

Abstract
Background
Congenital nephrogenic diabetes insipidus (NDI) is characterised by an inability to concentrate urine despite normal or elevated plasma levels of the antidiuretic hormone arginine vasopressin. We report a Japanese extended family with NDI caused by an 11.2-kb deletion that includes the entire AVPR2 locus and approximately half of the Rho GTPase-activating protein 4 (ARHGAP4) locus. ARHGAP4 belongs to the RhoGAP family, Rho GTPases are critical regulators of many cellular activities, such as motility and proliferation which enhances intrinsic GTPase activity.
ARHGAP4 is expressed at high levels in hematopoietic cells, and it has been reported that an NDI patient lacking AVPR2 and all of ARHGAP4 showed immunodeficiency characterised by a marked reduction in the number of circulating CD3+ cells and almost complete absence of CD8+ cells.
Methods
PCR and sequencing were performed to identify the deleted region in the Japanese NDI patients. Immunological profiles of the NDI patients were analysed by flow cytometry. We also investigated the gene expression profiles of peripheral blood mononuclear cells (PBMC) from NDI patients and healthy controls in microarray technique.
Results
We evaluated subjects (one child and two adults) with 11.2-kb deletion that includes the entire AVPR2 locus and approximately half of the ARHGAP4. Hematologic tests showed a reduction of CD4+ cells in one adult patient, a reduction in CD8+ cells in the paediatric patient, and a slight reduction in the serum IgG levels in the adult patients, but none of them showed susceptibility to infection. Gene expression profiling of PBMC lacking ARHGAP4 revealed that expression of RhoGAP family genes was not influenced greatly by the lack of ARHGAP4.
Conclusion
These results suggest that loss of ARHGAP4 expression is not compensated for by other family members. ARHGAP4 may play some role in lymphocyte differentiation but partial loss of ARHGAP4 does not result in clinical immunodeficiency.
Background
Maintenance of body fluid volume and composition is essential for proper physiologic function in humans. Under normal conditions, the glomerular filtration rate of the two kidneys is 180 L day-1, and up to 90% of the filtrate is reabsorbed in the proximal tubule and descending limb of Henle's loop. The key hormone that regulates reabsorption is the antidiuretic hormone arginine vasopressin (AVP), which is secreted by the posterior pituitary in response to hypovolemia or hypernatremia [1]. AVP is transported by the blood to the kidney and binds to arginine vasopressin receptor 2 (AVPR2), leading to an increase in intracellular cAMP levels via the stimulatory Gs protein and adenylate cyclase, and to subsequent activation of protein kinase A and phosphorylation of aquaporin 2 (AQP2) water channels [2]. This process is necessary for proper reabsorption of the water in the principal cells of the collecting duct under the control of AVPR2 [3].
Congenital nephrogenic diabetes insipidus (NDI) is characterised by an inability to concentrate urine despite a normal or elevated plasma level of AVP. Two genes have been reported to be associated with NDI; X-linked AVPR2 [4] and autosomal AQP2 [5,6]. The X-linked form of NDI is present in up to 90% of patients. Males with the disease-causing mutation are usually affected, and females heterozygous for the disease-causing mutation show various degrees of penetrance. Skewed X inactivation, which is preferential methylation of the normal allele of the AVPR2 gene, can cause NDI in female heterozygotes [7].
To date, 178 AVPR2 mutations, including 12 gross deletions [8-13], have been deposited in the BIOBASE database [14]. Large deletions that lead to complete loss of AVPR2 and parts of the neighboring genes ARHGAP4 [9,11,15] and L1 cell adhesion molecule (L1CAM) [16] have been reported. ARHGAP4, which is a member of the GTPase-activating protein family, is located telomeric to AVPR2 and is expressed at a high level in hematopoietic cells. Recently, an NDI patient lacking AVPR2 and all of ARHGAP4 showed immunodeficiency characterised by a marked reduction in the number of circulating CD3+ cells and almost complete absence of CD8+ cells [17]. Herein, we describe a Japanese extended family with multiple NDI patients lacking the entire AVPR2 locus and approximately half of ARHGAP4. Although none of the family members with NDI showed clinical signs of immunodeficiency, immunologic profiling showed slight abnormalities.
Results
Mutation screening
Two patients (IV-2 and IV-4 in Figure 1) were admitted to the hospital at the age of 2 months with fever of unknown origin. NDI was diagnosed on the basis of clinical symptoms and laboratory findings (dehydration, hypernatremia, and hypotonic urine) and failure to increase urine osmolarity in response to 1-2esamino-8-D-arginine vasopressin (dDAVP) (Table 1). The sister (IV-1) had no history of dehydration, but polyuria and polydipsia were noticed by her family members, and NDI was diagnosed on the basis of laboratory findings at the age of 2 years. None of the patients had any evidence of mental retardation or significant disease other than NDI. The pedigree of the Japanese NDI family is shown in Figure 1. Subjects indicated by a diagonal box (II-1, III-3, III-4, and II-4) have a history of polyuria and polydipsia since childhood, but NDI has not been diagnosed.
Figure 1.
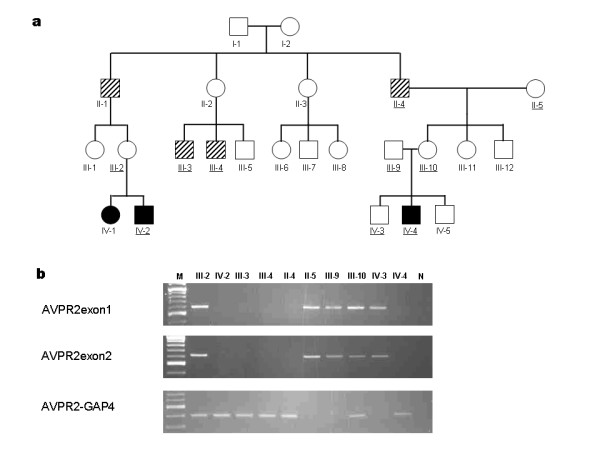
a; The pedigree of the family with NDI. A closed pox indicates affected members. Several family members (II-1, II-4, III-3, and III-4, indicated by a diagonal box) have histories of polyuria and polydipsia but have not been diagnosed with NDI. Family members marked with underlines are ones for PCR analysis. b; PCR amplification of the NDI family members. M, 100 bp marker, N, negative control. The regions amplified with the primer pairs (AVPR2exon1, AVPR2exon2, and AVPR2-GAP) are shown in Figure 2.
Table 1.
basal urine osmolality and plasma osmolality
| Basal urine osmolality | Urine osmolality response to AVP or dDAVP | Plasma osmolality | |
| IV-1 | 102 | 102→171 | NA |
| IV-2 | 30 | 30→26 | NA |
| IV-4 | 100 | 100→125 | 308 |
NA; data not available
Genomic DNA from patients and available family members was subjected to PCR analysis. We first used the primer pair (additional file 1) that amplifies the genomic region between exon 1 and exon 3 of AVPR2, and no PCR products were amplified from DNA of IV-2 and IV-4 (data not shown). Therefore, we hypothesised that a large sequence around the AVPR2 gene was deleted and performed PCR using the primers listed in the additional file 1. After narrowing the deleted region, we designed the primer pair AVPR2-GAP4 and obtained 386-bp PCR products for III-2, IV-2, III-3, III-4, II-4, III-10, and IV-4 (Figure 1), suggesting that these seven family members carry a large deletion that covers AVPR2 and part of ARHGAP4. The AVPR2exon1 and AVPR2exon2 primer pairs amplify the genomic region of AVPR2 exon 1 and exon 2, respectively. PCR was successful with DNAs from III-2, II-5, III-9, III-10, and IV-3, indicating that III-2 and III-10 are asymptomatic female carriers of the deletion. The physical map and identified deleted regions are shown in Figure 2. The deleted region includes all of the AVPR2 gene locus and approximately half of the ARHGAP4 gene. Sequencing of the 386-bp product from IV-4 revealed that the 5' and 3' break points were at positions 53,385 and 64,673 bp, respectively, relative to the published sequence (GenBank U52112.2) with an insertion of the GGGTACACCTC sequence in the break point (Figure 3).
Figure 2.
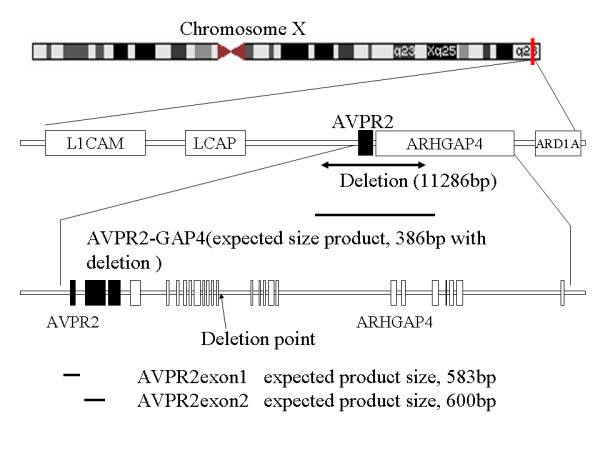
The physical map and the deleted region in the Japanese NDI family. 11,286 bp deletion was observed around AVPR2 gene. Bold lines indicated amplified regions with primers (AVPR2-GAP, 386 bp with deletion, AVPR2exon1, expected product size 583 bp with no deletion, and AVPR2exon2, expected product size 600 bp with no deletion). 5' deleted point occurred between LCAP and AVPR2, and 3' deleted point was in the intron 10 of ARHGAP4.
Figure 3.
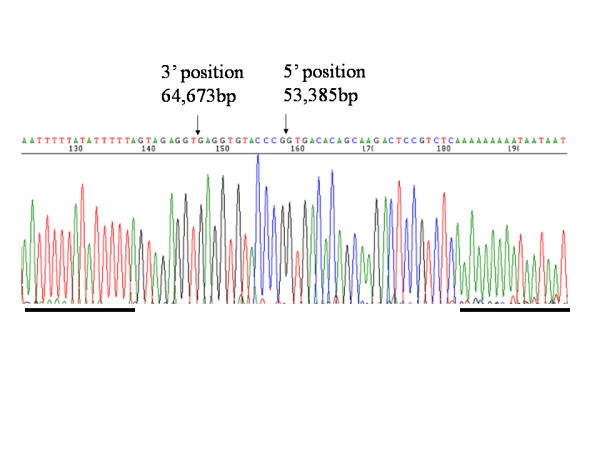
Sequence of deletion breakpoint in the NDI family. 5'and 3' breakpoints at positions 53,385 and 64,673 bp, respectively, relative to the published sequence (GenBank U52112.2) are indicated with arrows. The sequence is shown in antisense orientation. There is an insertion of the "GAGGTGTACCC" sequence in the break point. Bold lines indicate palindromic sequence which could cause the interstitial deletion.
Characteristics of immune function
We evaluated the immune function of 3 family members lacking ARHGAP4. None of the patients in the present study had a clinical history of recurrent or opportunistic infections. Differential leukocyte counts were within normal range (data not shown), however, a reduction of CD4+ cells was observed in III-4, and a reduction of CD8+ cells was observed in IV-4 (Table 2). Normal levels of CD3+CD4+CD45RA+ and CD3+CD4+CD45RO+ T cells were observed [18] and T cell receptor excision circles (TREC) of whole blood were within normal range in three patients tested. Serum IgG levels were relatively low in adult patients with slight reduction of CD19+ cells in III-3 and III-4. However differentiation of B cells using IgD, IgM and CD27 was not different from normal controls (data not shown).
Table 2.
Immunological evaluation of NDI patients
| III-3 32 yr |
III-4 29 yr |
IV-4 5 yr |
Normal range (adult) | Normal range (children) | |
| CD3+ [%] | 74.3 | 68.9 | 85.9 | 55–78 | 55–83 |
| CD4+/CD3+ [%] | 54.9 | 17.7 | 69.6 | 28–57 | 27–53 |
| CD8+/CD3+ [%] | 34.5 | 61.8 | 14 | 10–39 | 19–34 |
| CD4/CD8 | 1.6 | 0.3 | 5 | 1.0–3.6 | 0.9–2.6 |
| CD19+ [%] | 5.2 | 5.7 | 6.7 | 6–19 | 10–31 |
| IgG (mg/dl) | 810.0 | 741.0 | 754.0 | 870–1700 | 600–1450 |
| IgA (mg/dl) | 147.0 | 72.0 | 105.0 | 110–410 | 39–233 |
| IgM (mg/dl) | 59.0 | 127.0 | 72.0 | 33–190 | 69–268 |
| TREC/μgDNA | 1.9 × 102 | 2.8 × 102 | 1.6 × 103 | 3.4 ± 3.6 × 102 | 3.5 ± 2.8 × 103 |
The normal range of the lymphocyte subpopulation was lower and the upper confidence limit at a confidence level of 0.95. The normal range of IgG, IgA, and IgM are calculated as mean - 2SD (lower) and mean + 2SD (upper). The normal range of TREC is expressed as mean ± SD.
Microarray analysis
We examined the gene expression profiles of peripheral blood mononuclear cells (PBMCs) from subjects lacking ARHGAP4 (III-3 and III-4) and 6 healthy male volunteers (age, 23–39 years). Transcripts satisfying all of the following criteria were identified as up- or down-regulated by microarray analysis: (1) transcripts that were expressed in at least 2 of 8 samples, (2) transcripts that showed statistically significant differences between subjects lacking ARHGAP4 and, controls (Welch t-test, q < 0.05; multiple tests were corrected by using the Benjamini and Hochberg false discovery rate [19]), and (3) transcripts that showed an average increase/decrease of more than 1.5-fold. Transcripts of ARHGAP4 in PBMCs in III-3 and III-4 were absent, whereas strong expression was observed in all of the controls. Up- and down-regulated genes in PBMCs lacking ARHGAP4 are listed in Table 3. We hypothesized that lack of ARHGAP4 could be compensated for by upregulation of other RhoGAP family members. Fifty-seven transcripts belonging to the RhoGAP family were included in the microarray analysis, and 25, including ARHGAP4 were expressed in at least 2 of 8 samples. Expression levels of the transcripts other than ARHGAP4 are shown in Table 4. None of the transcripts of the RhoGAP family members were statistically significantly up/down regulated in the PBMCs.
Table 3.
Up/down regulated genes in PBMC lacking ARHGAP4
| Gene Name | Description | aFold change | b q value | cAccession |
| Up-regulation | ||||
| PLK3 | Polo-like kinase 3 | 4.35 | 0.0402 | NM_004073 |
| NHSL2 | NHS-like 2 | 2.38 | 0.0453 | NM_001013627 |
| PTAFR | Platelet-activating factor receptor | 2.27 | 0.0288 | NM_000952 |
| KLHL26 | Kelch-like 26 | 1.99 | 0.0272 | NM_018316 |
| MAPKAPK2 | Mitogen-activated protein kinase- activated protein kinase 2 | 1.96 | 0.0442 | NM_032960 |
| CKAP4 | Cytoskeleton-associated protein 4 | 1.85 | 0.0272 | NM_006825 |
| CYB5R3 | Cytochrome b5 reductase 3 | 1.82 | 0.0328 | NM_007326 |
| ZC3H12A | Zinc finger CCCH-type containing | 1.81 | 0.0428 | NM_025079 |
| FURIN | Furin | 1.79 | 0.0448 | NM_002569 |
| GRN | Granulin | 1.76 | 0.0453 | NM 002087 |
| PPOX | Protoporphyrinogen oxidase | 1.75 | 0.0307 | NM_000309 |
| NQO2 | NAD(P)H dehydrogenase, quinone 2 | 1.74 | 0.0453 | NM_000904 |
| DPH3 | Diphthamide biosynthesis protein 3 | 1.70 | 0.0307 | NM_206831 |
| PGD | Phosphogluconate dehydrogenase | 1.68 | 0.00562 | NM_002631 |
| RIPK5 | Receptor interacting protein kinase 5 | 1.66 | 0.0386 | NM_015375 |
| CD14 | CD14 molecule | 1.62 | 0.0272 | NM_000591 |
| PGD | Phosphogluconate dehydrogenase | 1.57 | 0.0272 | NM_002631 |
| Down-regulation | ||||
| TUSC4 | Tumor suppressor candidate 4 | 0.63 | 0.0453 | NM_006545 |
| PLEKHG4 | Pleckstrin homology domain containing, family G (with RhoGef | 0.62 | 0.0272 | NM_015432 |
| SFXN1 | Sideroflexin 1 | 0.61 | 0.0272 | NM_022754 |
| GNRH1 | Gonadotropin-releasing hormone 1 | 0.60 | 0.0328 | NM_000825 |
| PPP1R3E | Protein phosphatase 1, regulatory | 0.60 | 0.0383 | XM_940069 |
| MC1R | Melanocortin 1 receptor | 0.56 | 0.0272 | NM_002386 |
| LRCH4 | Leucine-rich repeats and calponin homology (CH) domain containing 4 | 0.55 | 0.0411 | NM_002319 |
| LOC642787 | Hypothetical protein LOC642787 | 0.44 | 0.0288 | XM_926202 |
| SGPP2 | Sphingosine-1-phosphate phosphotase | 0.09 | 0.0307 | XM_938742 |
| BNIPL | BCL2/adenovirus E1B 19 kD | 0.06 | 0.0226 | NM_138278 |
| ARHGAP4 | Rho GTPase activating protein 4 | 0.00 | 0.000567 | NM_001666 |
aThe fold change was determined by calculating the ratio of global normalized signals from the PBMCs of NDI patients to those from the PBMCs of normal subjects. bP values corrected by the Benjamini and Hochberg false discovery rate, and expressed as q values cGenBank accession numbers
Table 4.
RhoGAP family gene expressions in PBMC lacking ARHGAP4
| Gene Name | Description | aFold change | bq value | cAccession |
| ARHGAP1 | Rho GTPase activating protein 1 | 0.83 | 0.473 | NM_004308 |
| ARHGAP9 | Rho GTPase activating protein 9 | 1.23 | 0.473 | NM_032496 |
| ARHGAP10 | Rho GTPase activating protein10 | 1.01 | 0.914 | NM_024605 |
| ARHGAP12 | Rho GTPase activating protein 12 | 1.03 | 0.655 | NM_018287 |
| ARHGAP17 | Rho GTPase activating protein 17 | 0.67 | 0.473 | NM_018054 |
| ARHGAP19 | Rho GTPase activating protein 19 | 1.16 | 0.473 | NM_032900 |
| ARHGAP21 | Rho GTPase activating protein 21 | 0.98 | 0.655 | NM_020824 |
| ARHGAP24 | Rho GTPase activating protein 24 | 1.33 | 0.891 | NM_001025616 |
| ARHGAP25 | Rho GTPase activating protein 25 | 0.90 | 0.565 | NM_001007231 |
| ARHGAP25 | Rho GTPase activating protein 25 | 0.93 | 0.83 | NM 014882 |
| ARHGAP27 | Rho GTPase activating protein 27 | 0.81 | 0.473 | NM_199282 |
| ARHGAP30 | Rho GTPase activating protein 30 | 1.16 | 0.473 | NM_001025598 |
| ARHGAP30 | Rho GTPase activating protein 30 | 0.86 | 0.565 | NM_181720 |
| BM046 | Rho GTPase activating protein 15 | 0.92 | 0.655 | NM 018460 |
| CDGAP | Cdc42 GTPase-activating protein | 0.86 | 0.891 | NM 020754 |
| CENTD2 | Centaurin, delta 2 | 1.26 | 0.473 | NM_139181 |
| CENTD3 | Centaurin, delta 3 | 1.12 | 0.655 | NM_022481 |
| GMIP | GEM interacting protein | 1.17 | 0.473 | NM_016573 |
| MYO9B | Myosin IXB | 1.20 | 0.473 | NM_004145 |
| OCRL | Oculocerebrorenal syndrome of Lowe | 1.00 | 0.655 | NM_000276 |
| PIK3R1 | Phosphoinositide-3-kinase, regulatory | 0.94 | 0.571 | NM_181504 |
| PIK3R2 | Phosphoinositide-3-kinase, regulatory subunit 2 (beta) | 0.86 | 0.655 | NM_005027 |
| RACGAP1 | Rac GTPase activating protein 1 | 0.83 | 0.473 | NM 013277 |
| RALBP1 | RalA binding protein 1 | 0.84 | 0.565 | NM_006788 |
aFold change was determined by calculating the ratio of global normalized signals from PBMC of NDI patients to that of normal subjects. bP values corrected by the Benjamini and Hochberg false discovery rate, and expressed as q values cGenBank accession number
Discussion
In this study, we identified an extended Japanese family with X-linked NDI whose affected and carrier members have an 11.2-kb deletion and 11-bp insertion that leads to complete loss of the AVPR2 gene and part of the ARHGAP4 gene.
X-linked congenital NDI is caused by loss of or decreased function of AVPR2, and large deletions that lead to complete loss of AVPR2 and parts of the neighboring genes ARHGAP4 and L1CAM [9,11,15] have been reported.ARHGAP4 is expressed at high levels in hematopoietic cells. Because of the predominant expression pattern in hematopoietic cells, Schoneberg et al [11] performed immunologic analysis of blood from an NDI patient with complete loss of the AVPR2 gene and most of ARHGAP4. They observed a slight reduction in numbers of CD4+ cells and normal white blood cell counts, proliferation, cytokine and immunoglobulin production, respiratory burst, and phagocytosis. In addition to the patients in the present study, five NDI patients with partial deletion of ARHGAP4 have been reported [9,11,15], and none showed clinical signs of immunodeficiency. However, Broides et al. reported a pediatric NDI patient lacking AVPR2 and all of ARHGAP4 who showed immunodeficiency characterised by a marked reduction in the number of circulating CD3+ cells and an almost complete absence of CD8+ cells [17]. In this patient, the deleted region included 1.4 kb of intron 1, the first exon of ARHGAP4 and a 2.8 kb sequence between ARHGAP4 and ARD1A; this sequence is highly conserved across species, and Broides et al, speculated that this conserved region was the cause of the immunodeficiency phenotype observed in their patient. Hematologic tests of our patients showed a reduction of CD4+ cells in III-4, a reduction of CD8+ cells in IV-4, and a slight reduction of serum IgG levels in adult patients, but none of our patients showed susceptibility to infection. Taken together with the results of previous studies, our present results suggest that ARHGAP4 plays some role in lymphocyte differentiation, but does not cause the immunodeficiency phenotype. ARHGAP4 belongs to the RhoGAP family which enhances intrinsic GTPase activity. RhoGTPases are critical regulators of many cellular activities, such as motility and proliferation. We used the microarray technique to examine the possible effects on gene expression patterns in PBMCs by lacking ARHGAP4; several up-/down- regulated genes were identified. Our microarray study has several limitations. Firstly, it should be noted that microarray analysis is a screening method for detecting genes that may be regulated. Bosotti et al., reported that genes detected as differentially expressed by microarray platforms were also found to be differentially expressed by real-time quantitative PCR, although differences in the magnitudes of individual expression ratios were observed. [20] Secondly, PBMCs comprise a heterogeneous population of cells, and the possibility remains that the gene expression changes observed may merely reflect changes in the population of cells. However, because these patients lack ARHGAP4 gene expression in all tissues and cells, it is likely that gene expression changes may be detected even in the heterogeneous cell populations. Gene expression profiling of PBMCs lacking ARHGAP4 revealed that the expression of RhoGAP family genes was not influenced greatly by the lack of ARHGAP4, suggesting that loss of ARHGAP4 expression is not compensated for by other gene family members.
In the present study, one girl (IV-1) was affected by NDI. Female carriers do not usually show symptoms of NDI as observed in IV-2 and IV-4, but a small proportion of female carriers develop varying degrees of polyuria and polydipsia that can be explained by skewed inactivation of the X chromosome containing the functional AVPR2 allele. Although genetic testing has not been done for IV-1, it is very likely that she is a carrier of the deletion and that skewed X inactivation caused her symptoms.
A recent study showed that ARHGAP4 inhibits the migration of NIH/3T3 cells and the outgrowth of hippocampal axons and that the N-terminus and C-terminus of ARHGAP4 play different but essential roles in the potent inhibition of cell and axon motility [21]. The patients in the present study lack the C-terminus of ARHGAP4 (amino acids 515–965 according to the reference sequence NM_001666), but none of them showed clinical signs of immunodeficiencies or neurologic disorders. Further studies are needed to elucidate the role of ARHGAP4 in immune function and neuronal development.
Conclusion
Our study indicated that loss of ARHGAP4 expression is not compensated for by other family members. ARHGAP4 may play some role in lymphocyte differentiation but partial loss of ARHGAP4 dose not result in clinical immunodeficiency.
Methods
Patients and mutation detection
The pedigree of the Japanese NDI family is shown in Figure 1. A full verbal and written explanation of the study was given to family members, and informed consent for the genetic study was obtained from members who participated in this study. Informed consent for the children was provided by their parents. This study was approved by the Committee of Ethics of the University of Tsukuba and was performed in accordance with the ethical standards of the 1964 Declaration of Helsinki. DNA was extracted from peripheral blood leukocytes collected in ethylenediaminetetraacetic acid (EDTA). To identify the deleted region around AVPR2, primers were designed according to the published sequence (GenBank accession number U52112.2) with Primer3 software [22]. The primer sequences are listed in the additional file 1. Sequencing was performed with the Big Dye Terminator kit (Applied Biosystems, Foster City, California, USA) on an ABI PRISM 3100 DNA Sequencer (Applied Biosystems).
Immunological analysis
Peripheral blood samples from III-3, III-4 and IV-4 in Figure 1 were labeled with monoclonal antibodies according to the manufacturer's instructions and analysed with a FACSCalibur system (BD Biosciences, San Jose, California, USA). Monoclonal antibodies were purchased from BD Biosciences or Beckman-Coulter (Fullerton, California, USA). Quantification of TRECs was performed as described previously [23]. In brief, total DNA was isolated from 100 μl of peripheral blood with the use of a QIAamp DNA Micro Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. The DNA concentration was determined with a GeneQuant pro system (GE Healthcare Bio-Sciences Corp. Piscataway, New Jersey, USA). Quantitative real-time polymerase chain reaction (PCR) for δRec-ψJα sjTRECs was performed with the following primers and probe: δRec primer, 5'-TCGTGAGAACGGTGAATGAAG-3', ψJα primer, 5'-CCATGCTGACACC'TCTGGTT-3', and δRec probe, FAM-5'-CACGGTGATGCATAGGCACCTGC-3'-TAMRA. As an internal control, RNase P genes were amplified in each sample tested. In each experiment, serial dilutions (109, 108, 107, 106, 105, 103, 101, and 100) of the subcloned sjTREC-plasmid (pCR4-TOPO, Invitrogen Corp., Carlsbad, California, USA) and RNase P-plasmid were used as standards for absolute quantification of TRECs and RNase P copies. Each 20-μl reaction contained 1 μl DNA, and the final concentration of each component for TREC-PCR was as follows: 2× TaqMan Universal PCR Master Mix (Applied Biosystems), 500 nM of each primer, and 250 nM TaqMan probe. The final concentration of each component for RNase P-PCR was as follows: 2× TaqMan Universal PCR Master Mix and 20× TaqMan RNase P Primer Probe (VIC dye) Mix (Applied Biosystems). PCR conditions were 50°C for 2 minutes, 95°C for 10 minutes, and 40 cycles at 95°C for 15 seconds and 60°C for 1 minute. Experiments were performed and analysed with an ABI PRISM 7300 Sequence Detection System (Applied Biosystems). Results were extrapolated to the number of TRECs per μg DNA and that of RNase P copies per μg DNA. To confirm that the DNA yields were adequately purified, TRECs were also normalised to copies of RNase P genomic DNA.
Microarray analysis
PBMCs from III-3, III-4, and six healthy male volunteers (age, 23–39 years) were purified with a Ficoll-Paque™ gradient (GE Healthcare Bio-Sciences Corp.). RNA from PBMCs was extracted with an RNeasy Mini Kit (Qiagen) according to the manufacturer's instructions. cRNA was synthesized with an Illumina® RNA Amplification Kit (Ambion, Austin, Texas, USA) according to the manufacturer's instructions. In brief, 500 ng total RNA from PBMCs was reverse transcribed to synthesise first and second strand cDNA and purified on spin columns, and in vitro transcription was performed to synthesise biotin-labeled cRNA. A total of 1500 ng biotin-labelled cRNA was hybridised to a Sentrix Human-6 Expression BeadChip version 2 (Illumina, San Diego, California, USA) at 55°C for 18 hours. The hybridised BeadChip was washed and labelled with streptavidin-Cy3 (GE Healthcare Bio-Sciences Corp.) and then scanned with the Illumina BeadStation 500 System (Illumina). The scanned image was imported into BeadStudio (Illumina) for analysis. Forty-eight thousand transcripts representing six whole-genome samples can be analysed on a single BeadChip. We included at least two technical replicates (i.e., the same cRNA samples) for each BeadChip.
Background-corrected values for each probe on the BeadChip array were extracted using BeadStudio version 1.5.1.3 (Illumina). This is based on the average of negative control genes and is called the method of background normalization by Illumina. These extracted values were exported to the software GeneSpring version 7.3.1 (Silicon Genetics, Redwood, CA), and per chip and per gene normalization were performed. The statistical significance of the microarray data was calculated using the Welch t-test, and multiple tests were corrected by the Benjamini and Hochberg false discovery rate [19]. Significance was defined as q < 0.05.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
MF, KH, RK, KK and SS carried out molecular genetic study, participated in the study design and coordination and wrote the draft of the manuscript. KI, YM and SN carried out immunologic studies. EN and TA participated in the design of the study and performed the statistical analysis. All authors read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Acknowledgments
Acknowledgements
We are grateful to the family members who participated in this study.
Contributor Information
Masaya Fujimoto, Email: m0620547@md.tsukuba.ac.jp.
Kohsuke Imai, Email: kimai@ndmc.ac.jp.
Kenji Hirata, Email: svtc_hira005@hotmail.com.
Reiichi Kashiwagi, Email: ryouyantree@yahoo.co.jp.
Yoichi Morinishi, Email: grd1711@ndmc.ac.jp.
Katsuhiko Kitazawa, Email: kitazawa@hospital.asahi.chiba.jp.
Sei Sasaki, Email: ssasaki.kid@tmd.ac.jp.
Tadao Arinami, Email: tarinami@md.tsukuba.ac.jp.
Shigeaki Nonoyama, Email: nonoyama@ndmc.ac.jp.
Emiko Noguchi, Email: enoguchi@md.tsukuba.ac.jp.
References
- McKinley MJ, Johnson AK. The physiological regulation of thirst and fluid intake. News Physiol Sci. 2004;19:1–6. doi: 10.1152/nips.01470.2003. [DOI] [PubMed] [Google Scholar]
- Fushimi K, Sasaki S, Marumo F. Phosphorylation of serine 256 is required for cAMP-dependent regulatory exocytosis of the aquaporin-2 water channel. J Biol Chem. 1997;272:14800–14804. doi: 10.1074/jbc.272.23.14800. [DOI] [PubMed] [Google Scholar]
- Robben JH, Knoers NV, Deen PM. Cell biological aspects of the vasopressin type-2 receptor and aquaporin 2 water channel in nephrogenic diabetes insipidus. Am J Physiol Renal Physiol. 2006;291:F257–70. doi: 10.1152/ajprenal.00491.2005. [DOI] [PubMed] [Google Scholar]
- Rosenthal W, Seibold A, Antaramian A, Lonergan M, Arthus MF, Hendy GN, Birnbaumer M, Bichet DG. Molecular identification of the gene responsible for congenital nephrogenic diabetes insipidus. Nature. 1992;359:233–235. doi: 10.1038/359233a0. [DOI] [PubMed] [Google Scholar]
- Deen PM, Verdijk MA, Knoers NV, Wieringa B, Monnens LA, van Os CH, van Oost BA. Requirement of human renal water channel aquaporin-2 for vasopressin-dependent concentration of urine. Science. 1994;264:92–95. doi: 10.1126/science.8140421. [DOI] [PubMed] [Google Scholar]
- Mulders SM, Bichet DG, Rijss JP, Kamsteeg EJ, Arthus MF, Lonergan M, Fujiwara M, Morgan K, Leijendekker R, van der Sluijs P, van Os CH, Deen PM. An aquaporin-2 water channel mutant which causes autosomal dominant nephrogenic diabetes insipidus is retained in the Golgi complex. J Clin Invest. 1998;102:57–66. doi: 10.1172/JCI2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura Y, Onigata K, Nagashima T, Yutani S, Mochizuki H, Nagashima K, Morikawa A. Detection of skewed X-inactivation in two female carriers of vasopressin type 2 receptor gene mutation. J Clin Endocrinol Metab. 1997;82:3434–3437. doi: 10.1210/jc.82.10.3434. [DOI] [PubMed] [Google Scholar]
- Bichet DG, Birnbaumer M, Lonergan M, Arthus MF, Rosenthal W, Goodyer P, Nivet H, Benoit S, Giampietro P, Simonetti S, et al. Nature and recurrence of AVPR2 mutations in X-linked nephrogenic diabetes insipidus. Am J Hum Genet. 1994;55:278–286. [PMC free article] [PubMed] [Google Scholar]
- Demura M, Takeda Y, Yoneda T, Furukawa K, Usukura M, Itoh Y, Mabuchi H. Two novel types of contiguous gene deletion of the AVPR2 and ARHGAP4 genes in unrelated Japanese kindreds with nephrogenic diabetes insipidus. Hum Mutat. 2002;19:23–29. doi: 10.1002/humu.10011. [DOI] [PubMed] [Google Scholar]
- Jinnouchi H, Araki E, Miyamura N, Kishikawa H, Yoshimura R, Isami S, Yamaguchi K, Iwamatsu H, Shichiri M. Analysis of vasopressin receptor type II (V2R) gene in three Japanese pedigrees with congenital nephrogenic diabetes insipidus: identification of a family with complete deletion of the V2R gene. Eur J Endocrinol. 1996;134:689–698. doi: 10.1530/eje.0.1340689. [DOI] [PubMed] [Google Scholar]
- Schoneberg T, Pasel K, von Baehr V, Schulz A, Volk HD, Gudermann T, Filler G. Compound deletion of the rhoGAP C1 and V2 vasopressin receptor genes in a patient with nephrogenic diabetes insipidus. Hum Mutat. 1999;14:163–174. doi: 10.1002/(SICI)1098-1004(1999)14:2<163::AID-HUMU8>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- van Lieburg AF, Verdijk MA, Schoute F, Ligtenberg MJ, van Oost BA, Waldhauser F, Dobner M, Monnens LA, Knoers NV. Clinical phenotype of nephrogenic diabetes insipidus in females heterozygous for a vasopressin type 2 receptor mutation. Hum Genet. 1995;96:70–78. doi: 10.1007/BF00214189. [DOI] [PubMed] [Google Scholar]
- Wildin RS, Antush MJ, Bennett RL, Schoof JM, Scott CR. Heterogeneous AVPR2 gene mutations in congenital nephrogenic diabetes insipidus. Am J Hum Genet. 1994;55:266–277. [PMC free article] [PubMed] [Google Scholar]
- http://www.hgmd.cf.ac.uk/ac/index.php
- Dong Y, Sheng H, Chen X, Yin J, Su Q. Deletion of the V2 vasopressin receptor gene in two Chinese patients with nephrogenic diabetes insipidus. BMC Genet. 2006;7:53. doi: 10.1186/1471-2156-7-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegay DH, Lane AH, Roohi J, Hatchwell E. Contiguous gene deletion involving L1CAM and AVPR2 causes X-linked hydrocephalus with nephrogenic diabetes insipidus. Am J Med Genet A. 2007;143:594–598. doi: 10.1002/ajmg.a.31536. [DOI] [PubMed] [Google Scholar]
- Broides A, Ault BH, Arthus MF, Bichet DG, Conley ME. Severe combined immunodeficiency associated with nephrogenic diabetes insipidus and a deletion in the Xq28 region. Clin Immunol. 2006;120:147–155. doi: 10.1016/j.clim.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Huenecke S, Behl M, Fadler C, Zimmermann SY, Bochennek K, Tramsen L, Esser R, Klarmann D, Kamper M, Sattler A, von Laer D, Klingebiel T, Lehrnbecher T, Koehl U. Age-matched lymphocyte subpopulation reference values in childhood and adolescence: application of exponential regression analysis. Eur J Haematol. 2008 doi: 10.1111/j.1600-0609.2008.01052.x. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B. 1995;57:289–300. [Google Scholar]
- Bosotti R, Locatelli G, Healy S, Scacheri E, Sartori L, Mercurio C, Calogero R, Isacchi A. Cross platform microarray analysis for robust identification of differentially expressed genes. BMC Bioinformatics. 2007;8 Suppl 1:S5. doi: 10.1186/1471-2105-8-S1-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt DL, Gray CD, Young WS, 3rd, Orellana SA, Malouf AT. ARHGAP4 is a novel RhoGAP that mediates inhibition of cell motility and axon outgrowth. Mol Cell Neurosci. 2007 doi: 10.1016/j.mcn.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi
- Hazenberg MD, Otto SA, Cohen Stuart JW, Verschuren MC, Borleffs JC, Boucher CA, Coutinho RA, Lange JM, Rinke de Wit TF, Tsegaye A, van Dongen JJ, Hamann D, de Boer RJ, Miedema F. Increased cell division but not thymic dysfunction rapidly affects the T-cell receptor excision circle content of the naive T cell population in HIV-1 infection. Nat Med. 2000;6:1036–1042. doi: 10.1038/79549. [DOI] [PubMed] [Google Scholar]