/EB, n=3). A significant interaction (**p=0.016) exists between the 4-day testosterone treatment and control group. The semi-quantitative RT-PCR results for PRA+B are normalized to the housekeeping gene RPL19 and are represented as mean ± SEM. (From Foecking and Levine, 2005).
Previous studies have also implicated PRs in the mechanism by which androgens suppress pituitary responsiveness to GnRH stimulation. Progesterone augments GnRH-induced LH secretion from pituitary cells of female rats (Attardi and Happe, 1986; Emons et al., 1992), and this effect can be attenuated by DHT (Turgeon and Waring, 1999). At least part of this action may be mediated by a suppression of progesterone-stimulated transcriptional activity within the pituitary, as we have also found that the expression of PR itself in pituitary tissues is inhibited by androgen treatments in vivo (Figure 8) (Foecking, unpublished).
Figure 8. Androgen treatment in adulthood suppresses progesterone receptor mRNA expression in the pituitary.
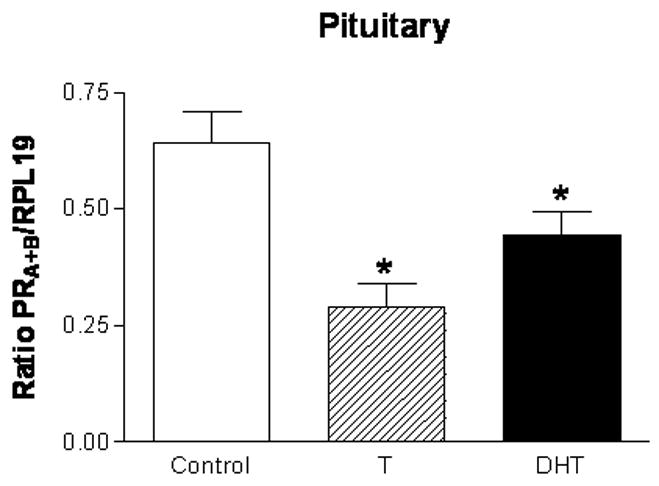
Cycling female rats were implanted with either empty Silastic capsules or capsules filled with either crystalline T, or DHT on proestrus. After four consecutive days of treatment, animals were sacrificed on presumptive proestrus and the pituitary of each animal was dissected. RNA was isolated from the pituitary, reverse transcribed, and processed for PRA+B mRNA expression via semi-quantitative PCR. PRA+B mRNA expression is significantly decreased in the pituitary of both the T- and DHT-treated rats compared to controls (n=5 for all groups). The semi-quantitative RT-PCR results for PRA+B are normalized to the housekeeping gene RPL19 and are represented as mean ± SEM.
In summary, the effects of androgens in vivo in the adult female rat appear to be uniformly inhibitory, with pronounced suppressive effects on both basal and cyclic GnRH and gonadotropin secretion (Foecking and Levine, 2005) and previously unpublished data presented here. We have obtained circumstantial evidence to suggest that these inhibitory effects are mediated in part by suppression of PR expression in the preoptic-hypothalamus and possibly also the pituitary gland (Figures 6, 7, and 8). When combined with previous findings regarding the obligatory role of PRs in the release of GnRH surges, these data make a particularly compelling case for the idea that androgens block the positive feedback actions of estrogen by attenuating estrogen-induced PR expression. However, we do not know if this attenuation results from AR mediated interference with binding of estrogen to its receptor, associated ligands, or other aspects ER-mediated transcriptional regulation at the PR promoter.
The effectiveness of DHT in exerting the foregoing effects also strongly implicates the AR in these androgen actions. It has recently been shown that DHT can be metabolized to an estrogenic metabolite, 3βAdiol, and thus some involvement of ER in mediating androgen actions remains possible (Lund et al., 2004). However, the relatively robust effects of DHT compared to T on LH levels in the female suggest that this metabolite would play little, if any role in the inhibitory effects of DHT. Moreover, only a small fraction of DHT would be subjected to metabolism via this route and therefore, it would be bioavailable for exceedingly short periods. In addition, previous studies have employed even more robust DHT treatment paradigms than those used in these studies, and have unequivocally revealed no apparent ER-mediated alterations in gonadotropin secretion, even as there were concurrent alterations via unambiguous AR activation (Lindzey et al., 1998). The importance of the AR in mediating suppressive effects on LH is well established in the male (Nansel and Trent, 1979), where DHT is highly effective in suppressing LH secretion. Most convincingly, the tfm mouse, which lacks normally functioning ARs, exhibits an elevation of LH secretion that is comparable to levels reached post-castration in WT males (Naik et al., 1984) and DHT is without effect on LH secretion in these animals. It is thus possible that the presence of androgens, specifically at male-typical levels in the female, activates the same receptor (AR) and signaling pathways as are stimulated by androgens in the male to manifest a suppression of LH secretion.
Neonatal Androgen Exposure: Effects on Basal Gonadotropin Secretions
The preponderance of studies on sex differences in hormone secretions, and the organizational effects of androgens on GnRH and LH secretions, have been performed in female rats receiving neonatal (postnatal days 1–5) androgen treatments (Barraclough, 1961; Barraclough and Gorski, 1961). Neonatal androgen treatments render female rats permanently sterile (Barraclough, 1961), exhibiting a persistent estrous syndrome in adulthood that includes anovulation, acyclic LH secretion (Goomer et al., 1977), and attenuated pituitary responsiveness to GnRH (Liaw and Barraclough, 1993b). Basal LH levels in neonatally androgenized females have been reported to be decreased (Korenbrot et al., 1975) or unchanged (Kubo et al., 1975) in the ovary-intact state. Following ovariectomy, LH levels rise at a significantly slower rate and remain chronically lower in neonatally androgenized females compared to their control counterparts (Moguilevsky et al., 1979). We have recently assessed the characteristics of LH pulsatility in long-term ovariectomized females that received neonatal testosterone or control treatments (Figure 9) (Foecking, unpublished). Our findings reveal that androgenization during this period almost completely prevents the post-ovariectomy rise in LH pulse frequency, amplitude, and mean levels in adulthood. Thus, neonatal androgen exposure appears to impair the post-ovariectomy GnRH neurosecretory process in a manner that is indistinguishable from the effects of androgen exposure in adulthood. Although neonatal androgenization does not appear to reduce the number of GnRH neurons or alter their distribution or morphology (King, 1985), it would appear to be the case that androgens can permanently alter the potency of some critically important neural input, e.g. afferent kisspeptin neurons (Kauffman et al., 2007); (Smith et al., 2005a; Smith et al., 2005b).
Figure 9. Neonatal androgen treatment blocks LH responses to ovariectomy in adulthood.
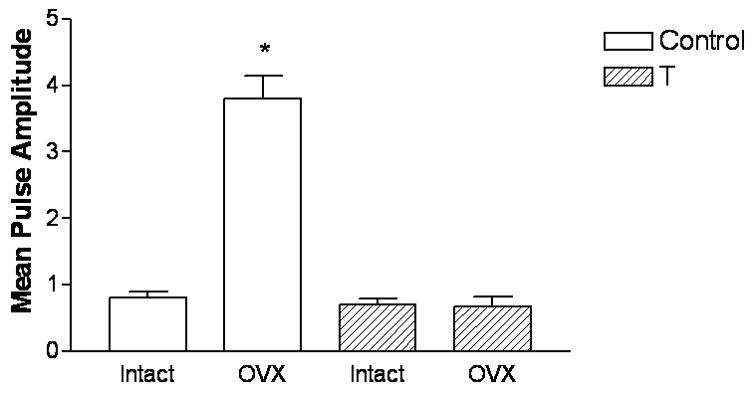
Female rat pups were subcutaneously injected with oil or 2.5 mg of testosterone propionate on the day of birth. On PND 60, rats were either ovariectomized (OVX) or sham-operated and left intact. Six days later, these rats were fitted with jugular catheters. The following day, blood samples were collected every 30 minutes for 3 hours from 1200h to 2100h. Serum LH levels were determined by RIA. Both mean LH levels (ng/ml) and mean pulse amplitude of LH was significantly increased (*p<0.001) by ovariectomy in control-treated females as compared to the SHAM control group (SHAM, n=3; OVX, n=5). Ovariectomy did not alter LH levels or pulse amplitude in the T-treated (n=5 for both groups) females.
Neonatal Androgen Exposure: Effects on Preovulatory Gonadotropin Surges
Neonatal androgen exposure also programs hormonal acyclicity in adulthood, at least in part by promoting the development of neuronal circuitries that are resistant to the positive feedback actions of estrogen (Gogan et al., 1980; Watts and Fink, 1984; Weiland and Barraclough, 1984). Classical experiments demonstrated that exposure of female rat pups to testosterone, but not DHT on postnatal days 1–10 permanently renders them incapable of releasing LH surges (Korenbrot et al., 1975). Findings such as these have given rise to the prevailing dogma regarding the establishment of sex differences in gonadotropin secretion: androgen released perinatally in males is aromatized in the AVPV and other neuronal groups, and the derived estrogen activates ERs; rates of cell proliferation, differentiation, and/or apoptosis are modulated to establish a neuronal circuitry that will not respond to estrogen in adulthood. In the absence of the perinatal androgen surge, the pathway leads to the development of an AVPV (and likely other hypothalamic areas) that is responsive to the positive feedback actions of estrogen in adulthood. It is generally believed that the treatment of female neonates with testosterone or estrogen recapitulates the events that take place in the male, in which the neurosecretory control system is structurally and functionally defeminized, and thereby incapable of supporting cyclic GnRH and gonadotropin secretion in adulthood (Robinson, 2006). Some evidence has suggested that the activation of androgen receptors may also play a role in this defeminization process, as neonatal treatment with the AR antagonist, flutamide, can sex-reverse the volume of the AVPV (Lund et al., 2000). It is possible that AR activation may serve to enhance the expression of aromatase, and thereby potentiate the local accumulation of estrogen and the activation of ERs.
There is some evidence to suggest that neonatal androgen exposure impairs the development of afferent neurons innervating the GnRH neurons that control the preovulatory LH surge. Electrochemical stimulation of the medial preoptic area (Chappel and Barraclough, 1976), as well as the icv infusion of norepinephrine (Handa et al., 1986; Liaw and Barraclough, 1993a; Liaw and Barraclough, 1993b), result in release of significantly less LH secretion in neonatally androgenized rats as compared to their control counterparts. Other studies have shown that neonatal androgenization alters the activity of afferent cell groups: noradrenergic turnover is reduced (Siddiqui and Shah, 1997) and opiatergic tone is increased (Petersen and Barraclough, 1986) in the hypothalamus of neonatally androgenized rats. Most compelling is the case for the programming of kisspeptin neuronal development by neonatal androgen exposure. The expression of kisspeptin mRNA is greater in females than in males in the AVPV, and is stimulated by estrogen treatment in the adult female (Smith et al., 2005a; Smith et al., 2005b). Neonatal androgenization of females results in the development of a male-typical (lower) kisspeptin expression pattern, and a resistance to the stimulatory effects of estrogen on kisspeptin expression (Kauffman et al., 2007). Because norepinephrine, endogenous opiates, and kisspeptin are released within the neuronal circuitries that control release of GnRH resulting in the LH surge, it is reasonable to hypothesize that neonatal androgen excess programs the development of these particular cell groups such that they fail to respond to the positive feedback actions of estrogen in adulthood.
Prenatal Androgen Exposure: Effects on Basal Gonadotropin Secretions
Past studies have examined effects of prenatal androgen exposure on ovulatory cyclicity in female rodents (Fels and Bosch, 1971; Huffman and Hendricks, 1981; Slob et al., 1983; Swanson and van der Werff ten Bosch, 1964; Swanson and van der Werff ten Bosch, 1965; Whalen and Luttge, 1971), yet remarkably few have directly assessed the effects of prenatal androgens on GnRH or gonadotropin secretion (Foecking et al., 2005; Rhees et al., 1997; Sullivan and Moenter, 2004). In these studies, pregnant dams have been administered various regimens of testosterone propionate, free testosterone, or DHT, thereby exposing the fetuses to levels of androgens that are capable of partially masculinizing the external genitalia.
One recent study demonstrated that prenatal exposure to DHT leads to elevated LH and testosterone secretion, as well as irregular estrous cyclicity in female mice (Sullivan and Moenter, 2004). These effects were associated with the development of resistance to progesterone’s effects on excitatory GABAergic inputs to GnRH neurons in hypothalamic tissue slices. The results of this study are consistent with findings in other experimental animal species, most notably sheep, and rhesus monkeys, in which prenatal androgen exposure was found to produce hypersecretion of LH and resistance to the negative feedback actions of gonadal steroids (Dumesic et al., 2002; Sarma et al., 2005; Steiner et al., 1976). One study in sheep specifically detailed the development of resistance to progesterone’s negative feedback actions in the prenatally androgenized ewe (Robinson et al., 1999). In both sheep and monkeys, critical periods of sensitivity to these specific androgenic effects were found to be present at specific early gestational periods, most likely during an important maturational stage of the GnRH pulse generating mechanism (Abbott et al., 1998; Dumesic et al., 1997; Goy et al., 1988; Masek et al., 1999; Robinson et al., 1999; Thornton, 1983). These and other studies reveal that distinct requirements exist for differentiation of basal versus cyclic reproductive hormone control systems, whereby aromatization is necessary to prevent the surge control of GnRH from operating, but nonaromatizable androgens such as DHT can differentiate the tonic control to permit high GnRH secretion earlier in life (Masek et al., 1999).
We have investigated the effects of prenatal androgen exposure on LH pulsatility in adult intact, ovariectomized, and ovariectomized, estrogen- and progesterone- treated rats. Our studies have revealed that prenatal androgen exposure does not produce an increase in basal LH levels in ovary-intact animals. However, the pulse frequency was significantly increased in ovariectomized animals exposed to T or DHT in utero (Figure 10) (Foecking et al., 2005). Mean levels of LH and the amplitude of the LH pulses were significantly increased by only DHT exposure in utero in adult rats following ovariectomy (Figure 10) (Foecking et al., 2005). We have not observed any significant alteration induced by prenatal androgens in the responsiveness to estrogen or progesterone’s negative feedback actions on LH pulsatility (unpublished observations). It is possible that the androgen treatment paradigm that we have utilized does not fully evoke the hypersecretory phenotype seen in other species, perhaps due to less-than-optimal timing, magnitude, and/or duration of the androgen exposure. In any event, the effects of prenatal androgen exposure on GnRH pulse generator activity appear to be positive, at least under open-loop feedback conditions. This suggests that androgens may program the development of a GnRH pulse generator that runs at a more rapid rate in adulthood. We suspect that this programming effect represents a partial masculinization of the pulse generating mechanism; following gonadectomy, the frequency of GnRH and LH pulsatility is increased more rapidly, and is sustained at a higher rate in the male than in the female rat (Blackwell and Amoss, 1971; Eldridge et al., 1974; Ellis et al., 1983; Hiatt et al., 1987; Leipheimer and Gallo, 1983). Although the physiological significance of this apparent sex difference is not known, it may nevertheless explain the consistent observations among several species that prenatal androgenization of the female leads to an acceleration of the GnRH pulse generator and/or resistance to the negative feedback actions of gonadal steroids.
Figure 10. Prenatal androgen exposure accelerates LH pulsatility in adulthood.
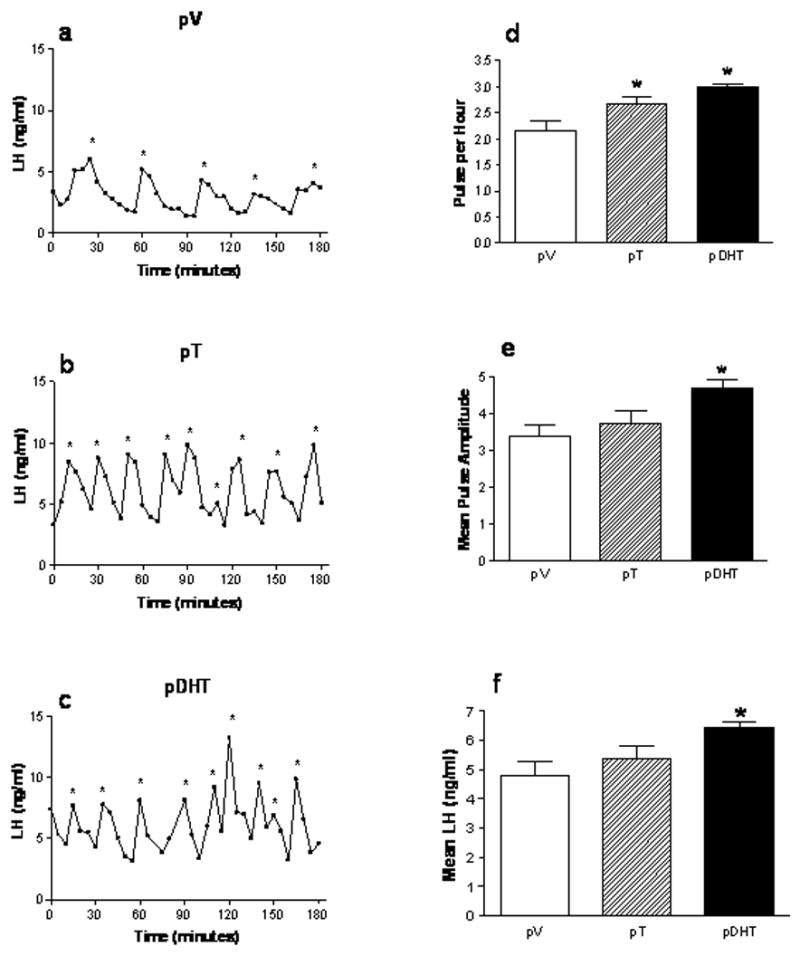
Pregnant female Sprague-Dawley rats were subcutaneously injected with oil vehicle (V), 5mg of T, or 5mg of DHT daily on embryonic days 16–19. Female offspring were ovariectomized in adulthood (approximately PND 60–70). Six days later, these rats were fitted with jugular catheters. The following day, blood samples were collected every 5 minutes for 3 hours from 1200h to 1500h. LH pulsatility is accelerated in pT (B; n=3) and pDHT (C; n=6) rats compared to pV females (A; n=6) (*significant pulse by ULTRA pulse analysis program). Prenatal exposure to DHT and T results in a significant increase (*p<0.05) in LH pulse frequency (D) compared to pV animals. Mean pulse amplitude (E) and mean LH levels (F) were significantly increased in the pDHT rats compared to pV animals. The data are represented as mean ± SEM. (From Foecking et.al., 2005)
Prenatal Androgen Exposure: Effects on Preovulatory Gonadotropin Surges
A classic study demonstrated that in utero exposure to elevated T or diethylstilbestrate (DES) from gestational day 15 onward produced a state of anovulatory infertility in female rats similar to that seen in neonatally androgenized animals (Dohler et al., 1984). Combined prenatal and postnatal androgen exposure was also found to prompt development of a sexually dimorphic nucleus (SDN) in the preoptic area in females that was comparable in size to the SDN in males (Tarttelin and Gorski, 1988). A more recent study showed that both prenatal and postnatal androgen exposures can significantly reduce the volume of the AVPV in female rats (Lund et al., 2000). Interestingly, the administration of a single dose of free T to pregnant rats on a single day of late pregnancy failed to impair the response of adult female offspring to the positive feedback actions of estrogen and progesterone (Rhees et al., 1997). Given that the former study required multiple prenatal injections to “masculinize” the volume of the AVPV, we theorized that a continued exposure of the female fetus to T, mimicking more closely the duration of exposure to endogenous T in the male, would be required to program resistance to the positive feedback actions of estrogen. We therefore examined the effects of free T or DHT injections given on gestational days 16–19 on the ability of the female adult offspring to respond to the positive feedback actions of estrogen. As shown in Figure 11, estrogen treatments evoked robust LH surges in control ovariectomized rats, but were without effect in animals prenatally exposed to T or DHT (Foecking et al., 2005). Thus, androgen exposure in utero during these gestational days renders offspring unresponsive to estrogen’s positive feedback actions, and this effect is likely an underlying reason for the hormonal acyclicity that is exhibited by prenatally androgenized, ovary-intact rats. The equal ability of T and DHT to program refractoriness to estrogen implicates the AR in mediating at least some of these effects.
Figure 11. Prenatal androgen exposure blocks the EB-induced preovulatory LH surge in adulthood.
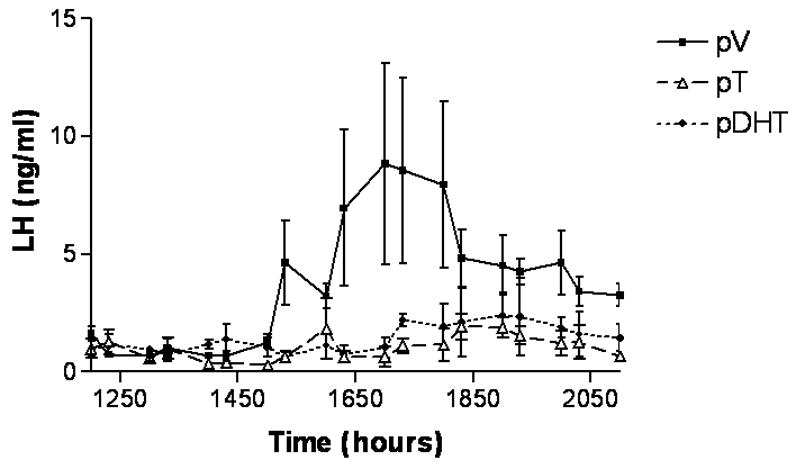
Pregnant female Sprague-Dawley rats were subcutaneously injected with oil vehicle (V), 5mg of T, or 5mg of DHT daily on embryonic days 16–19. Female offspring were ovariectomized (OVX) in adulthood at 0900h and treated with estradiol benzoate (EB; 30 μg sc). At the same time, these rats were fitted with jugular catheters. Twenty-four hours later, blood samples were collected every 30 minutes from 1200h to 2100h to capture the release of the LH surge. Serum LH levels were determined via RIA. Statistically significant LH surges were detected in females treated prenatally with vehicle (n=4) but not in pT-(n=3) or pDHT-treated (n=3) females. The data are represented as mean ± SEM. (From Foecking et.al., 2005)
How does prenatal androgenization program resistance to the positive feedback actions of estrogen? We have provided evidence that the positive feedback actions of estrogen on GnRH and LH surges are absolutely dependent upon the induction of PRs in the AVPV (Chappell et al., 2000; Chappell and Levine, 2000; Chappell et al., 1997; Chappell et al., 1999), and that androgen treatments in adult animals blocks both surge- and PR-induction by estrogen (Foecking and Levine, 2005). It thus appears that androgens may block the induction of GnRH and LH surges in adult female rats by impairing the ability of estrogen to induce PR expression. Based on these observations, we hypothesized that prenatal androgen exposure can program a permanent impairment of ER-mediated PR induction. To test this hypothesis, we assessed the ability of prenatal androgen treatments to render adult female rats resistant to the stimulatory effects of estrogen on PR expression. These studies revealed that exposure to either T and DHT in utero program complete refractoriness to estrogen’s ability to induce PR expression in adulthood (Figure 12) (Foecking et al., 2005). In a parallel study, we also found that PR mRNA expression is dramatically reduced in prenatally androgenized ovary-intact rats, compared to their control counterparts during metestrus or proestrus (Foecking et al., 2005) These findings are consistent with the idea that androgens program refractoriness of PR gene expression to estrogen’s inductive effects. However, causality in this hypothetical molecular mechanism remains to be documented.
Figure 12. Prenatal androgen exposure suppresses EB-induced PR mRNA expression in the POA of adult female rats.
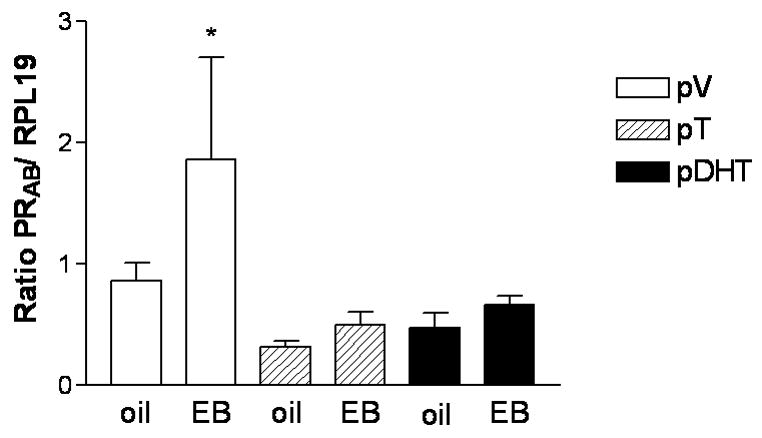
Pregnant female Sprague-Dawley rats were subcutaneously injected with oil vehicle (V), 5mg of T, or 5mg of DHT daily on embryonic days 16–19.Female offspring were ovariectomized (OVX) in adulthood and treated with estradiol benzoate (EB; 30 μg sc) or sesame oil vehicle (oil). The POA was dissected from the brain of each animal, and RNA was isolated, reverse transcribed, and processed for PRA+B mRNA expression via semi-quantitative PCR. PRA+B mRNA expression is significantly increased in the POA of EB-treated, ovariectomized control rats (pV, n=6 for both groups) compared to their oil-treated counterparts (*p<0.05). PRA+B mRNA expression is not induced by EB in pT- (n=7 for both groups) or pDHT-treated (OVX/oil, n=7; OVX/EB, n=9) females. The semi-quantitative RT-PCR results for PRA+B are normalized to the housekeeping gene RPL19 and are represented as mean ± SEM. (From Foecking et.al., 2005)
The preceding information demonstrates that androgens exert effects on female reproductive function at many stages of development. It is particularly interesting that treatment of females either in utero or in adulthood with androgens can prevent the induction of PR by estrogen in adult female rats (Figure 12, (Foecking et al., 2005) and Figure 6 (Foecking unpublished)). These results suggest androgens may be acting by similar mechanisms throughout life to alter the activity of the hypothalamic-pituitary-gonadal axis (HPG) in females. Allostasis is the term used to describe the alteration of homeostatic relationships in response to environmental perturbations in order to reestablish homeostatic stability, but when sustained for extended periods of time the physiological adjustments can lead to disease or malfunction (allostatic overload) (Landys et al., 2006; McEwen and Wingfield, 2003). We hypothesize that some effects of androgens on the female neuroendocrine system may result from the cumulative exposure to androgens resulting in allostatic adjustment of the HPG axis of females.
Summary of Androgen Effects
There are clearly multiple effects of androgens on GnRH and LH secretion, and these are differentially manifested depending upon the age at which the androgens are administered to the animal, and the mode of secretion that is being examined. Where there is sufficient consistency in the literature, we draw some basic conclusions and mechanistic interpretations.
Adult Treatments, Basal GnRH and LH pulsatility
The administration of androgens at male-typical levels in adulthood results in a suppression of LH secretion, which in turn is mediated, at least in part, by a reduction in pulsatile GnRH release. These effects may represent the activation of negative feedback pathways that normally operate in the male in response to the higher level of ligand present in the male versus female circulation. Thus, they appear to be mediated by activation of ARs, and possibly ERs, in both the hypothalamus and pituitary gland. Androgen treatments that recapitulate T levels normally observed in proestrous females can enhance GnRH-stimulated LHβ subunit gene expression and LH secretion, suggesting a role for endogenous T in shaping the course of the proestrous LH surge.
Neonatal treatments, Basal GnRH and LH pulsatility
Androgen excess in neonates leads to a reduced gonadotropin response to ovariectomy in adulthood, suggesting sensitivity during the neonatal period resulting in long-term impairment of GnRH and LH secretion by androgen excess.
Prenatal treatments, Basal GnRH and LH pulsatility
In contrast to neonatal and adult effects of androgens, prenatal androgenization can program subsequent acceleration of the GnRH pulse generator. This programming may reflect the sensitivity of the nascent GnRH neurosecretory system during this critical stage of development to the “masculinizing” (e.g. accelerating) effects of fetal androgens that would normally be present only in the male. That DHT induces the greatest effect in both rats (Foecking et al., 2005) and mice (Pielecka and Moenter, 2006) suggests a dependence upon AR activation.
Adult treatments, gonadotropin surges
Androgen treatments in adulthood block release of gonadotropin surges, most likely by combined effects on GnRH release and GnRH-stimulated gonadotropin secretion. The mechanism for this blockade appears to involve androgenic suppression of estrogen-induced PR expression in the AVPV.
Neonatal and prenatal treatments, gonadotropin surges
Both neonatal and prenatal androgen excess program for resistance to the positive feedback actions of estrogen in adulthood. These actions appear to be mediated, at least in part, via the programming of resistance to the estrogen-dependent induction of PRs in adulthood. While many classic studies have implicated the ER in mediating the effects of neonatal androgenization on hormonal cyclicity, evidence has also been obtained that reveal a role for AR in the prenatal programming of resistance to estrogen’s positive feedback actions. It is possible that androgens may program for acyclicity in adulthood by a combination of both receptor-mediated mechanisms.
How Do Androgens “Program” Neuroendocrine Function? Do PRs Provide a Clue?
The prevailing paradigm in research on hormone-dependent sex differences has held that the prenatal/perinatal surge of androgen in the male masculinizes and defeminizes neuroendocrine neuronal circuitries that govern GnRH neurosecretion and sexual behavior (Becu-Villalobos et al., 1997; Simerly, 2002; Simerly, 2005). As a corollary, it has also been widely accepted that androgen excess in the female can exert some or all of the same masculinizing and defeminizing effects in the preoptic area and hypothalamus (Becu-Villalobos et al., 1997). Much of this research has been directed towards an understanding of the morphological and neurochemical differences that emerge in adulthood as a consequence of the presence or absence of androgens during the prenatal or perinatal “critical periods” of sensitivity to their organizational actions. Sexually differentiated neuronal nuclei and projections have been elegantly described (Simerly, 2002), and some are clearly linked to the subsequent capacity to produce preovulatory LH surges and support estrous cyclicity (Becu-Villalobos et al., 1997). In general, the organizational effects of early T and/or estrogen exposure (or their absence), include differences in cell number, axonal fiber density, dendrite morphology, and neuronal transmitter phenotype (Simerly, 2002). Steroids may induce these morphological changes through a variety of mechanisms, including the induction or suppression of apoptosis (Chung et al., 2000; Forger et al., 2004; Zup et al., 2003), cell migration (Henderson et al., 1999), and synaptogenesis (Gu et al., 2003; Rudick and Woolley, 2001), as well as trophic interactions with growth factor signaling (Cardona-Gomez et al., 2000; Garcia-Segura et al., 2001), induction of paracrine mediators such as prostaglandin E2 (Amateau and McCarthy, 2002; Amateau and McCarthy, 2004), and effects on glial-neuronal apposition (Mong et al., 1999). Differences in neurotransmitter synthesis, release, and postsynaptic signal transduction are believed to be among the many functional manifestations of these structural differences in adulthood (Becu-Villalobos et al., 1997).
The development of sexually dimorphic circuits under the organizational influence of steroids has been reviewed by Simerly (Simerly, 2002) and others (Becu-Villalobos et al., 1997), and is well beyond the scope of this review. For our purposes, we note that the AVPV of the preoptic area is larger and contains more dopaminergic (tyrosine hydroxylase containing) and peptidergic neurons in female than in male rats (Bloch and Gorski, 1988; Simerly, 1998; Simerly et al., 1985). Recent studies extend the identity of these neurons to include a population of kisspeptin neurons in the AVPV that is distinct from the dopaminergic neurons (Kauffman et al., 2007). Moreover, efferent projections from the AVPV to the arcuate nucleus are denser in the female, while afferent projections from basal forebrain nuclei such as the bed nucleus of the stria terminalis (BNST) are more robust in the male (Gu and Simerly, 1997). The AVPV receives projections from the SCN, and extends axonal projections that form direct synaptic connections with GnRH neurons (Simerly, 1998). Moreover, lesions of this nucleus render animals unable to produce gonadotropin surges (Wiegand and Terasawa, 1982), and ER and PR expression here is obligatory for release of surges (Chappell and Levine, 2000; Petersen and Barraclough, 1989; Watson et al., 1995). Therefore, the AVPV of the preoptic area is essential in the regulation of GnRH surges.
In the absence of prenatal and perinatal androgen exposure, the AVPV nucleus acquires the female typical cell number, volume, and architecture, as well as responsiveness to the positive feedback actions of estrogen (Corbier, 1985; Henderson et al., 1976; Lund et al., 2000). The presence of androgens during the perinatal period defeminizes the morphological characteristics of the AVPV, and programs subsequent resistance to estrogen’s positive feedback actions (Becu-Villalobos et al., 1997). There is no evidence to challenge the view that the formation of the female-typical nuclear architecture per se is a necessary prerequisite for the acquisition of the capacity to release GnRH and LH surges. It is also reasonable to assume that the presence or absence of androgens evokes divergent patterns of gene expression that initially establish the sexually dimorphic cellular architecture of the AVPV, as well as programs constitutive gene expression to permanently sustain the sexually dimorphic morphology into adulthood.
It is equally probable, that the perinatal milieu also programs gene expression in ways that may also be essential in establishing male- or female-typical hormone secretions in adulthood, but may not be manifest as a microscopically observable structural or cytoarchitectural characteristics of the AVPV. Thus, the programming process may occur at the level of genes that are responsible for mediating estrogen’s positive feedback actions, such as the PR gene. The burgeoning field of epigenetics has revealed a variety of common mechanisms by which genes may be stably silenced or up-regulated. These include histone modifications and DNA methylation at specific sites in gene promoters (Yan et al., 2001). We hypothesize that prenatal androgen exposure programs refractoriness of AVPV neurons to estrogen’s positive feedback actions by one or more of these epigenetic mechanisms. Thus, programming of resistance to estrogen could occur via androgen-induced hypermethylation of CpG islands in the ER and/or PR promoters in AVPV neurons, or by modifications in the methylation of promoters of other genes that are essential in mediating estrogen’s positive feedback effects (Leader et al., 2006).
A number of observations support the model of epigenetic programming of AVPV neurons by androgens. First there is evidence that PRs are required for the induction of GnRH surges, that these PRs must be induced by estrogen, but exposure to androgens either prenatally or in adulthood blocks estrogen-induced expression of PRs (Chappell and Levine, 2000; Chappell et al., 1999; Corbier, 1985; Foecking and Levine, 2005; Henderson et al., 1976). Second, there are the observations demonstrating that both male and female rats express PRs in the AVPV to similar degree under basal conditions, but PR expression is only increased in females in response to estrogen (Brown et al., 1987; Lauber et al., 1991; Rainbow et al., 1982). Third, are the data which demonstrate that androgens also block both GnRH surges and PR induction by estrogen when given to adult females (Foecking and Levine, 2005). Activated ARs and ERs can program expression of target genes through epigenetic mechanisms (Cui et al., 2004; Yamane et al., 2006). The promotors of ER (Weigel and deConinck, 1993) and PR (Lapidus et al., 1996) genes can be regulated by epigenetic mechanisms. Androgens can suppress PR gene expression in a variety of tissues (Foecking, 2006). Although these pieces of evidence have been acquired in different experimental systems, they demonstrate the potential for epigenetic modifications to genes in AVPV neurons. They also suggest a mechanism for the allostatic responses of the female neuroendocrine system following prolonged exposure to androgens. This model of epigenetic programming of PRs and other estrogen target genes by androgens in the AVPV remains to be demonstrated directly, as it may clarify some fundamental mechanisms mediating the establishment of sexually differentiated hormone response characteristics.
Implications for Hyperandrogenic Disorders in Women
It is possible that programming of neuroendocrine function by androgens may also play an important role in the pathophysiology of PCOS and other hyperandrogenic disorders in women. The core symptoms of PCOS include hyperandrogenemia and anovulation, and these are often accompanied by LH excess, polcystic ovaries, insulin resistance, hyperinsulinemia, and obesity (Sam and Dunaif, 2003). There is one report (Cattrall et al., 2005) documenting that women with PCOS have a slight reduction in the ratio of their second to fourth finger lengths, suggesting they were exposed to androgens in utero. The ratio of the lengths of these fingers is sexually dimorphic and influenced by exposure to androgens prenatally (Brown et al., 2002). A current theory holds that PCOS has a fetal origin, in which one or more genetic mutations leads to prenatal androgen excess (Abbott et al., 2006; Manneras et al., 2007; Xita and Tsatsoulis, 2006), which in turn reprograms multiple tissues (ovary, brain, pancreas, liver, and others) to exhibit the features of PCOS in adolescence and adulthood. This theory has been bolstered by the findings of several studies showing that prenatal, perinatal, or postnatal androgen exposure in monkeys (Abbott et al., 2002), sheep (West et al., 2001), rats (Foecking and Levine, 2005; Foecking et al., 2005; Manneras et al., 2007), and mice (Sullivan and Moenter, 2004) can prompt the development of many of the symptoms of PCOS. Exposing female monkey and sheep fetuses to excess androgens, for example results in subsequent development of LH excess and enlarged ovaries that are polyfollicular, anovulatory, and hyperandrogenic (Abbott et al., 2005; Birch et al., 2003; Masek et al., 1999).
Hypothalamic-pituitary function is deranged in most prenatally androgenized experimental animals, mimicking the reproductive neuroendocrine phenotype of PCOS. The LH excess in these animals is driven by increases in the frequency or amplitude of pulsatile GnRH and LH secretion (Abbott et al., 2006; Foecking et al., 2005). In monkeys and sheep, the disturbances in GnRH and LH secretion appear to be dependent in part on desensitization to the negative feedback effects of estrogen and progesterone (Abbott et al., 2005; Sarma et al., 2005). We have not observed gross disturbances of negative feedback in the prenatally androgenized female rat (Foecking and Levine, 2005; Foecking et al., 2005), although GnRH pulsatility is moderately increased in the open-loop condition (Foecking et al., 2005). Others have observed increased LH secretion in prenatally androgenized mice, even in the presence of hyperandrogenemia (Sullivan and Moenter, 2004). In general, the observed changes in basal GnRH pulsatility and/or sensitivity to ovarian feedback in prenatally androgenized animals are similar to the LH secretory phenotypes in PCOS; women with this common endocrine disorder show accelerated and/or exaggerated LH and are relatively resistant to suppression by estrogen and progesterone (Blank et al., 2006; Pastor et al., 1998).
Prenatal and neonatal androgenization of female rats also results in anovulation, which is in part due to the inability of these animals to release gonadotropin surges (Foecking and Levine, 2005; Foecking et al., 2005; Manneras et al., 2007). A similar effect was observed in ewes exposed to T early in gestation, where failure to generate LH surges occurs by the second breeding season (Birch et al., 2003; Unsworth et al., 2005). Prenatal T in female monkeys, however, does not disrupt the positive feedback actions of estrogen (Dumesic et al., 1997; Steiner et al., 1976), and it is not known if the LH surge mechanism is impaired in PCOS patients. It is possible that androgen exposure is less effective in disrupting the LH surge in female primates versus non-primate mammals, given that the LH surge mechanism does not appear to be sexually differentiated in primates (Resko and Roselli, 1997) compared to rats (Becu-Villalobos et al., 1997), sheep (Wood and Foster, 1998) and other species. Anovulation in PCOS patients may be precipitated by several factors, which may include acyclic and elevated LH secretion (Blank et al., 2006). The rapid frequency of GnRH pulses causes hypersecretion of LH and testosterone, lowered levels of FSH, and failure of the later stages of follicular maturation. The acceleration of the GnRH pulse generator may in turn be caused by refractoriness to steroid negative feedback (Blank et al., 2007). The altered glucose homeostasis and hyperinsulinemia in PCOS women may additionally disrupt ovarian follicular development and ovulation (Legro et al., 1999; Sam and Dunaif, 2003). However, it is not clear how much of the altered ovarian follicular morphology and ovarian function in PCOS is a consequence of intraovarian androgen action, absence of LH surges, LH hypersecretion, altered LH/FSH ratio, or metabolic hormone effects.
We have not considered in this review the many observations that have been made on the metabolic consequences of androgen exposure, and how these may also mimic the metabolic phenotype presented by many PCOS women. We briefly note here, however, that prenatal androgenization of sheep (Recabarren et al., 2005) and monkeys (Bruns et al., 2007) results in the development of several of these PCOS characteristics, such as visceral adiposity, insulin resistance, and hyperinsulinemia. In female rats, prenatal (our unpublished observations) and postnatal androgenization (Manneras et al., 2007) produce some of these characteristics, such as increased body fat and insulin resistance.
Collectively, the parallel observations made in experimental animals and in PCOS women suggest that reproductive consequences of prenatal and postnatal androgen exposure may be mediated in part by androgenic programming of hypothalamic and pituitary function. The finding that androgens may program expression of PR suggests the intriguing possibility that the androgenic programming of the PR gene and other genes that confer responsiveness to steroid actions may underlie the etiology of PCOS.
Acknowledgments
This work was supported by NIH grants P01 HD44405, R01 HD20677, and T32 HD07068
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- Abbott D, et al. Insights into the development of PCOS from studies of prenatally androgenized female rhesus monkeys. Trends Endocrinol Metab. 1998;9:62l–67. doi: 10.1016/s1043-2760(98)00019-8. [DOI] [PubMed] [Google Scholar]
- Abbott DH, et al. Androgen excess fetal programming of female reproduction: a developmental aetiology for polycystic ovary syndrome? Hum Reprod Update. 2005;11:357–74. doi: 10.1093/humupd/dmi013. [DOI] [PubMed] [Google Scholar]
- Abbott DH, et al. Developmental origin of polycystic ovary syndrome - a hypothesis. J Endocrinol. 2002;174:1–5. doi: 10.1677/joe.0.1740001. [DOI] [PubMed] [Google Scholar]
- Abbott DH, et al. Contributions of androgen and estrogen to fetal programming of ovarian dysfunction. Reprod Biol Endocrinol. 2006;4:17. doi: 10.1186/1477-7827-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amateau SK, McCarthy MM. A novel mechanism of dendritic spine plasticity involving estradiol induction of prostaglandin-E2. J Neurosci. 2002;22:8586–96. doi: 10.1523/JNEUROSCI.22-19-08586.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amateau SK, McCarthy MM. Induction of PGE2 by estradiol mediates developmental masculinization of sex behavior. Nat Neurosci. 2004;7:643–50. doi: 10.1038/nn1254. [DOI] [PubMed] [Google Scholar]
- Attardi B, Happe HK. Modulation of the estradiol-induced luteinizing hormone surge by progesterone or antiestrogens: effects on pituitary gonadotropin-releasing hormone receptors. Endocrinology. 1986;119:274–83. doi: 10.1210/endo-119-1-274. [DOI] [PubMed] [Google Scholar]
- Auger AP, et al. Expression of the nuclear receptor coactivator, cAMP response element-binding protein, is sexually dimorphic and modulates sexual differentiation of neonatal rat brain. Endocrinology. 2002;143:3009–16. doi: 10.1210/endo.143.8.8975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barraclough CA. Production of anovulatory, sterile rats by single injections of testosterone propionate. Endocrinology. 1961;68:62–7. doi: 10.1210/endo-68-1-62. [DOI] [PubMed] [Google Scholar]
- Barraclough CA, Gorski RA. Evidence that the hypothalamus is responsible for androgen-induced sterility in the female rat. Endocrinology. 1961;68:68–79. doi: 10.1210/endo-68-1-68. [DOI] [PubMed] [Google Scholar]
- Barraclough CA, Haller EW. Positive and negative feedback effects of estrogen on pituitary LH synthesis and release in normal and androgen-sterilized rats. Endocrinology. 1970;86:542–51. doi: 10.1210/endo-86-3-542. [DOI] [PubMed] [Google Scholar]
- Bauer-Dantoin AC, et al. Roles of estrogen, progesterone, and gonadotropin-releasing hormone (GnRH) in the control of pituitary GnRH receptor gene expression at the time of the preovulatory gonadotropin surges. Endocrinology. 1995;136:1014–9. doi: 10.1210/endo.136.3.7867555. [DOI] [PubMed] [Google Scholar]
- Baum MJ, et al. Immediate postnatal rise in whole body androgen content in male rats: correlation with increased testicular content and reduced body clearance of testosterone. Biol Reprod. 1988;38:980–6. doi: 10.1095/biolreprod38.5.980. [DOI] [PubMed] [Google Scholar]
- Baum MJ, et al. Sex difference in whole-body androgen content in rats on fetal days 18 and 19 without evidence that androgen passes from males to females. Biol Reprod. 1991;44:747–51. doi: 10.1095/biolreprod44.5.747. [DOI] [PubMed] [Google Scholar]
- Becker JB, et al. Strategies and methods for research on sex differences in brain and behavior. Endocrinology. 2005;146:1650–73. doi: 10.1210/en.2004-1142. [DOI] [PubMed] [Google Scholar]
- Becu-Villalobos D, et al. Brain sexual differentiation and gonadotropins secretion in the rat. Cell Mol Neurobiol. 1997;17:699–715. doi: 10.1023/A:1022542221535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsham DD, et al. Regulation of gonadotropin-releasing hormone (GnRH) gene expression by 5alpha-dihydrotestosterone in GnRH-secreting GT1–7 hypothalamic neurons. Endocrinology. 1998;139:1108–14. doi: 10.1210/endo.139.3.5846. [DOI] [PubMed] [Google Scholar]
- Birch RA, et al. Prenatal programming of reproductive neuroendocrine function: fetal androgen exposure produces progressive disruption of reproductive cycles in sheep. Endocrinology. 2003;144:1426–34. doi: 10.1210/en.2002-220965. [DOI] [PubMed] [Google Scholar]
- Blackwell RE, Amoss MS., Jr A sex difference in the rate of rise of plasma LH in rats following gonadectomy. Proc Soc Exp Biol Med. 1971;136:11–4. doi: 10.3181/00379727-136-35181. [DOI] [PubMed] [Google Scholar]
- Blank SK, et al. Neuroendocrine effects of androgens in adult polycystic ovary syndrome and female puberty. Semin Reprod Med. 2007;25:352–9. doi: 10.1055/s-2007-984741. [DOI] [PubMed] [Google Scholar]
- Blank SK, et al. The origins and sequelae of abnormal neuroendocrine function in polycystic ovary syndrome. Hum Reprod Update. 2006;12:351–61. doi: 10.1093/humupd/dml017. [DOI] [PubMed] [Google Scholar]
- Bloch GJ, Gorski RA. Estrogen/progesterone treatment in adulthood affects the size of several components of the medial preoptic area in the male rat. J Comp Neurol. 1988;275:613–22. doi: 10.1002/cne.902750409. [DOI] [PubMed] [Google Scholar]
- Bousios S, et al. Effects of gender and stress on the regulation of steroid receptor coactivator-1 expression in the rat brain and pituitary. J Steroid Biochem Mol Biol. 2001;78:401–7. doi: 10.1016/s0960-0760(01)00123-6. [DOI] [PubMed] [Google Scholar]
- Brann DW, et al. gamma-Aminobutyric acid-opioid interactions in the regulation of gonadotropin secretion in the immature female rat. Neuroendocrinology. 1992;56:445–52. doi: 10.1159/000126281. [DOI] [PubMed] [Google Scholar]
- Brown TJ, et al. Regional sex differences in progestin receptor induction in the rat hypothalamus: effects of various doses of estradiol benzoate. J Neurosci. 1987;7:2529–36. [PMC free article] [PubMed] [Google Scholar]
- Brown WM, et al. Masculinized finger length patterns in human males and females with congenital adrenal hyperplasia. Horm Behav. 2002;42:380–6. doi: 10.1006/hbeh.2002.1830. [DOI] [PubMed] [Google Scholar]
- Bruns CM, et al. Prenatal androgen excess negatively impacts body fat distribution in a nonhuman primate model of polycystic ovary syndrome. Int J Obes (Lond) 2007;31:1579–85. doi: 10.1038/sj.ijo.0803638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger LL, et al. The regulation of FSHbeta transcription by gonadal steroids: testosterone and estradiol modulation of the activin intracellular signaling pathway. Am J Physiol Endocrinol Metab. 2007;293:E277–85. doi: 10.1152/ajpendo.00447.2006. [DOI] [PubMed] [Google Scholar]
- Caraty A, Locatelli A. Effect of time after castration on secretion of LHRH and LH in the ram. J Reprod Fertil. 1988;82:263–9. doi: 10.1530/jrf.0.0820263. [DOI] [PubMed] [Google Scholar]
- Cardona-Gomez GP, et al. Estrogen receptors and insulin-like growth factor-I receptors mediate estrogen-dependent synaptic plasticity. Neuroreport. 2000;11:1735–8. doi: 10.1097/00001756-200006050-00027. [DOI] [PubMed] [Google Scholar]
- Carruth LL, et al. Sex chromosome genes directly affect brain sexual differentiation. Nat Neurosci. 2002;5:933–4. doi: 10.1038/nn922. [DOI] [PubMed] [Google Scholar]
- Cattrall FR, et al. Anatomical evidence for in utero androgen exposure in women with polycystic ovary syndrome. Fertil Steril. 2005;84:1689–92. doi: 10.1016/j.fertnstert.2005.05.061. [DOI] [PubMed] [Google Scholar]
- Chappel SC, Barraclough CA. Plasma concentration changes in LH and FSH following electrochemical stimulation of the medial preoptic are or dorsal anterior hypothalamic area of estrogen- or androgen-sterilized rats. Biol Reprod. 1976;15:661–9. doi: 10.1095/biolreprod15.5.661. [DOI] [PubMed] [Google Scholar]
- Chappell PE, et al. Stimulation of gonadotropin-releasing hormone surges by estrogen. II. Role of cyclic adenosine 3′5′-monophosphate. Endocrinology. 2000;141:1486–92. doi: 10.1210/endo.141.4.7427. [DOI] [PubMed] [Google Scholar]
- Chappell PE, Levine JE. Stimulation of gonadotropin-releasing hormone surges by estrogen. I. Role of hypothalamic progesterone receptors. Endocrinology. 2000;141:1477–85. doi: 10.1210/endo.141.4.7428. [DOI] [PubMed] [Google Scholar]
- Chappell PE, et al. Endocrine defects in mice carrying a null mutation for the progesterone receptor gene. Endocrinology. 1997;138:4147–52. doi: 10.1210/endo.138.10.5456. [DOI] [PubMed] [Google Scholar]
- Chappell PE, et al. Absence of gonadotropin surges and gonadotropin-releasing hormone self-priming in ovariectomized (OVX), estrogen (E2)-treated, progesterone receptor knockout (PRKO) mice. Endocrinology. 1999;140:3653–8. doi: 10.1210/endo.140.8.6895. [DOI] [PubMed] [Google Scholar]
- Chung WC, et al. Apoptosis during sexual differentiation of the bed nucleus of the stria terminalis in the rat brain. J Neurobiol. 2000;43:234–43. [PubMed] [Google Scholar]
- Clay CM, et al. Transcriptional repression of the glycoprotein hormone alpha subunit gene by androgen may involve direct binding of androgen receptor to the proximal promoter. J Biol Chem. 1993;268:13556–64. [PubMed] [Google Scholar]
- Colciago A, et al. Dimorphic expression of testosterone metabolizing enzymes in the hypothalamic area of developing rats. Brain Res Dev Brain Res. 2005;155:107–16. doi: 10.1016/j.devbrainres.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Corbier P. Sexual differentiation of positive feedback: effect of hour of castration at birth on estradiol-induced luteinizing hormone secretion in immature male rats. Endocrinology. 1985;116:142–7. doi: 10.1210/endo-116-1-142. [DOI] [PubMed] [Google Scholar]
- Cui Y, et al. Phosphorylation of estrogen receptor alpha blocks its acetylation and regulates estrogen sensitivity. Cancer Res. 2004;64:9199–208. doi: 10.1158/0008-5472.CAN-04-2126. [DOI] [PubMed] [Google Scholar]
- Dohler KD, et al. Pre- and postnatal influence of testosterone propionate and diethylstilbestrol on differentiation of the sexually dimorphic nucleus of the preoptic area in male and female rats. Brain Res. 1984;302:291–5. doi: 10.1016/0006-8993(84)90242-7. [DOI] [PubMed] [Google Scholar]
- Dohler KD, Wuttke W. Changes with age in levels of serum gonadotropins, prolactin and gonadal steroids in prepubertal male and female rats. Endocrinology. 1975;97:898–907. doi: 10.1210/endo-97-4-898. [DOI] [PubMed] [Google Scholar]
- Drouin J, Labrie F. Selective effect of androgens on LH and FSH release in anterior pituitary cells in culture. Endocrinology. 1976;98:1528–34. doi: 10.1210/endo-98-6-1528. [DOI] [PubMed] [Google Scholar]
- Drouva SV, et al. Effects of ovarian steroids on in vitro release of LHRH from mediobasal hypothalamus. Neuroendocrinology. 1983;37:336–41. doi: 10.1159/000123572. [DOI] [PubMed] [Google Scholar]
- Dumesic DA, et al. Prenatal exposure of female rhesus monkeys to testosterone propionate increases serum luteinizing hormone levels in adulthood. Fertil Steril. 1997;67:155–63. doi: 10.1016/s0015-0282(97)81873-0. [DOI] [PubMed] [Google Scholar]
- Dumesic DA, et al. Impaired developmental competence of oocytes in adult prenatally androgenized female rhesus monkeys undergoing gonadotropin stimulation for in vitro fertilization. J Clin Endocrinol Metab. 2002;87:1111–9. doi: 10.1210/jcem.87.3.8287. [DOI] [PubMed] [Google Scholar]
- Dunaif A, Thomas A. Current concepts in the polycystic ovary syndrome. Annu Rev Med. 2001;52:401–19. doi: 10.1146/annurev.med.52.1.401. [DOI] [PubMed] [Google Scholar]
- Dunlap KD, Sridaran R. Plasma levels of dihydrotestosterone in the cycling rat: implications for the regulation of lordosis behavior. Physiol Behav. 1988;42:199–202. doi: 10.1016/0031-9384(88)90298-3. [DOI] [PubMed] [Google Scholar]
- Eagleson CA, et al. Polycystic ovarian syndrome: evidence that flutamide restores sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by estradiol and progesterone. J Clin Endocrinol Metab. 2000;85:4047–52. doi: 10.1210/jcem.85.11.6992. [DOI] [PubMed] [Google Scholar]
- Eldridge JC, et al. Effects of castration of immature rats on serum FSH and LH, and of various steroid treatments after castration. Biol Reprod. 1974;10:438–46. doi: 10.1095/biolreprod10.4.438. [DOI] [PubMed] [Google Scholar]
- Ellis GB, et al. Control of pulsatile LH release in male rats. Neuroendocrinology. 1983;37:177–83. doi: 10.1159/000123540. [DOI] [PubMed] [Google Scholar]
- Emons G, et al. Effects of progesterone on gonadotropin-releasing hormone receptor concentration in cultured estrogen-primed female rat pituitary cells. J Steroid Biochem Mol Biol. 1992;42:831–9. doi: 10.1016/0960-0760(92)90091-v. [DOI] [PubMed] [Google Scholar]
- Fels E, Bosch L. Effect of prenatal administration of testosterone on ovarian function in rats. Am J Obstet Gynecol. 1971;111:964–969. doi: 10.1016/0002-9378(71)90954-9. [DOI] [PubMed] [Google Scholar]
- Fink G. The self-priming effect of LHRH: a unique servomechanism and possible cellular model for memory. Front Neuroendocrinol. 1995;16:183–90. doi: 10.1006/frne.1995.1006. [DOI] [PubMed] [Google Scholar]
- Foecking E. Neurobiology and Physiology, Vol PhD. Northwestern University; Evanston: 2006. The Pathophysiological Effects of Androgens on Female Reproduction and Metabolism in the Rat; p. 230. [Google Scholar]
- Foecking EM, Levine JE. Effects of experimental hyperandrogenemia on the female rat reproductive axis: suppression of progesterone-receptor messenger RNA expression in the brain and blockade of luteinizing hormone surges. Gend Med. 2005;2:155–65. doi: 10.1016/s1550-8579(05)80044-0. [DOI] [PubMed] [Google Scholar]
- Foecking EM, et al. Neuroendocrine consequences of prenatal androgen exposure in the female rat: absence of luteinizing hormone surges, suppression of progesterone receptor gene expression, and acceleration of the gonadotropin-releasing hormone pulse generator. Biol Reprod. 2005;72:1475–83. doi: 10.1095/biolreprod.105.039800. Epub 2005 Mar 2. [DOI] [PubMed] [Google Scholar]
- Forger NG, et al. Deletion of Bax eliminates sex differences in the mouse forebrain. Proc Natl Acad Sci U S A. 2004;101:13666–71. doi: 10.1073/pnas.0404644101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Segura LM, et al. Neuroprotection by estradiol. Prog Neurobiol. 2001;63:29–60. doi: 10.1016/s0301-0082(00)00025-3. [DOI] [PubMed] [Google Scholar]
- Gaveriaux-Ruff C, Kieffer BL. Conditional gene targeting in the mouse nervous system: Insights into brain function and diseases. Pharmacol Ther. 2007;113:619–34. doi: 10.1016/j.pharmthera.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Giguere V, et al. Androgens decrease LHRH binding sites in rat anterior pituitary cells in culture. Endocrinology. 1981;108:350–2. doi: 10.1210/endo-108-1-350. [DOI] [PubMed] [Google Scholar]
- Glidewell-Kenney C, et al. Nonclassical estrogen receptor alpha signaling mediates negative feedback in the female mouse reproductive axis. Proc Natl Acad Sci U S A. 2007;104:8173–7. doi: 10.1073/pnas.0611514104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogan F, et al. Effect of neonatal administration of steroids or gonadectomy upon oestradiol-induced luteinizing hormone release in rats of both sexes. J Endocrinol. 1980;85:69–74. doi: 10.1677/joe.0.0850069. [DOI] [PubMed] [Google Scholar]
- Goomer N, et al. Effect of neonatal testosterone and oestradiol treatment on the development of the hypothalamo-hypophysial axis in the female rat. J Reprod Fertil. 1977;50:239–43. doi: 10.1530/jrf.0.0500239. [DOI] [PubMed] [Google Scholar]
- Gorski RA. Sexual dimorphisms of the brain. J Anim Sci. 1985;61:38–61. doi: 10.1093/ansci/61.supplement_3.38. [DOI] [PubMed] [Google Scholar]
- Gorski RA, et al. Evidence for a morphological sex difference within the medial preoptic area of the rat brain. Brain Res. 1978;148:333–46. doi: 10.1016/0006-8993(78)90723-0. [DOI] [PubMed] [Google Scholar]
- Goy RW, et al. Behavioral masculinization is independent of genital masculinization in prenatally androgenized female rhesus macaques. Horm Behav. 1988;22:552–71. doi: 10.1016/0018-506x(88)90058-x. [DOI] [PubMed] [Google Scholar]
- Gu G, et al. Sexual differentiation of projections from the principal nucleus of the bed nuclei of the stria terminalis. J Comp Neurol. 2003;460:542–62. doi: 10.1002/cne.10677. [DOI] [PubMed] [Google Scholar]
- Gu GB, Simerly RB. Projections of the sexually dimorphic anteroventral periventricular nucleus in the female rat. J Comp Neurol. 1997;384:142–64. [PubMed] [Google Scholar]
- Haisenleder DJ, et al. Gonadotropin subunit and gonadotropin-releasing hormone receptor gene expression are regulated by alterations in the frequency of calcium pulsatile signals. Endocrinology. 1997;138:5227–30. doi: 10.1210/endo.138.12.5611. [DOI] [PubMed] [Google Scholar]
- Handa RJ, et al. Gonadotropin secretion following intraventricular norepinephrine infusion into neonatally androgenized female rats. Neuroendocrinology. 1986;43:269–72. doi: 10.1159/000124539. [DOI] [PubMed] [Google Scholar]
- Hart BL. Neonatal castration: influence on neural organization of sexual reflexes in male rats. Science. 1968;160:1135–6. doi: 10.1126/science.160.3832.1135. [DOI] [PubMed] [Google Scholar]
- Hassani M, et al. Action of testosterone propionate on the gonadotrophic function of the pituitary gland in the cyclic female rat. Acta Endocrinol (Copenh) 1978;89:551–6. doi: 10.1530/acta.0.0890551. [DOI] [PubMed] [Google Scholar]
- Henderson RG, et al. Sex differences in cell migration in the preoptic area/anterior hypothalamus of mice. J Neurobiol. 1999;41:252–66. doi: 10.1002/(sici)1097-4695(19991105)41:2<252::aid-neu8>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Henderson SR, et al. Proceedings: Effect of precise oestradiol-17 beta exposure on the gonadotrophin response to luteinizing hormone releasing factor and the spontaneous secretion of luteinizing hormone in acutely and chronically ovariectomized rats. J Endocrinol. 1976;69:1P–2P. [PubMed] [Google Scholar]
- Herbison AE. Neurochemical identity of neurones expressing oestrogen and androgen receptors in sheep hypothalamus. J Reprod Fertil Suppl. 1995;49:271–83. [PubMed] [Google Scholar]
- Hiatt ES, et al. Sex differences following gonadectomy in basal gonadotropin secretion rate of rat pituitary fragments in vitro. Biol Reprod. 1987;37:1114–20. doi: 10.1095/biolreprod37.5.1114. [DOI] [PubMed] [Google Scholar]
- Hrabovszky E, et al. Detection of estrogen receptor-beta messenger ribonucleic acid and 125I-estrogen binding sites in luteinizing hormone-releasing hormone neurons of the rat brain. Endocrinology. 2000;141:3506–9. doi: 10.1210/endo.141.9.7788. [DOI] [PubMed] [Google Scholar]
- Huffman L, Hendricks SE. Prenatally injected testosterone propionate and sexual behavior of female rats. Physiol Behav. 1981;26:773–8. doi: 10.1016/0031-9384(81)90097-4. [DOI] [PubMed] [Google Scholar]
- Jackson GL, et al. Testosterone inhibits gonadotropin-releasing hormone pulse frequency in the male sheep. Biol Reprod. 1991;45:188–94. doi: 10.1095/biolreprod45.1.188. [DOI] [PubMed] [Google Scholar]
- Jakacka M, et al. An estrogen receptor (ER)alpha deoxyribonucleic acid-binding domain knock-in mutation provides evidence for nonclassical ER pathway signaling in vivo. Mol Endocrinol. 2002;16:2188–201. doi: 10.1210/me.2001-0174. [DOI] [PubMed] [Google Scholar]
- Kamel F, et al. Gonadal steroids modulate pulsatile luteinizing hormone secretion by perifused rat anterior pituitary cells. Endocrinology. 1987;120:1651–7. doi: 10.1210/endo-120-4-1651. [DOI] [PubMed] [Google Scholar]
- Kamel F, Krey LC. Gonadal steroid modulation of LH secretion stimulated by LHRH, Ca2+ and cAMP. Mol Cell Endocrinol. 1983;32:285–300. doi: 10.1016/0303-7207(83)90089-8. [DOI] [PubMed] [Google Scholar]
- Kamel F, Kubajak CL. Modulation of gonadotropin secretion by corticosterone: interaction with gonadal steroids and mechanism of action. Endocrinology. 1987;121:561–8. doi: 10.1210/endo-121-2-561. [DOI] [PubMed] [Google Scholar]
- Kamel F, Kubajak CL. Gonadal steroid effects on LH response to arachidonic acid and protein kinase C. Am J Physiol. 1988;255:E314–21. doi: 10.1152/ajpendo.1988.255.3.E314. [DOI] [PubMed] [Google Scholar]
- Kauffman AS, et al. Sexual differentiation of Kiss1 gene expression in the brain of the rat. Endocrinology. 2007;148:1774–83. doi: 10.1210/en.2006-1540. [DOI] [PubMed] [Google Scholar]
- King JCTSA, Snavely FL, Arimura AA. The LHRH system in normal and neonatally androgenized female rats. Peptides. 1985;1:85–100. [Google Scholar]
- Knobil E. Patterns of hypophysiotropic signals and gonadotropin secretion in the rhesus monkey. Biol Reprod. 1981;24:44–9. doi: 10.1095/biolreprod24.1.44. [DOI] [PubMed] [Google Scholar]
- Korenbrot CC, et al. Effects of testosterone propionate or dihydrotestosterone propionate on plasma FSH and LH levels in neonatal rats and on sexual differentiation of the brain. Endocrinology. 1975;97:709–17. doi: 10.1210/endo-97-3-709. [DOI] [PubMed] [Google Scholar]
- Kubo K, et al. Similarity of plasma LH release in androgenized and normal rats following electrochemical stimulation of the basal forebrain. Endocrinology. 1975;96:492–500. doi: 10.1210/endo-96-2-492. [DOI] [PubMed] [Google Scholar]
- Kudwa AE, et al. Roles of estrogen receptors alpha and beta in differentiation of mouse sexual behavior. Neuroscience. 2006;138:921–8. doi: 10.1016/j.neuroscience.2005.10.018. [DOI] [PubMed] [Google Scholar]
- Landys MM, et al. Actions of glucocorticoids at a seasonal baseline as compared to stress-related levels in the regulation of periodic life processes. Gen Comp Endocrinol. 2006;148:132–49. doi: 10.1016/j.ygcen.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Lapidus RG, et al. Methylation of estrogen and progesterone receptor gene 5′ CpG islands correlates with lack of estrogen and progesterone receptor gene expression in breast tumors. Clin Cancer Res. 1996;2:805–10. [PubMed] [Google Scholar]
- Larsson K. Effects of neonatal castration upon the development of the mating behavior of the male rat. Z Tierpsychol. 1966;23:867–73. doi: 10.1111/j.1439-0310.1966.tb01716.x. [DOI] [PubMed] [Google Scholar]
- Lauber AH, et al. Sex difference in estradiol regulation of progestin receptor mRNA in rat mediobasal hypothalamus as demonstrated by in situ hybridization. Neuroendocrinology. 1991;53:608–13. doi: 10.1159/000125781. [DOI] [PubMed] [Google Scholar]
- Leader JE, et al. Epigenetic regulation of nuclear steroid receptors. Biochem Pharmacol. 2006;72:1589–96. doi: 10.1016/j.bcp.2006.05.024. [DOI] [PubMed] [Google Scholar]
- Legan SJ, et al. Role of estrogen as initiator of daily LH surges in the ovariectomized rat. Endocrinology. 1975;96:50–6. doi: 10.1210/endo-96-1-50. [DOI] [PubMed] [Google Scholar]
- Legan SJ, Karsch FJ. A daily signal for the LH surge in the rat. Endocrinology. 1975;96:57–62. doi: 10.1210/endo-96-1-57. [DOI] [PubMed] [Google Scholar]
- Legro RS, et al. Prevalence and predictors of risk for type 2 diabetes mellitus and impaired glucose tolerance in polycystic ovary syndrome: a prospective, controlled study in 254 affected women. J Clin Endocrinol Metab. 1999;84:165–9. doi: 10.1210/jcem.84.1.5393. [DOI] [PubMed] [Google Scholar]
- Leipheimer RE, Gallo RV. Acute and long-term changes in central and pituitary mechanisms regulating pulsatile luteinizing hormone secretion after ovariectomy in the rat. Neuroendocrinology. 1983;37:421–6. doi: 10.1159/000123587. [DOI] [PubMed] [Google Scholar]
- Levine JE. New concepts of the neuroendocrine regulation of gonadotropin surges in rats. Biol Reprod. 1997;56:293–302. doi: 10.1095/biolreprod56.2.293. [DOI] [PubMed] [Google Scholar]
- Levine JE, Duffy MT. Simultaneous measurement of luteinizing hormone (LH)-releasing hormone, LH, and follicle-stimulating hormone release in intact and short-term castrate rats. Endocrinology. 1988;122:2211–21. doi: 10.1210/endo-122-5-2211. [DOI] [PubMed] [Google Scholar]
- Levine JE, Ramirez VD. Luteinizing hormone-releasing hormone release during the rat estrous cycle and after ovariectomy, as estimated with push-pull cannulae. Endocrinology. 1982;111:1439–48. doi: 10.1210/endo-111-5-1439. [DOI] [PubMed] [Google Scholar]
- Liaw JJ, Barraclough CA. N-Methyl-D, L-Aspartic Acid Differentially Affects Lh-Release and Lhrh Messenger-Rna Levels in Estrogen-Treated Ovariectomized Control and Androgen-Sterilized Rats. Molecular Brain Research. 1993a;17:112–118. doi: 10.1016/0169-328x(93)90079-5. [DOI] [PubMed] [Google Scholar]
- Liaw JJ, Barraclough CA. Pituitary gland responsiveness to LHRH and LHRH neuronal responsiveness to excitatory stimuli are severely impaired in female rats treated neonatally with high doses of androgen. Brain Res. 1993b;607:233–40. doi: 10.1016/0006-8993(93)91511-p. [DOI] [PubMed] [Google Scholar]
- Lindzey J, et al. Effects of castration and chronic steroid treatments on hypothalamic gonadotropin-releasing hormone content and pituitary gonadotropins in male wild-type and estrogen receptor-alpha knockout mice. Endocrinology. 1998;139:4092–101. doi: 10.1210/endo.139.10.6253. [DOI] [PubMed] [Google Scholar]
- Lund TD, et al. Dihydrotestosterone may inhibit hypothalamo-pituitary-adrenal activity by acting through estrogen receptor in the male mouse. Neurosci Lett. 2004;365:43–7. doi: 10.1016/j.neulet.2004.04.035. [DOI] [PubMed] [Google Scholar]
- Lund TD, et al. Pre- or postnatal testosterone and flutamide effects on sexually dimorphic nuclei of the rat hypothalamus. Brain Res Dev Brain Res. 2000;120:261–6. doi: 10.1016/s0165-3806(00)00013-4. [DOI] [PubMed] [Google Scholar]
- Mani SK. Signaling mechanisms in progesterone-neurotransmitter interactions. Neuroscience. 2006;138:773–81. doi: 10.1016/j.neuroscience.2005.07.034. [DOI] [PubMed] [Google Scholar]
- Manneras L, et al. A new rat model exhibiting both ovarian and metabolic characteristics of polycystic ovary syndrome. Endocrinology. 2007;148:3781–91. doi: 10.1210/en.2007-0168. [DOI] [PubMed] [Google Scholar]
- Masek KS, et al. Prenatal dihydrotestosterone differentially masculinizes tonic and surge modes of luteinizing hormone secretion in sheep. Endocrinology. 1999;140:3459–66. doi: 10.1210/endo.140.8.6913. [DOI] [PubMed] [Google Scholar]
- McAbee MD, DonCarlos LL. Ontogeny of region-specific sex differences in androgen receptor messenger ribonucleic acid expression in the rat forebrain. Endocrinology. 1998;139:1738–45. doi: 10.1210/endo.139.4.5940. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Wingfield JC. The concept of allostasis in biology and biomedicine. Horm Behav. 2003;43:2–15. doi: 10.1016/s0018-506x(02)00024-7. [DOI] [PubMed] [Google Scholar]
- McGinnis MY, et al. In vitro measurement of cytosol and cell nuclear androgen receptors in male rat brain and pituitary. Brain Res. 1983;275:75–82. doi: 10.1016/0006-8993(83)90418-3. [DOI] [PubMed] [Google Scholar]
- Meredith JM, Levine JE. Effects of castration on LH-RH patterns in intrahypophysial microdialysates. Brain Res. 1992;571:181–8. doi: 10.1016/0006-8993(92)90653-q. [DOI] [PubMed] [Google Scholar]
- Moguilevsky JA, et al. Effect of castration on serum gonadotropin levels in androgenized male and female rats. Neuroendocrinology. 1979;29:323–8. doi: 10.1159/000122940. [DOI] [PubMed] [Google Scholar]
- Mong JA, et al. Gonadal steroids promote glial differentiation and alter neuronal morphology in the developing hypothalamus in a regionally specific manner. J Neurosci. 1999;19:1464–72. doi: 10.1523/JNEUROSCI.19-04-01464.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik SI, et al. Pituitary gonadotropin-releasing hormone receptor regulation in mice. I: Males. Endocrinology. 1984;115:106–13. doi: 10.1210/endo-115-1-106. [DOI] [PubMed] [Google Scholar]
- Nansel DD, Trent DF. Frequency modulation of pulsatile luteinizing hormone-releasing hormone stimulation can alter the effectiveness of direct androgen feedback on luteinizing hormone-releasing hormone-induced luteinizing hormone release. Endocrinology. 1979;104:532–5. doi: 10.1210/endo-104-2-532. [DOI] [PubMed] [Google Scholar]
- Navarro CE, et al. Regulation of cyclic adenosine 3′,5′-monophosphate signaling and pulsatile neurosecretion by Gi-coupled plasma membrane estrogen receptors in immortalized gonadotropin-releasing hormone neurons. Mol Endocrinol. 2003;17:1792–804. doi: 10.1210/me.2003-0040. Epub 2003 Jun 20. [DOI] [PubMed] [Google Scholar]
- Pang SF, et al. Serum concentrations of testosterone, oestrogens, luteinizing hormone and follicle-stimulating hormone in male and female rats during the critical period of neural sexual differentiation. J Endocrinol. 1979;80:103–10. doi: 10.1677/joe.0.0800103. [DOI] [PubMed] [Google Scholar]
- Pastor CL, et al. Polycystic ovary syndrome: evidence for reduced sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by estradiol and progesterone. J Clin Endocrinol Metab. 1998;83:582–90. doi: 10.1210/jcem.83.2.4604. [DOI] [PubMed] [Google Scholar]
- Pelletier G, et al. Localization of oestrogen receptor alpha, oestrogen receptor beta and androgen receptors in the rat reproductive organs. J Endocrinol. 2000;165:359–70. doi: 10.1677/joe.0.1650359. [DOI] [PubMed] [Google Scholar]
- Petersen SL, Barraclough CA. Effect of naloxone and morphine on LH and prolactin release in androgen-sterilized rats. Neuroendocrinology. 1986;44:84–8. doi: 10.1159/000124626. [DOI] [PubMed] [Google Scholar]
- Petersen SL, Barraclough CA. Suppression of spontaneous LH surges in estrogen-treated ovariectomized rats by microimplants of antiestrogens into the preoptic brain. Brain Res. 1989;484:279–89. doi: 10.1016/0006-8993(89)90371-5. [DOI] [PubMed] [Google Scholar]
- Petersen SL, et al. Direct and indirect regulation of gonadotropin-releasing hormone neurons by estradiol. Biol Reprod. 2003;69:1771–8. doi: 10.1095/biolreprod.103.019745. [DOI] [PubMed] [Google Scholar]
- Pielecka J, Moenter SM. Effect of steroid milieu on gonadotropin-releasing hormone-1 neuron firing pattern and luteinizing hormone levels in male mice. Biol Reprod. 2006;74:931–7. doi: 10.1095/biolreprod.105.049619. [DOI] [PubMed] [Google Scholar]
- Poletti A, et al. Steroid binding and metabolism in the luteinizing hormone-releasing hormone-producing neuronal cell line GT1-1. Endocrinology. 1994;135:2623–8. doi: 10.1210/endo.135.6.7988451. [DOI] [PubMed] [Google Scholar]
- Rainbow TC, et al. Sex differences in rat brain oestrogen and progestin receptors. Nature. 1982;300:648–9. doi: 10.1038/300648a0. [DOI] [PubMed] [Google Scholar]
- Recabarren SE, et al. Postnatal developmental consequences of altered insulin sensitivity in female sheep treated prenatally with testosterone. Am J Physiol Endocrinol Metab. 2005;289:E801–6. doi: 10.1152/ajpendo.00107.2005. [DOI] [PubMed] [Google Scholar]
- Resko JA, Roselli CE. Prenatal hormones organize sex differences of the neuroendocrine reproductive system: observations on guinea pigs and nonhuman primates. Cell Mol Neurobiol. 1997;17:627–48. doi: 10.1023/A:1022534019718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhees RW, et al. Effects of prenatal testosterone on sexual behavior, reproductive morphology and LH secretion in the female rat. Dev Neurosci. 1997;19:430–7. doi: 10.1159/000111240. [DOI] [PubMed] [Google Scholar]
- Rhoda J, et al. Gonadal steroid concentrations in serum and hypothalamus of the rat at birth: aromatization of testosterone to 17 beta-estradiol. Endocrinology. 1984;114:1754–60. doi: 10.1210/endo-114-5-1754. [DOI] [PubMed] [Google Scholar]
- Robinson J. Prenatal programming of the female reproductive neuroendocrine system by androgens. Reproduction. 2006;132:539–47. doi: 10.1530/rep.1.00064. [DOI] [PubMed] [Google Scholar]
- Robinson JE, et al. In utero exposure of female lambs to testosterone reduces the sensitivity of the gonadotropin-releasing hormone neuronal network to inhibition by progesterone. Endocrinology. 1999;140:5797–805. doi: 10.1210/endo.140.12.7205. [DOI] [PubMed] [Google Scholar]
- Ronnekleiv OK, Kelly MJ. Diversity of ovarian steroid signaling in the hypothalamus. Front Neuroendocrinol. 2005;26:65–84. doi: 10.1016/j.yfrne.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Roos J, et al. Mechanisms of action of testosterone propionate on LH and FSH release by the pituitary gland in cyclic female rats. Acta Endocrinol (Copenh) 1980;95:158–65. doi: 10.1530/acta.0.0950158. [DOI] [PubMed] [Google Scholar]
- Roselli CE, et al. Quantitative distribution of nuclear androgen receptors in microdissected areas of the rat brain. Neuroendocrinology. 1989;49:449–53. doi: 10.1159/000125151. [DOI] [PubMed] [Google Scholar]
- Rosenfield RL. Current concepts of polycystic ovary syndrome. Baillieres Clin Obstet Gynaecol. 1997;11:307–33. doi: 10.1016/s0950-3552(97)80039-9. [DOI] [PubMed] [Google Scholar]
- Roy D, et al. Estrogen directly respresses gonadotropin-releasing hormone (GnRH) gene expression in estrogen receptor-alpha (ERalpha)- and ERbeta-expressing GT1–7 GnRH neurons. Endocrinology. 1999;140:5045–53. doi: 10.1210/endo.140.11.7117. [DOI] [PubMed] [Google Scholar]
- Rudick CN, Woolley CS. Estrogen regulates functional inhibition of hippocampal CA1 pyramidal cells in the adult female rat. J Neurosci. 2001;21:6532–43. doi: 10.1523/JNEUROSCI.21-17-06532.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush ME, Blake CA. Serum testosterone concentrations during the 4-day estrous cycle in normal and adrenalectomized rats. Proc Soc Exp Biol Med. 1982;169:216–21. doi: 10.3181/00379727-169-41334. [DOI] [PubMed] [Google Scholar]
- Ryan BC, Vandenbergh JG. Intrauterine position effects. Neurosci Biobehav Rev. 2002;26:665–78. doi: 10.1016/s0149-7634(02)00038-6. [DOI] [PubMed] [Google Scholar]
- Sam S, Dunaif A. Polycystic ovary syndrome: syndrome XX? Trends Endocrinol Metab. 2003;14:365–70. doi: 10.1016/j.tem.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Sarma HN, et al. Fetal programming: excess prenatal testosterone reduces postnatal luteinizing hormone, but not follicle-stimulating hormone responsiveness, to estradiol negative feedback in the female. Endocrinology. 2005;146:4281–91. doi: 10.1210/en.2005-0322. Epub 2005 Jun 23. [DOI] [PubMed] [Google Scholar]
- Shakil T, et al. Differential regulation of gonadotropin-releasing hormone secretion and gene expression by androgen: membrane versus nuclear receptor activation. Mol Endocrinol. 2002;16:2592–602. doi: 10.1210/me.2002-0011. [DOI] [PubMed] [Google Scholar]
- Shiina H, et al. Premature ovarian failure in androgen receptor-deficient mice. Proc Natl Acad Sci U S A. 2006;103:224–9. doi: 10.1073/pnas.0506736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shupnik MA. Gonadotropin gene modulation by steroids and gonadotropin-releasing hormone. Biol Reprod. 1996;54:279–86. doi: 10.1095/biolreprod54.2.279. [DOI] [PubMed] [Google Scholar]
- Siddiqui A, Shah BH. Neonatal androgen manipulation differentially affects the development of monoamine systems in rat cerebral cortex, amygdala and hypothalamus. Brain Res Dev Brain Res. 1997;98:247–52. doi: 10.1016/s0165-3806(96)00171-x. [DOI] [PubMed] [Google Scholar]
- Simerly RB. Organization and regulation of sexually dimorphic neuroendocrine pathways. Behav Brain Res. 1998;92:195–203. doi: 10.1016/s0166-4328(97)00191-5. [DOI] [PubMed] [Google Scholar]
- Simerly RB. Wired for reproduction: organization and development of sexually dimorphic circuits in the mammalian forebrain. Annu Rev Neurosci. 2002;25:507–36. doi: 10.1146/annurev.neuro.25.112701.142745. [DOI] [PubMed] [Google Scholar]
- Simerly RB. Wired on hormones: endocrine regulation of hypothalamic development. Curr Opin Neurobiol. 2005;15:81–5. doi: 10.1016/j.conb.2005.01.013. [DOI] [PubMed] [Google Scholar]
- Simerly RB, et al. Distribution of androgen and estrogen receptor mRNA-containing cells in the rat brain: an in situ hybridization study. J Comp Neurol. 1990;294:76–95. doi: 10.1002/cne.902940107. [DOI] [PubMed] [Google Scholar]
- Simerly RB, et al. The distribution of monoaminergic cells and fibers in a periventricular preoptic nucleus involved in the control of gonadotropin release: immunohistochemical evidence for a dopaminergic sexual dimorphism. Brain Res. 1985;330:55–64. doi: 10.1016/0006-8993(85)90007-1. [DOI] [PubMed] [Google Scholar]
- Skynner MJ, et al. Detection of estrogen receptor alpha and beta messenger ribonucleic acids in adult gonadotropin-releasing hormone neurons. Endocrinology. 1999;140:5195–201. doi: 10.1210/endo.140.11.7146. [DOI] [PubMed] [Google Scholar]
- Slob AK, et al. Prenatal testosterone propionate and postnatal ovarian activity in the rat. Acta Endocrinol (Copenh) 1983;103:420–7. doi: 10.1530/acta.0.1030420. [DOI] [PubMed] [Google Scholar]
- Slob AK, et al. Prenatal and early postnatal sex differences in plasma and gonadal testosterone and plasma luteinizing hormone in female and male rats. J Endocrinol. 1980;87:81–7. doi: 10.1677/joe.0.0870081. [DOI] [PubMed] [Google Scholar]
- Smith JT, et al. Regulation of Kiss1 gene expression in the brain of the female mouse. Endocrinology. 2005a;146:3686–92. doi: 10.1210/en.2005-0488. [DOI] [PubMed] [Google Scholar]
- Smith JT, et al. Differential regulation of KiSS-1 mRNA expression by sex steroids in the brain of the male mouse. Endocrinology. 2005b;146:2976–84. doi: 10.1210/en.2005-0323. [DOI] [PubMed] [Google Scholar]
- Smith JT, et al. Kiss1 neurons in the forebrain as central processors for generating the preovulatory luteinizing hormone surge. J Neurosci. 2006;26:6687–94. doi: 10.1523/JNEUROSCI.1618-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MJ, Jennes L. Neural signals that regulate GnRH neurones directly during the oestrous cycle. Reproduction. 2001;122:1–10. doi: 10.1530/rep.0.1220001. [DOI] [PubMed] [Google Scholar]
- Steiner RA, et al. Sexual differentiation and feedback control of luteinizing hormone secretion in the rhesus monkey. Biol Reprod. 1976;15:206–12. doi: 10.1095/biolreprod15.2.206. [DOI] [PubMed] [Google Scholar]
- Sullivan SD, Moenter SM. Prenatal androgens alter GABAergic drive to gonadotropin-releasing hormone neurons: implications for a common fertility disorder. Proc Natl Acad Sci U S A. 2004;101:7129–34. doi: 10.1073/pnas.0308058101. Epub 2004 Apr 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson H, van der Werff ten Bosch J. The “early-androgen” syndrome; differences in response to pre-natal and postnatal adminstration of various doses of testosterone propionate in female and male rats. Acta Endocrinol (Copenh) 1964;47:37–50. [PubMed] [Google Scholar]
- Swanson H, van der Werff ten Bosch J. The “early-androgen” syndrome: effects of pre-natal testosterone propionate. Acta Endocrinol (Copenh) 1965;50:379–390. doi: 10.1530/acta.0.0500379. [DOI] [PubMed] [Google Scholar]
- Taga M, et al. Effects of sex steroid hormones on rat anterior pituitary LH-RH receptor. Nippon Sanka Fujinka Gakkai Zasshi. 1982;34:627–33. [PubMed] [Google Scholar]
- Tarttelin MF, Gorski RA. Postnatal influence of diethylstilbestrol on the differentiation of the sexually dimorphic nucleus in the rat is as effective as perinatal treatment. Brain Res. 1988;456:271–4. doi: 10.1016/0006-8993(88)90227-2. [DOI] [PubMed] [Google Scholar]
- Temple JL, et al. Direct action of estradiol on gonadotropin-releasing hormone-1 neuronal activity via a transcription-dependent mechanism. J Neurosci. 2004;24:6326–33. doi: 10.1523/JNEUROSCI.1006-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton J. Effects of Prenatal Androgens on Adult Ovarian Cyclicity and Femal Sexual Behavior in the Rhesus Monkey. PhD. University of Wisconsin; Madison: 1983. [Google Scholar]
- Tsukahara S. Increased Fos immunoreactivity in suprachiasmatic nucleus before luteinizing hormone surge in estrogen-treated ovariectomized female rats. Neuroendocrinology. 2006;83:303–12. doi: 10.1159/000095341. [DOI] [PubMed] [Google Scholar]
- Turgeon JL, Waring DW. Androgen modulation of luteinizing hormone secretion by female rat gonadotropes. Endocrinology. 1999;140:1767–74. doi: 10.1210/endo.140.4.6642. [DOI] [PubMed] [Google Scholar]
- Unsworth WP, et al. Prenatal programming of reproductive neuroendocrine function: the effect of prenatal androgens on the development of estrogen positive feedback and ovarian cycles in the ewe. Biol Reprod. 2005;72:619–27. doi: 10.1095/biolreprod.104.035691. [DOI] [PubMed] [Google Scholar]
- Urban JH, et al. Steroid modulation of neuropeptide Y-induced luteinizing hormone releasing hormone release from median eminence fragments from male rats. Neuroendocrinology. 1996;63:112–9. doi: 10.1159/000126947. [DOI] [PubMed] [Google Scholar]
- Van der Beek EM, et al. Evidence for a direct neuronal pathway from the suprachiasmatic nucleus to the gonadotropin-releasing hormone system: combined tracing and light and electron microscopic immunocytochemical studies. J Comp Neurol. 1997;384:569–79. doi: 10.1002/(sici)1096-9861(19970811)384:4<569::aid-cne6>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Watson RE, Jr, et al. Estrogen-receptive neurons in the anteroventral periventricular nucleus are synaptic targets of the suprachiasmatic nucleus and peri-suprachiasmatic region. Brain Res. 1995;689:254–64. doi: 10.1016/0006-8993(95)00548-5. [DOI] [PubMed] [Google Scholar]
- Watts AG, Fink G. Pulsatile luteinizing hormone release, and the inhibitory effect of estradiol-17 beta in gonadectomized male and female rats: effects of neonatal androgen or exposure to constant light. Endocrinology. 1984;115:2251–9. doi: 10.1210/endo-115-6-2251. [DOI] [PubMed] [Google Scholar]
- Weigel RJ, de Coninck EC. Transcriptional control of estrogen receptor in estrogen receptor-negative breast carcinoma. Cancer Res. 1993;53:3472–4. [PubMed] [Google Scholar]
- Weiland NG, Barraclough CA. Neonatal castration of male and female rats affects luteinizing hormone responses to estrogen during adulthood. Biol Reprod. 1984;31:942–9. doi: 10.1095/biolreprod31.5.942. [DOI] [PubMed] [Google Scholar]
- Weisz J, Ward IL. Plasma testosterone and progesterone titers of pregnant rats, their male and female fetuses, and neonatal offspring. Endocrinology. 1980;106:306–16. doi: 10.1210/endo-106-1-306. [DOI] [PubMed] [Google Scholar]
- West C, et al. Intra-follicular activin availability is altered in prenatally-androgenized lambs. Mol Cell Endocrinol. 2001;185:51–9. doi: 10.1016/s0303-7207(01)00632-3. [DOI] [PubMed] [Google Scholar]
- Whalen RE, Luttge WG. Perinatal administration of dihydrotestosterone to female rats and the development of reproductive function. Endocrinology. 1971;89:1320–2. doi: 10.1210/endo-89-5-1320. [DOI] [PubMed] [Google Scholar]
- Wiegand SJ, Terasawa E. Discrete lesions reveal functional heterogeneity of suprachiasmatic structures in regulation of gonadotropin secretion in the female rat. Neuroendocrinology. 1982;34:395–404. doi: 10.1159/000123335. [DOI] [PubMed] [Google Scholar]
- Wierman ME, et al. Selective failure of androgens to regulate follicle stimulating hormone beta messenger ribonucleic acid levels in the male rat. Mol Endocrinol. 1988;2:492–8. doi: 10.1210/mend-2-6-492. [DOI] [PubMed] [Google Scholar]
- Wierman ME, et al. Divergent regulation of gonadotropin subunit mRNA levels by androgens in the female rat. Biol Reprod. 1990;43:191–5. doi: 10.1095/biolreprod43.2.191. [DOI] [PubMed] [Google Scholar]
- Winters SJ, et al. Effects of testosterone on gonadotropin subunit messenger ribonucleic acids in the presence or absence of gonadotropin-releasing hormone. Endocrinology. 1992;130:726–34. doi: 10.1210/endo.130.2.1370794. [DOI] [PubMed] [Google Scholar]
- Wood RI, Foster DL. Sexual differentiation of reproductive neuroendocrine function in sheep. Rev Reprod. 1998;3:130–40. doi: 10.1530/ror.0.0030130. [DOI] [PubMed] [Google Scholar]
- Xita N, Tsatsoulis A. Review: fetal programming of polycystic ovary syndrome by androgen excess: evidence from experimental, clinical, and genetic association studies. J Clin Endocrinol Metab. 2006;91:1660–6. doi: 10.1210/jc.2005-2757. [DOI] [PubMed] [Google Scholar]
- Yamane K, et al. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell. 2006;125:483–95. doi: 10.1016/j.cell.2006.03.027. [DOI] [PubMed] [Google Scholar]
- Yan L, et al. Role of DNA methylation and histone acetylation in steroid receptor expression in breast cancer. J Mammary Gland Biol Neoplasia. 2001;6:183–92. doi: 10.1023/a:1011308707512. [DOI] [PubMed] [Google Scholar]
- Yasin M, et al. Testosterone is required for gonadotropin-releasing hormone stimulation of luteinizing hormone-beta messenger ribonucleic acid expression in female rats. Endocrinology. 1996;137:1265–71. doi: 10.1210/endo.137.4.8625898. [DOI] [PubMed] [Google Scholar]
- Yeh S, et al. Generation and characterization of androgen receptor knockout (ARKO) mice: an in vivo model for the study of androgen functions in selective tissues. Proc Natl Acad Sci U S A. 2002;99:13498–503. doi: 10.1073/pnas.212474399. Epub 2002 Oct 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zup SL, et al. Overexpression of bcl-2 reduces sex differences in neuron number in the brain and spinal cord. J Neurosci. 2003;23:2357–62. doi: 10.1523/JNEUROSCI.23-06-02357.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]