

Abstract
The neurotrophin brain-derived neurotrophic factor (BDNF) inhibits food intake, and rodent models of BDNF disruption all exhibit increased food intake and obesity, as well as hyperactivity. We report an 8-year-old girl with hyperphagia and severe obesity, impaired cognitive function, and hyperactivity who harbored a de novo chromosomal inversion, 46,XX,inv(11)(p13p15.3), a region encompassing the BDNF gene. We have identified the proximal inversion breakpoint that lies 850 kb telomeric of the 5′ end of the BDNF gene. The patient’s genomic DNA was heterozygous for a common coding polymorphism in BDNF, but monoallelic expression was seen in peripheral lymphocytes. Serum concentration of BDNF protein was reduced compared with age- and BMI-matched subjects. Haploinsufficiency for BDNF was associated with increased ad libitum food intake, severe early-onset obesity, hyper-activity, and cognitive impairment. These findings provide direct evidence for the role of the neurotrophin BDNF in human energy homeostasis, as well as in cognitive function, memory, and behavior.
Genetic studies in patients with severe obesity have established the importance of signaling through the leptinmelanocortin axis in the regulation of human energy homeostasis (1–3). Recently, the concept that hypothalamic neuronal networks involved in energy homeostasis are “hardwired” has been challenged. In mice, hypothalamic neurones projecting from the arcuate nucleus to the paraventricular nucleus develop after birth, and their development is regulated by leptin (4). In addition, synaptic plasticity in the mature rodent brain has been identified as a component of the neuronal regulation of energy homeostasis, as leptin has been shown to acutely modulate excitatory and inhibitory synaptic inputs at the level of first-order arcuate neurones (5). However, it is difficult to establish whether synaptic plasticity plays a role in the physiological regulation of energy homeostasis in humans and whether, under pathological conditions, hypothalamic neuronal networks and plasticity may be impaired and contribute to human obesity.
Brain-derived neurotrophic factor (BDNF) regulates the development, survival, and differentiation of neurons through its high-affinity receptor, tropomyosin-related kinase B (TrkB) (6). Unlike other neurotrophins, BDNF is secreted in an activity-dependent manner that allows for highly controlled release (7). Recently, BDNF has been implicated in the regulation of body weight, as its expression is reduced by fasting (8), and BDNF administration causes weight loss in wild-type mice through a reduction in food intake (9). BDNF has also been implicated in memory and a range of behaviors using a number of conditional knockout models (10–14).
To date, no humans have been described with null alleles of BDNF. A common polymorphism in the human BDNF gene, Val66Met (dbSNP no. rs6265), which appears to result in impairment of intracellular trafficking and reduced activity-dependent secretion of BDNF by cultured hippocampal neurons, is associated with poorer episodic memory and abnormal hippocampal activation using functional magnetic resonance imaging (15,16). One study (17) has associated this single nucleotide polymorphism with eating disorders. In 5,614 U.K. Caucasian subjects, we found no association between the Val66Met polymorphism and BMI (J.G., I.S.F., S.O.R., personal observations). We previously reported a child with severe obesity, impaired short-term memory, and developmental delay who had a de novo missense mutation impairing the function of TrkB, the tyrosine kinase receptor that mediates the effects of both BDNF and the neurotrophin, NT4/5 (18). We now report a patient with severe hyperphagia and obesity and a complex neurobehavioral phenotype, including impaired cognitive function and memory and distinctive hyperactive behavior. Interestingly, this patient has a de novo paracentric inversion, 46,XX,inv(11)(p13p15.3), which encompasses the BDNF locus. We went on to fine map this rearrangement with the use of fluorescence in situ hybridization (FISH).
RESEARCH DESIGN AND METHODS
DNA isolation from bacterial artificial chromosomes
Clones from the RPCI-11 libraries were obtained from BACPAC Resources Center (http://bacpac.chori.org/home.htm). Clones were amplified on Luria-Bertani agar plates with 20 μg/ml chloramphenicol and incubated overnight at 37°C. Single colonies were picked and inoculated into 5-ml cultures of Luria-Bertani broth, supplemented with the appropriate antibiotic, and grown overnight at 37°C at 215 rpm. DNA was isolated using a QIAprep Spin Miniprep kit (Qiagen).
FISH
A total of 500 ng miniprep DNA was labeled with SpectrumOrange dUTP by nick translation (Vysis). After incubation at 15°C for 15 h, the probe was ethanol precipitated and resuspended in 6 μl Tris-EDTA buffer. Then, 80 ng of probe was hybridized to metaphase chromosomes with 1 μg cot−1 DNA, 15 μl hybrisol VI (Appligene-oncor), and 10 ng chromosome 11–specific probes (Vysis). Slides were analyzed using a Leica DMRB epifluorescence light microscope and images recorded using SmartCapture 2 software.
Genomic DNA extraction from lymphoblastoid cell lines
Genomic DNA was extracted from lymphoblastoid cell lines using a DNeasy Tissues kit (Qiagen), according to the manufacturer’s instructions. DNA was then ethanol precipitated and resuspended in Elution Buffer (Qiagen).
Southern blots
PCR was used to generate ~500 bp probes mapping to restriction fragments of the genomic region of interest, using bacterial artificial chromosome (BAC)-derived miniprep DNA as a template. Primers were designed using Blast-like alignment tool (BLAT) analysis (http://www.genome.ucsc.edu/cgi-bin/hgBlat) and optimized to avoid nonspecific binding to genomic DNA. PCR products (10 ng/μl) were labeled with P32 using Rediprime Randon Prime labeling kit (Amersham Biosciences).
Control or patient genomic DNA (10 μg) was digested at 37°C for 8 h with 1 μl NEB HindIII restriction enzyme (100,000 units/ml). Samples were loaded onto agarose gels, electrophoresed overnight, and transferred onto Imobilon-P transfer membranes (Milipore). Membranes were neutralized and ultraviolet cross-linked before incubation with prehybe solution (containing 10 μg/μl salmon testes DNA) at 65°C for 4–6 h, with fresh prehybe solution after 2 h. Labeled probe was added to fresh hybe buffer and hybridized to membrane-bound DNA overnight at 65°C. Membranes were washed with stringency washes and exposed to film at −80°C for 3–7 days.
Serum BDNF measurement
BDNF serum protein levels were measured after an overnight fast using a commercially available enzyme-linked immunosorbent assay. Serum BDNF in the patient was compared with age-matched normal weight control subjects and age-matched obese girls and boys (>95th percentile; mean BMI SDs 2.3 for boys, 2.6 for girls).
Clinical phenotyping
The patients and their relatives were invited to the Wellcome Trust Clinical Research Facility at Addenbrooke’s Hospital, Cambridge, U.K. The clinical studies were performed after approval by the local regional ethics committee of Cambridge. All clinical studies were conducted in accordance with the principles of the Declaration of Helsinki and performed as previously described (18). A number of cognitive and behavioral tests were used in the BDNF patient and compared with the previously reported patient with a TrkB mutation and normal control subjects of the same age (see online appendix 1 for details and references [available at http://diabetes.diabetesjournals.org]).
RESULTS
We identified a patient with severe obesity and hyperphagia, as well as a complex neurobehavioral phenotype, who harbored a chromosomal inversion, 46,XX,inv(11) (p13p15.3), determined by chromosome banding analysis (Fig. 1A). Both parents were of normal intelligence and body weight and had a normal karyotype. Paternity was confirmed by conventional haplotype analysis, indicating that the inversion in the patient occurs de novo. There are ~100 known transcripts in the inversion region, extending from the proximal breakpoint in 11p14.1 to 11p15.3 (NCBI MapViewer, Human Build 36). Of these, ~15 are known to be predominantly centrally expressed. For six of these, mice knockouts or hypomorphs have been generated, and, in a few cases, human mutations have been associated with disease. None of these rodent models or human mutations have been associated with a comparable neurobehavioral and obesity phenotype when disrupted. BDNF is located within this chromosomal region. As BDNF deficiency in rodents results in severe obesity, impaired memory, and hyperactivity, we investigated whether this inversion disrupted the BDNF gene itself or BDNF expression.
FIG. 1.
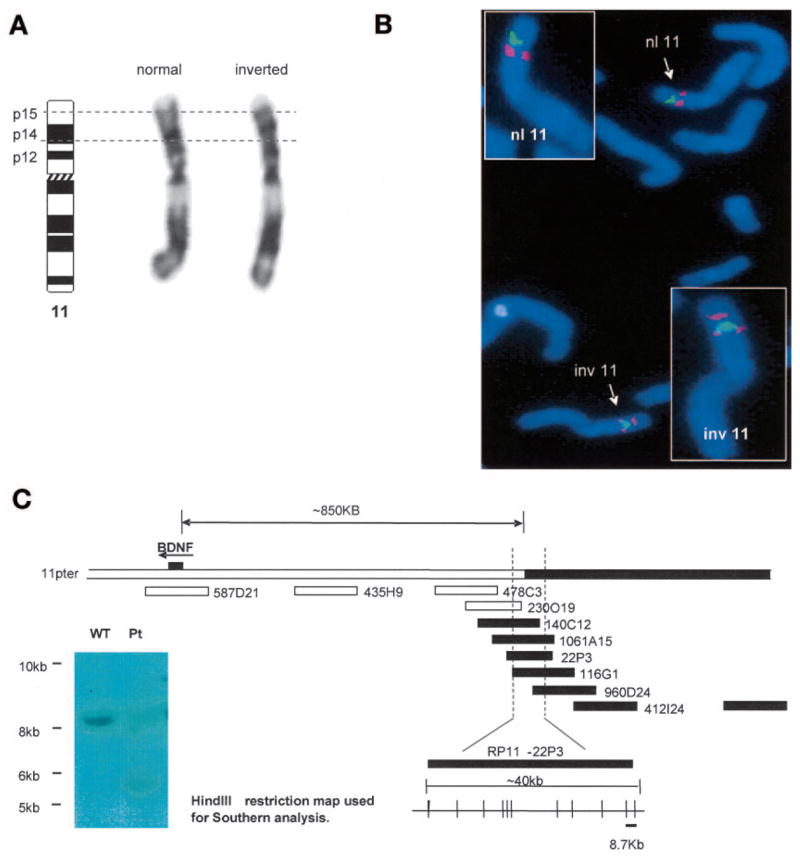
Mapping of a de novo chromosome 11 paracentric inversion in an 8-year-old girl with severe obesity and developmental delay. A: G-banding of the patient’s metaphase chromosomes revealed a paracentric inversion extending 11p13 to 11p15.3. B: FISH mapping of the breakpoint-spanning BAC, RP11-1061A15 (red), and a midinversion BAC, RP11–580I19 (green). In the normal chromosome 11 (nl 11), RP11-580I19 (green) lies ~8-Mb distal to RP11-1061A15 (red). In the abnormal chromosome (inv 11), red signal from RP11-1061A15 is seen both proximal and distal to the midinversion BAC (green). BAC clones were obtained from BACPAC Resources Center (http://bacpac.chori.org/home.htm). C: Schematic map of the proximal breakpoint region, showing relative positions of BACs used for FISH analysis, the BDNF gene, and mapping of the region that was subsequently analyzed using Southern hybridizations. BACs (1-Mb apart) were initially used to identify the approximate region of the breakpoint. Further FISH analysis using overlapping BACs saturating the identified region allowed for further mapping of the breakpoint to an ~40-kb region, as shown. Subsequent Southern hybridization analysis used known BAC sequence (RP11–22P3, AC015518) to design probes against HindIII fragments across this ~40-kb proximal breakpoint region. Patient and control genomic DNA was digested with HindIII and incubated with the PCR-derived P32-labeled probes. The probe, as indicated on the schematic, revealed an aberrant ~5.5-kb junction fragment in the patient (inset).
FISH with a locus-specific BAC probe determined that BDNF itself was not disrupted, and there was no evidence of deletion or duplication of BDNF (data not shown). Subsequently, FISH studies using a number of probes from BAC clones in the 11p14 region (online appendix Table 1) enabled the identification of the proximal breakpoint of the inversion to lie within BAC clone RP11-1061A15 (accession no. AQ672557) (Fig. 1B). This clone is ~850 kb upstream of BDNF (Fig. 1C). This breakpoint was further localized by Southern hybridization analysis. In the patient, a 5.5-kb HindIII junction fragment was observed in addition to the expected 8.7-kb wild-type fragment (Fig. 1C). There are no known genes within the 8.7-kb fragment (genomic localization 28,536,297–28,545,001 bp), confirming that none have been physically disrupted by the proximal breakpoint.
Since the BDNF gene itself was not physically interrupted, we hypothesized that a position effect resulting from the inversion may have disrupted its expression. The patient was heterozygous for the common BDNF coding polymorphism, Val66Met. Therefore, we examined whether both alleles of her BDNF gene were expressed equally in patient-derived cells expressing BDNF. We examined Epstein-Barr virus–transformed lymphoblastoid cells from normal-weight subjects who were also heterozygous for the Val66Met polymorphism and showed, firstly, that BDNF mRNA was readily detected in these cells and, secondly, that both alleles were always detectable in expressed RNA from heterozygous normal subjects (data not shown). In contrast, RNA from the proband’s lymphoblastoid cell line consistently showed exclusively monoallelic expression confirming the hypothesis that the chromosomal inversion disrupted the expression of BDNF (Fig. 2A). Supporting evidence for this was provided by the finding of low serum levels of BDNF in the proband compared with subjects matched for age alone or for age and degree of obesity (Fig. 2B). The low serum concentration of BDNF in this patient was not explained by platelet count, which is the only factor, other than age, reported to influence serum BDNF in children (19).
FIG. 2.
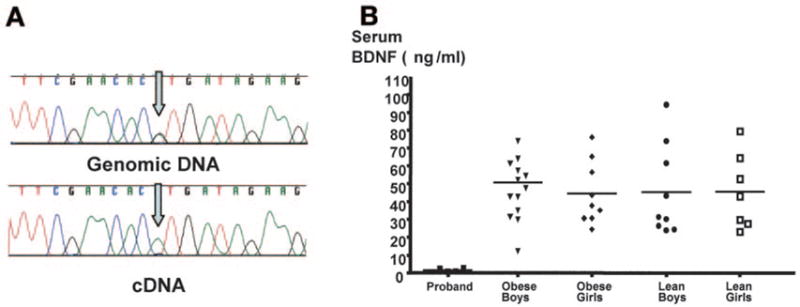
Monoallelic expression and low serum BDNF concentrations in the proband. A: Genomic and cDNA sequences from the patient’s lymphoblastoid cell line. The patient is heterozygous for the BDNF coding polymorphism, Val66Met. However, her cDNA sequence reveals RNA expression of the 66Met allele only, indicating monoallelic expression of BDNF in this patient. B: BDNF serum protein levels were measured after an overnight fast using a commercially available enzyme-linked immunosorbent assay. Serum BDNF in the patient was compared with that in age-matched normal-weight control subjects and age-matched obese girls and boys (>95th percentile; mean BMI SDs 2.3 for boys, 2.6 for girls).
We went on to study the metabolic and neurobehavioral phenotypes associated with BDNF haploinsufficiency after informed consent was obtained from the family. The patient was born at term (birth weight 3.3 kg) but experienced breathing abnormalities shortly after birth and was admitted to a neonatal intensive care unit. Developmental milestones were delayed with a specific impairment of speech and language development. The child rapidly gained weight from age 4 months, deviating from the predicted percentiles in the 1st year of life (Fig. 3A). One notable aspect of the patient’s behavior in early childhood was extreme hyperactivity, an unusual finding in severe obesity. The patient also had “no concept of danger” and impaired nociception on clinical examination.
FIG. 3.
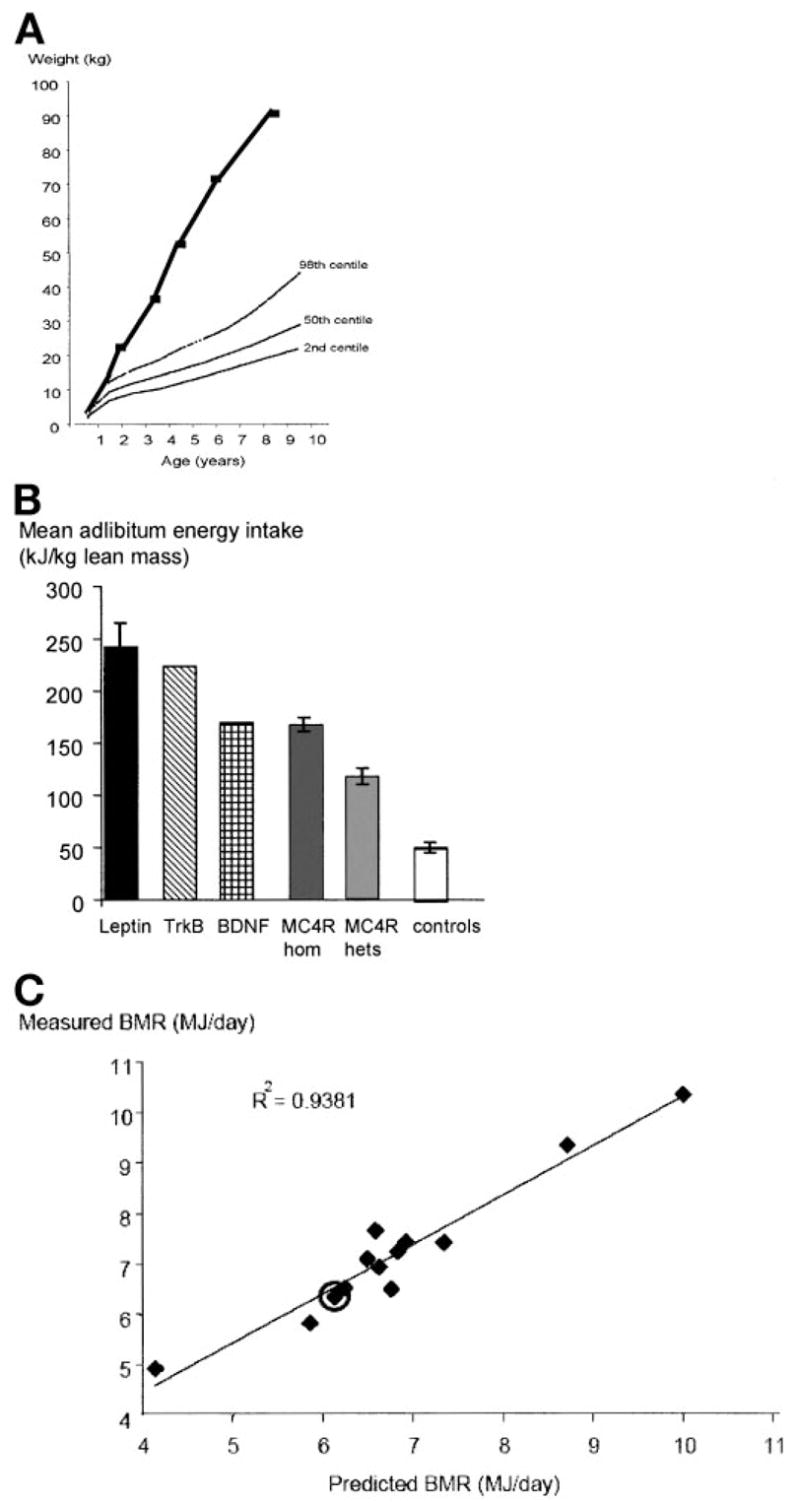
Obesity phenotype of an 8-year-old girl with haploinsufficiency at the BDNF locus. A: Weight gain compared with normal U.K. percentiles for girls. B: Food intake at an 18-MJ ad libitum test meal, expressed per kilogram lean body mass, compared with subjects with mutations in other genes responsible for obesity syndromes and controls. Food intake is expressed per kilogram lean body mass measured by dual-energy X-ray absorptiometry to allow comparison between subjects of different ages. C: Measured basal metabolic rate measured by indirect calorimetry compared with predicted values. The BDNF-haploinsufficient patient is highlighted and compared with age- and BMI-matched obese control subjects.
The BDNF-haploinsufficient patient was reported to be hyperphagic under free-living conditions, and the hyperphagia was confirmed using an 18-MJ ad libitum test meal administered after an overnight fast. The subject ate 153 kJ/kg lean body mass, three times that of control subjects (Fig. 3B). At age 8 years, her BMI was 4.3 SD above the age-related population-based mean. Basal metabolic rate measured by indirect calorimetery was comparable to that predicted by age- and sex-specific equations (Fig. 3C). The patient had a normal blood glucose (4.3 mmol/l) but had fasting hyperinsulinemia (332 pmol/l; 0–60 pmol/l). Full serum lipid profile, thyroid function tests, and insulin-like growth factor-1 concentrations were in the normal range (data not shown).
When investigated at 9 years of age, the proband was shown to have evidence of a global impairment of IQ to a level that was comparable with the patient with a mutation in TrkB (Table 1). The proband showed some repetitive behaviors as indicated by parental reports on the social communication questionnaire. Commensurate with a general impairment of IQ, both patients showed deficits on tests of language, numeracy, attention, and memory, including short-term memory (Wechsler Intelligence Scale for Children-III digit span) (Table 1 and online appendix 1).
TABLE 1.
Neurobehavioural phenotype of an 8-year-old girl with haploinsufficiency at the BDNF locus
|
TrkB (9-year-old boy) |
BDNF (9-year-old girl) |
|
|---|---|---|
| Social communication questionnaire | 25 (autism trait) | 7 (normal) |
| Intelligence (WISC-III) | ||
| Full-scale IQ | 42 (30.1) | 56 (0.2) |
| Verbal IQ | 48 (30.1) | 59 (0.3) |
| Performance IQ | 46 (30.1) | 59 (0.3) |
| Digit span (STM) subtest | 1 (very low) | 4 (very low) |
| Memory | ||
| Sutherland Memory Questionnaire | 139 (impaired) | 43 (normal) |
| CMS | ||
| General memory quotient | NA | 350 (30.1) |
| Visual immediate memory | NA | 75 (5) |
| Visual delayed memory | NA | 82 (12) |
| Verbal immediate memory | NA | 50 (30.1) |
| Verbal delayed memory | NA | 50 (30.1) |
| Attention/concentration | NA | 57 (0.2) |
| Learning | NA | 54 (30.1) |
| Delayed recognition | NA | 50 (30.1) |
| RBMT score | 4 (impaired) | 10 (impaired) |
| Language | ||
| BPVS | 341 (30.1) | 66 (1) |
| TROG (years [percentile]) | 4 (30.1) | 5 (1–5) |
| WORD | ||
| Reading | 70 (2) | 67 (1) |
| Spelling | NA | 63 (1) |
| Reading comprehension | NA | 58 (0.3) |
| Numeracy (WOND) | ||
| Mathematics reasoning | 59 (0.3) | 64 (1) |
| Numerical operations | 60 (0.4) | 59 (0.3) |
| Attention (TEA-Ch) | ||
| Selective visual attention | 1 (30.2) | 1 (30.2) |
| Sustained attention | NA | 3 (31.5) |
Data are scores (percentile) unless otherwise indicated. A summary of the performance of both the proband and the previously reported patient with a mutation in TrkB on a range of cognitive assessments. Average performance on the Wechsler Intelligence Scale for Children-III (WISC-III), Children’s Memory Scale (CMS), Wechsler Objective Reading Dimensions (WORD), Wechsler Objective Numerical Dimensions (WOND), Test for Reception and Grammar (TROG), Test for Everyday Attention for Children (TEA-Ch), and British Picture Vocabulary Scale (BPVS) is 100 ± 15 (normal range 85–115); average score for subtests, 10 ± 3 (7–13). Mean score (±SD) on the Sunderland Memory Questionnaire for healthy individuals aged between 10 and 27 years is 58.8 ± 8.3 (A.-L.R.A., J.R.H., unpublished observations). Percentiles and age-equivalent scores are shown where available. A percentile score of <10% indicates an impairment of clinical significance. NA, not available due to individual being unable to complete the test (full details and references in online appendix 1); RBMT, Rivermead Behavioral Memory Test.
Although poor memory performance may have been confounded by difficulties in focusing attention and/or comprehending task instructions, it is of interest to note the striking similarity in the performance of the two patients using this detailed cognitive assessment. Both the BDNF-haploinsufficient patient and the patient with the TrkB mutation ranged from exceptionally low to low performance on tests of IQ, memory (with the exception of visual memory subtests and Sunderland Memory Questionnaire in the BDNF-haploinsufficient patient [online appendix 1]), language, numerical abilities, and attention. Furthermore, the poor attention scores (in the Test of Everyday Attention for Children) correlated with the observed hyperactivity in both patients.
DISCUSSION
Given the unusual and distinctive combination of impaired cognitive function, hyperactivity, and severe obesity and the similarities in phenotype to a previously reported patient with a mutant TrkB and the strong evidence for loss of expression of one allele of BDNF, it would seem highly plausible that the clinical phenotype in this patient has resulted from a reduction in BDNF. However, the patient does harbor a chromosomal inversion and not a simple loss of function mutation, and it is possible that some aspects of her phenotype could relate to positional effects at other genes in the region. However, none of the rodent models or human mutations disrupting other genes in this region have been associated with a comparable neurobehavioral and obesity phenotype. Nonetheless, we cannot exclude the possibility of disruption of other genes of unknown function. As yet, we have not fully characterized the distal breakpoint and thus cannot exclude the possibility that a centrally expressed gene is disrupted either directly or indirectly. However, inspection of the human transcript map revealed no other obvious candidate genes near either inversion breakpoint.
There is evidence from other chromosomal rearrangements involving 11p to suggest that the 11p12–14 region harbors an important gene for the regulation of body weight. Chromosomal deletions spanning 11p11.2–p14.1 have been reported in WAGR syndrome associated with obesity (20). Borg et al. (21) recently reported a patient with mild mental retardation, dysmorphic features, and obesity who had a complex chromosomal translocation, t(3;12), and an additional balanced translocation, t(11;21)(p14;q22.1), with a chromosome breakpoint mapping to within 100 kb downstream of BDNF.
The cognitive findings reported here are consistent with the role of BDNF in learning and memory. Both the proband and the patient with a mutation in TrkB showed general impairments of intelligence, language, numeracy, attention, and memory. BDNF has been implicated in hippocampal function throughout the lifespan (22); therefore, everyday memory deficits were expected in these two patients. BDNF is known to play a role in the development and maturation of neurons in brain regions other than the hippocampus; thus, BDNF haploinsufficiency would be expected to be associated with the more general profile of cognitive dysfunction that we observed.
Interestingly, despite the similarities in their impaired cognitive function, the two patients showed different profiles on the Sunderland Memory Questionnaire and the Social Communication Questionnaire, with the BDNF-haploinsufficient patient performing in the normal range on both, while the TrkB patient was rated to be impaired on both. Although performance on the Sunderland Memory Questionnaire has been reported to be consistent with performance on standardized memory assessments (23), it is possible to obtain inconsistent results, as in the pro-band, as the tests tap different aspects of memory. Furthermore, poor performance on formal testing might also reflect difficulties in focusing attention and/or comprehending task instructions.
It is possible that a child with a TrkB mutation may be impaired on all tests including social communication (i.e., frontal lobe damage and more general brain damage) and, thus, be more severely affected than a child with BDNF haploinsufficiency, as TrkB is also the receptor for the neurotrophin NT4/5. However, as there is considerable overlap between BDNF and NT4 in terms of brain expression and in terms of effects on long-term potentiation and memory (24), it is impossible to reliably attribute these subtle differences between two patients to loss of one ligand versus loss of the receptor with any degree of confidence. Whereas the involvement of BDNF in the development of the forebrain and in functions of learning and memory has long been established, a possible role in energy homeostasis has emerged much more recently. In the hypothalamus, BDNF is expressed predominantly in the ventromedial hypothalamus (8) but also in the dorso-medial hypothalamus (25) and possibly other hypothalamic areas. Expression is nutritionally regulated (8) and, at least in the dorsomedial hypothalamus, appears to be regulated by leptin (25). There is some evidence to suggest that BDNF-expressing neurons may lie downstream of MC4R neuronal pathways, as ventromedial hypothalamus mRNA levels are reduced in mc4r-null mice and restored by administration of the melanocortin agonist MT-II (8). It is notable that both the patient with the TrkB missense mutation and this patient are markedly hyperphagic and obese (18). This is consistent with the increased food intake and obesity seen in the bdnf heterozygous–null mice (11) and the trkB hypomorphic mouse (8).
Neural growth factors such as BDNF may influence energy balance either through their effects on the development of the hypothalamus or by being dynamically regulated neurotransmitters involved in hypothalamic feeding circuits. In rodents, there are no gross morphological changes in the brains of bdnf heterozygous–null mice, and they remain responsive to the administration of BDNF with a reduction in food intake comparable with wild-type mice, suggesting that BDNF-responsive pathways are intact (11). Also, the postnatal conditional BDNF knockout exhibits increased food intake and is obese (12). Thus, the rodent data suggest that the effects of BDNF on energy homeostasis cannot be explained simply by a lack of development of BDNF-responsive circuits in the hypothalamus.
An alternative mechanism is that BDNF modulates synaptic plasticity. It is impossible to directly test whether synaptic plasticity is relevant in the human brain. However, it is worth noting that the most studied plasticity phenomenon, long-term potentiation in relation to learning and memory, is mediated by BDNF; thus, it is very plausible that BDNF may exert similar effects on hypothalamic feeding circuits.
Modulation of synaptic plasticity may offer a novel therapeutic approach for the treatment of human obesity. Flier and colleagues (26) recently demonstrated that another neuronal growth factor, ciliary neurotrophic factor, could promote neurogenesis in the adult mouse hypothalamus, and this mechanism may explain the long-term resetting of body weight “set points” seen in obese patients treated with the ciliary neurotrophic factor analog, axokine.
In summary, by describing the clinical and phenotypic features of a child with loss of one functional copy of BDNF, we have provided some of the first direct evidence for the functions of this important neurotrophin in the human brain. Understanding the mechanisms whereby BDNF regulates hypothalamic neuronal circuits may have potential therapeutic benefits for the treatment of human obesity.
Acknowledgments
This work was supported by the Wellcome Trust (G.S.H.Y., S.O.R., and I.S.F.) and the Medical Research Council (J.G., A.-L.R.A., J.M.K., J.R.H., and S.O.R.). J.A.Y. is supported by the Intramural Research Program of the National Institutes of Health Grant ZO1-HD-00641 (National Institute of Child Health and Human Development). J.A.Y. and J.C.H. are Commissioned Officers in the U.S. Public Health Service.
We are indebted to the patients and their families for their participation and to the physicians involved in the Genetics of Obesity Study. We thank Lionel Willat for support with FISH studies and Diane C. Adler-Wailes for assistance with the BDNF assay.
- BAC
bacterial artificial chromosome
- BDNF
brain-derived neurotrophic factor
- FISH
fluorescence in situ hybridization
- TrkB
tropomyosin-related kinase B
Footnotes
Additional information for this article can be found in an online appendix at http://diabetes.diabetesjournals.org.
References
- 1.Farooqi IS, O’Rahilly S. Monogenic obesity in humans. Annu Rev Med. 2005;56:443–458. doi: 10.1146/annurev.med.56.062904.144924. [DOI] [PubMed] [Google Scholar]
- 2.Schwartz MW, Morton GJ. Obesity: keeping hunger at bay. Nature. 2002;418:595–597. doi: 10.1038/418595a. [DOI] [PubMed] [Google Scholar]
- 3.Cone RD. Anatomy and regulation of the central melanocortin system. Nat Neurosci. 2005;8:571–578. doi: 10.1038/nn1455. [DOI] [PubMed] [Google Scholar]
- 4.Bouret SG, Draper SJ, Simerly RB. Trophic action of leptin on hypothalamic neurons that regulate feeding. Science. 2004;304:108–110. doi: 10.1126/science.1095004. [DOI] [PubMed] [Google Scholar]
- 5.Pinto S, Roseberry AG, Liu H, Diano S, Shanabrough M, Cai X, Friedman JM, Horvath TL. Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science. 2004;304:110–115. doi: 10.1126/science.1089459. [DOI] [PubMed] [Google Scholar]
- 6.Tapia-Arancibia L, Rage F, Givalois L, Arancibia S. Physiology of BDNF: focus on hypothalamic function. Front Neuroendocrinol. 2004;25:77–107. doi: 10.1016/j.yfrne.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 7.Bramham CR, Messaoudi E. BDNF function in adult synaptic plasticity: the synaptic consolidation hypothesis. Prog Neurobiol. 2005;76:99–125. doi: 10.1016/j.pneurobio.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 8.Xu B, Goulding EH, Zang K, Cepoi D, Cone RD, Jones KR, Tecott LH, Reichardt LF. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat Neurosci. 2003;6:736–742. doi: 10.1038/nn1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pelleymounter MA, Cullen MJ, Wellman CL. Characteristics of BDNF-induced weight loss. Exp Neurol. 1995;131:229–238. doi: 10.1016/0014-4886(95)90045-4. [DOI] [PubMed] [Google Scholar]
- 10.Snider WD. Functions of the neurotrophins during nervous system development: what the knockouts are teaching us. Cell. 1994;77:627–638. doi: 10.1016/0092-8674(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 11.Kernie SG, Liebl DJ, Parada LF. BDNF regulates eating behavior and locomotor activity in mice. EMBO J. 2000;19:1290–1300. doi: 10.1093/emboj/19.6.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rios M, Fan G, Fekete C, Kelly J, Bates B, Kuehn R, Lechan RM, Jaenisch R. Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol Endocrinol. 2001;15:1748–1757. doi: 10.1210/mend.15.10.0706. [DOI] [PubMed] [Google Scholar]
- 13.Monteggia LM, Barrot M, Powell CM, Berton O, Galanis V, Gemelli T, Meuth S, Nagy A, Greene RW, Nestler EJ. Essential role of brain-derived neurotrophic factor in adult hippocampal function. Proc Natl Acad Sci U S A. 2004;101:10827–10832. doi: 10.1073/pnas.0402141101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mamounas LA, Blue ME, Siuciak JA, Altar CA. Brain-derived neurotrophic factor promotes the survival and sprouting of serotonergic axons in rat brain. J Neurosci. 1995;15:7929–7939. doi: 10.1523/JNEUROSCI.15-12-07929.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen ZY, Patel PD, Sant G, Meng CX, Teng KK, Hempstead BL, Lee FS. Variant brain-derived neurotrophic factor (BDNF) (Met66) alters the intracellular trafficking and activity-dependent secretion of wild-type BDNF in neurosecretory cells and cortical neurons. J Neurosci. 2004;24:4401–4411. doi: 10.1523/JNEUROSCI.0348-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, Zaitsev E, Gold B, Goldman D, Dean M, Lu B, Weinberger DR. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. 2003;112:257–269. doi: 10.1016/s0092-8674(03)00035-7. [DOI] [PubMed] [Google Scholar]
- 17.Friedel S, Horro FF, Wermter AK, Geller F, Dempfle A, Reichwald K, Smidt J, Bronner G, Konrad K, Herpertz-Dahlmann B, Warnke A, Hemminger U, Linder M, Kiefl H, Goldschmidt HP, Siegfried W, Remschmidt H, Hinney A, Hebebrand J. Mutation screen of the brain derived neurotrophic factor gene (BDNF): identification of several genetic variants and association studies in patients with obesity, eating disorders, and attention-deficit/hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet. 2004;132:96–99. doi: 10.1002/ajmg.b.30090. [DOI] [PubMed] [Google Scholar]
- 18.Yeo GS, Connie Hung CC, Rochford J, Keogh J, Gray J, Sivaramakrishnan S, O’Rahilly S, Farooqi IS. A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat Neurosci. 2004;7:1187–1189. doi: 10.1038/nn1336. [DOI] [PubMed] [Google Scholar]
- 19.El-Gharbawy AH, Adler-Wailes DC, Mirch MC, Theim KR, Ranzenhofer L, Tanofsky-Kraff M, Yanovski JA. Serum brain derived neurotrophic factor concentrations in lean and overweight children and adolescents. J Clin Endocrinol Metab. 2006;91:3548–3552. doi: 10.1210/jc.2006-0658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gul D, Ogur G, Tunca Y, Ozcan O. Third case of WAGR syndrome with severe obesity and constitutional deletion of chromosome (11)(p12p14) Am J Med Genet. 2002;107:70–71. doi: 10.1002/ajmg.10013. [DOI] [PubMed] [Google Scholar]
- 21.Borg K, Stankiewicz P, Bocian E, Kruczek A, Obersztyn E, Lupski JR, Mazurczak T. Molecular analysis of a constitutional complex genome rearrangement with 11 breakpoints involving chromosomes 3, 11, 12, and 21 and a approximately 0.5-Mb submicroscopic deletion in a patient with mild mental retardation. Hum Genet. 2005;118:267–275. doi: 10.1007/s00439-005-0021-0. [DOI] [PubMed] [Google Scholar]
- 22.Webster MJ, Herman MM, Kleinman JE, Shannon Weickert C. BDNF and trkB mRNA expression in the hippocampus and temporal cortex during the human lifespan. Gene Expr Patterns. 2006;6:941–951. doi: 10.1016/j.modgep.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 23.Gadian DG, Aicardi J, Watkins KE, Porter DA, Mishkin M, Vargha-Khadem F. Developmental amnesia associated with early hypoxic-ischaemic injury. Brain. 2000;123:499–507. doi: 10.1093/brain/123.3.499. [DOI] [PubMed] [Google Scholar]
- 24.Xie CW, Sayah D, Chen QS, Wei WZ, Smith D, Liu X. Deficient long-term memory and long-lasting long-term potentiation in mice with a targeted deletion of neurotrophin-4 gene. Proc Natl Acad Sci U S A. 2000;97:8116–8121. doi: 10.1073/pnas.140204597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bariohay B, Lebrun B, Moyse E, Jean A. Brain-derived neurotrophic factor plays a role as an anorexigenic factor in the dorsal vagal complex. Endocrinology. 2005;146:5612–5620. doi: 10.1210/en.2005-0419. [DOI] [PubMed] [Google Scholar]
- 26.Kokoeva MV, Yin H, Flier JS. Neurogenesis in the hypothalamus of adult mice: potential role in energy balance. Science. 2005;310:679–683. doi: 10.1126/science.1115360. [DOI] [PubMed] [Google Scholar]