

Abstract
Changes in visual cortical responses that are induced by monocular visual deprivation are a widely studied example of competitive, experience-dependent neural plasticity. It has been thought that the deprived-eye pathway will fail to compete against the open-eye pathway for limited amounts of brain-derived neurotrophic factor, which acts on TrkB and is needed to sustain effective synaptic connections. We tested this model by using a chemical-genetic approach in mice to inhibit TrkB kinase activity rapidly and specifically during the induction of cortical plasticity in vivo. Contrary to the model, TrkB kinase activity was not required for any of the effects of monocular deprivation. When the deprived eye was re-opened during the critical period, cortical responses to it recovered. This recovery was blocked by TrkB inhibition. These findings suggest a more conventional trophic role for TrkB signaling in the enhancement of responses or growth of new connections, rather than a role in competition.
During development, experience strongly influences the formation and maturation of neuronal connectivity in the mammalian cortex. In the visual system, closing one eye for even a few days during a critical period of heightened plasticity in early postnatal life leads to a pronounced decrease in the cortical representation of the deprived eye, which is observed both physiologically and anatomically1. The experience-dependent changes following such monocular deprivation, termed ocular dominance plasticity (ODP), operate through a competitive interaction between inputs from the two eyes and depend on the activity state of the postsynaptic neurons1–5. These features of ODP have suggested that a retrograde signal released by the postsynaptic neurons may mediate plasticity by affecting afferents from the two eyes differently.
Brain-derived neurotrophic factor (BDNF), the cognate ligand for the TrkB receptor, has been proposed to be such a retrograde signal on the basis of a number of observations, including the activity-dependent expression of BDNF and trafficking of TrkB receptors6,7, as well as its established role for BDNF and TrkB in other brain region-specific forms of developmental plasticity8,9. In the visual cortex, BDNF expression is tightly regulated by visual experience10–13. Exogenous TrkB ligands block the effect of monocular deprivation physiologically14–16. Either applying exogenous TrkB ligand or blocking endogenous TrkB ligand inhibits the formation of ocular dominance columns17. Mice overexpressing BDNF in the postnatal brain show a precocious critical period for ODP18,19. The prevailing hypothesis8,17,20,21 has been that competition for limited amounts of BDNF, which acts on TrkB receptors, is the effector of activity-dependent plasticity in the cortex, and the conventional explanation for ODP is that the deprived eye fails to activate cortical cells as well as the open eye, thereby failing to stimulate them to release sufficient BDNF to sustain the deprived-eye pathway.
However, precise roles for TrkB-mediated signaling in ODP have not been determined because of the limitations associated with the current approaches for blocking TrkB. Standard genetic approaches, although offering high molecular specificity, lack the temporal control over gene inactivation that is required to determine whether the reported effects of TrkB/BDNF inactivation on synaptic plasticity are a consequence of remote events occurring earlier in development or are the direct result of the absence of TrkB signaling at the time plasticity is induced. For example, proper inhibitory circuitry is necessary for ocular dominance plasticity22, and the development of inhibitory neurons is closely regulated by BDNF-TrkB signaling19, suggesting a mechanism by which genetic manipulations of TrkB signaling for a prolonged period would indirectly affect cortical plasticity18,19. Pharmacological inhibitors of TrkB kinase activity provide temporal control, but have limited specificity, complicating interpretations of the data.
Here we employed a chemical-genetic approach that combines the specificity of gene-targeting with the temporal control offered by pharmacology to resolve the role of TrkB in ODP. A phenylalanine-to-alanine substitution (TrkBF616A) in the ATP binding pocket of the kinase rendered the receptor sensitive to specific inhibition by 4-amino-1-tertbutyl-3-(1-napthylmethyl)pyrazolo[3,4-D]pyrimidine (1NM-PP1), allowing for specific and temporally controlled inactivation of TrkB. Notably, this mutation is functionally silent in the absence of the cognate inhibitor, 1NM-PP1 (refs. 23,24). Also, we used chronic intrinsic-signal optical imaging to record from individual animals longitudinally, enabling us to examine changes in the absolute response magnitude, and not just the ratio, of deprived- and open-eye responses.
Our results demonstrate that the accepted role for BDNF-TrkB signaling in cortical plasticity is incorrect. We found that TrkB kinase activity was not required for the loss of cortical responses to deprived-eye stimulation. With or without TrkB activation, a decrease in response to the deprived eye occurred, followed by an increase in response to the open eye. The same techniques, however, revealed that TrkB kinase activation was essential for the subsequent recovery of deprived-eye responses induced by binocular vision. These findings indicate that TrkB kinase signaling is involved only in the enhancement of responses and is not a part of the plasticity machinery that is responsible for the loss of cortical responses in monocular deprivation. These findings are also of interest for their potential clinical importance for patients with amblyopia.
RESULTS
Inhibition of TrkBF616A phosphorylation by 1NM-PP1 in vivo
We first determined the effect of 1NM-PP1 on TrkBF616A kinase activity in the visual cortex in vivo using immunoprecipitation and immunoblotting (Fig. 1). Systemic administration of 1NM-PP1 significantly inhibited TrkBF616A kinase activity, either by a single intraperitoneal injection at a dose of 16.6 ng per g of body weight (P < 0.05; Fig. 1b)25 or by chronic subcutaneous infusion via osmotic minipump at 0.5 nmol per g per h (P < 0.01; Fig. 1c). We chose this infusion rate on the basis of a previous report24. Consistent with previous reports24,25, the inhibition by 1NM-PP1 was specific to mutant TrkB (Fig. 1b,c), reversible (Fig. 1b) and stable after 6 d of continuous infusion (Fig. 1c). In addition, area-specific TrkB inactivation was made possible by local infusion of 1NM-PP1 into one cortical hemisphere; TrkBF616A phosphorylation was greatly reduced in the infused side, but not in the contralateral hemisphere (Fig. 1d).
Figure 1.

Inhibition of TrkBF616A in the cortex by 1NM-PP1. (a) Basal and kainate (KA)-induced levels of TrkB autophosphorylation were similar in wild-type (WT) and TrkBF616A homozygous (A/A) animals. Because the basal level of phosphotyrosine in TrkB (p-Trk) was relatively low, we enhanced its level by intraperitoneal injection of kainate in b–d. (b) A single intraperitoneal injection of 1NM-PP1 (P) at a dose of 16.6 ng per g reduced TrkB kinase activity in the cortex of A/A, but not WT, animals at 30 min postinjection compared with vehicle (V) injection. The kinase function recovered by 80 min postinjection. (c) TrkBF616A kinase activity in the cortex was chronically inhibited by subcutaneous infusion of 1NM-PP1 using osmotic minipumps. Phospho-TrkB was examined at 1 or 6 d after implanting a minipump. (d) TrkBF616A kinase activity was locally inhibited by intracortical infusion of 1NM-PP1. We dissected the caudal-lateral quadrant of the left (L, infusion side) or right (R, control side) cortex for assays after 1 d of 1NM-PP1 infusion (P-1) or after 6 d of 1NM-PP1 (P-6) or vehicle (V-6) infusion. For quantification, phosphotyrosine signals were first normalized to those of total TrkB, which were then normalized to values of vehicle-treated WT animals. n = 3 in all groups, mean ± s.d. * P < 0.01, except for b where P < 0.05, compared with WT control.
Effects of TrkB inactivation on visual cortical function
Ntrk2F616A (TrkBF616A) homozygous mice have normal TrkB function in the absence of inhibitor and show growth and reproduction that is comparable to that observed in wild-type littermates24. Baseline synaptic transmission and plasticity in acute hippocampal slices were also normal in the absence of the inhibitor (P.M.E., unpublished data). Using intrinsic-signal imaging, we found that retinotopic organization and response magnitude in the visual cortex of TrkBF616A mice with normal visual experience were indistinguishable from those of wild-type littermates (Fig. 2a and Supplementary Fig. 1 online).
Figure 2.
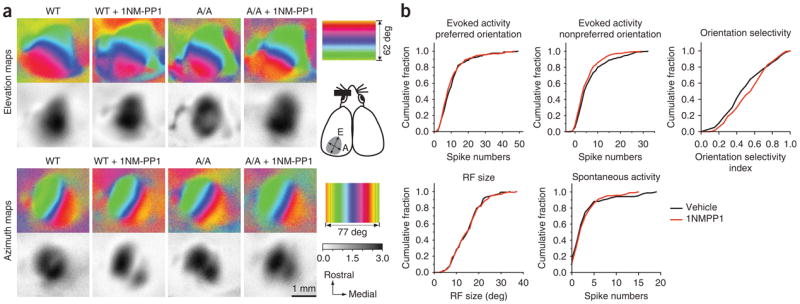
Effects of TrkB kinase inhibition during the critical period on visual cortical functions. (a) Functional visual cortical maps were normal in A/A mice. Both retinotopy and response magnitude of the visual cortex in A/A mice were indistinguishable from those in WT animals. TrkB inactivation by 1NM-PP1 for 6 d starting at P24–28 had no detectable effect on maps examined right after 6 d of treatment (at P30–34). The color scale represents the elevational or azimuthal position on the stimulus monitor. The cartoon on the right shows the dorsal view of the cortex; the approximate position of the left visual cortex where optical imaging maps were acquired is indicated in gray with elevational (E) and azimuth (A) axes (dotted lines). The gray scale represents the response magnitude as the fractional change in reflectance (× 104). (b) Single-unit extracellular recording showed no differences in neuronal responses to stimuli with preferred orientations, orientation selectivity, receptive field (RF) size and spontaneous activity between vehicle- (131 cells, 6 mice) and 1NM-PP1–treated (142 cells, 6 mice) animals. The distribution of responses to nonpreferred orientations in 1NM-PP1–treated animals shifted leftward from that of vehicle-treated animals (P = 0.041, Kolmogrov-Smirnov test).
Because treatments that strongly alter electrical activity in the cortex can affect ODP, we tested whether 1NM-PP1 alters the visual cortical responsiveness of TrkBF616A mice at the global and single-unit levels. Functional visual cortical maps (retinotopy and response magnitude) in animals treated with 1NM-PP1 for 6 d starting at postnatal day 24–28 (P24–28) were not different from those in vehicle-treated mice, as measured with intrinsic-signal imaging (Fig. 2a and Supplementary Fig. 1). Single-unit recording from neurons in the binocular area showed no difference between 1NM-PP1– and vehicle-treated mutant animals in spontaneous activity, evoked activity in response to stimuli with preferred orientations, and receptive field sizes (Fig. 2b).
Responses to bars with nonpreferred orientations were slightly, but significantly, lower in 1NM-PP1–treated animals than those in vehicle-treated animals (P = 0.041, Kolmogrov-Smirnov test), although the resulting difference in orientation selectivity, the relative responsiveness to preferred and nonpreferred orientations, did not reach significance (P = 0.053, Kolmogrov-Smirnov test). BDNF-TrkB signaling has been shown to be important in GABAergic neuron development19. Visual cortical neurons in mice carrying a targeted disruption of a GABA-synthesizing enzyme (Gad2−/− mice) show prolonged discharge to a light-bar stimulus moving out the cell’s receptive field, an indicator of reduced inhibition26. Cells having such prolonged discharge were rarely found in control animals (0–1 cells per animal) and did not increase after TrkB kinase inhibition for 6 d during the critical period (vehicle, 5 of 131 cells; 1NM-PP1, 6 of 142 cells). Acute inhibition of TrkB kinase also did not produce systematic changes in visually evoked and spontaneous activity in visual cortical neurons over 1 h following a single injection of 1NM-PP1 (Supplementary Fig. 2 online), a duration sufficient for the inhibitor to have its full effect (as demonstrated in Fig. 1b). These observations indicate that TrkB kinase inhibition does not grossly change the responsiveness of visual cortical neurons.
Ocular dominance plasticity is normal under TrkB inhibition
To determine the role of TrkB kinase activation in cortical plasticity, we deprived mice of vision in one eye by closing one eyelid for 4–5 d, starting at P25–26, during the peak of the critical period. We measured cortical responses to right- and left-eye stimulation that was restricted to the binocular portion of the visual field and computed the ocular dominance index (ODI) using intrinsic-signal imaging as described (Fig. 3a)27. This method readily detected the ocular dominance shift that was induced by monocular deprivation in wild-type and vehicle-treated TrkBF616A mice (Fig. 3b). Notably, inhibition of TrkB kinase activity did not affect the ocular dominance shift (Fig. 3b) or other features of cortical maps (Supplementary Fig. 3 online). The ODI after monocular deprivation in 1NM-PP1–treated TrkBF616A mice (−0.06 ± 0.05, mean ± s.d.) was indistinguishable from those in vehicle-treated TrkBF616A animals (−0.08 ± 0.05) or in wild-type littermates treated with 1NM-PP1 (−0.08 ± 0.04) or vehicle solution (−0.07 ± 0.07) (Fig. 3b). Single-unit recordings confirmed the optical imaging findings and showed that there was no significant difference between vehicle- and 1NM-PP1–treated TrkBF616A mice after monocular deprivation in either the ocular dominance distribution (P = 0.545, Fisher exact test) or the computed contralateral bias index (CBI) (P > 0.05) (Fig. 3c,d). These results in mutant mice are consistent with, but much stronger than, data obtained using conventional pharmacological approaches in wild-type animals; neither cortical infusion of K252a, a broad-spectrum tyrosine kinase inhibitor, nor TrkB-IgG fusion protein, a specific scavenger for extracellular TrkB ligands, impaired ODP during the normal critical period in wild-type mice (Supplementary Fig. 4 online).
Figure 3.

TrkB inactivation does not affect plasticity induced by monocular deprivation. (a) The visual stimulus pattern used for intrinsic-signal imaging of binocular visual cortex and the experimental schedule for acute recording are shown. Thin bars (2° × 20°) drifting vertically at 10° s−1 were displayed in the binocular visual field on the monitor. (b) ODIs in individual mice (circles) for different conditions, measured in acute imaging, are shown. ND, no visual deprivation; PP1, 1NM-PP1; Veh, vehicle solution. No significant difference was detected between groups with monocular deprivation. Horizontal lines indicate mean ODI in each group. (c,d) Single-unit recording showed no difference in ocular dominance score distribution (P = 0.545, Fisher exact test) or computed CBI between vehicle- and 1NM-PP1–treated A/A mice after 5-d monocular deprivation (MD). All recordings were made in A/A mice (n = 3 in each group; ND, 65 cells; MD-vehicle, 62 cells; MD–1NM-PP1, 67 cells). (e) Experimental schedule for repeated imaging of intrinsic signals and examples of amplitude maps. The scale represents the response magnitude as fractional change in reflectance (× 10− 4). M, medial; R, rostral. (f) The change in average response amplitude (± s.e.m.) over the course of MD was similar between vehicle- (n = 5) and 1NM-PP1–treated (n = 6) A/A mice. (g) ODIs in individual animals (circles) and group averages (horizontal bars) measured by repeated intrinsic-signal imaging. *P < 0.01 versus ND control (b,d), *P < 0.05 and **P < 0.01 versus pre-MD, repeated measure ANOVA (f,g).
The ocular dominance shift following monocular deprivation during the critical period is the product of two distinct changes: a decrease in deprived-eye responses and an increase in open-eye responses. To determine whether TrkB inactivation might affect the magnitude or time course of these changes, we recorded intrinsic signals repeatedly in individual animals before and after the eyelid suture (Fig. 3e–g). A robust decrease in response amplitude to deprived-eye stimulation occurred at 3 d of monocular deprivation and was maintained at 6 d of monocular deprivation, whereas an increase in the response magnitude to the open eye was detected at 6 d, but not at 3 d (Fig. 3f), resulting in a gradual change of the ODI (Fig. 3g). These observations are consistent with previous reports28,29. The degree and the time course of both these changes were not different between vehicle- and 1NM-PP1–treated TrkBF616A mice (Fig. 3f,g). Together, these results indicate that TrkB kinase signaling is not required for any aspect of the plasticity process by which monocular deprivation leads to changes in visual cortical responses to the two eyes during the critical period.
TrkB inactivation inhibits recovery from monocular deprivation
A second manifestation of cortical plasticity is the recovery of responsiveness to the deprived eye when normal vision is restored. Previous experiments have suggested that the loss and recovery of response to the deprived eye may share molecular mediators30. However, recent data suggest that different mechanisms contribute to these two processes31,32. Therefore, we examined whether TrkB kinase inhibition affects recovery. Using repeated imaging of intrinsic signals, visually evoked responses were recorded before the lid suture at P25, after 5 d of monocular deprivation (P30) and after another 4 d of binocular vision (P34) in TrkBF616A mice treated with systemic 1NM-PP1 or vehicle solution during the recovery period (Fig. 4a). In vehicle-treated animals, opening the previously deprived eye to allow binocular vision restored responses to both eyes to their baseline levels, as measured before the initial monocular deprivation (Fig. 4b,c); as a result, the ODI was shifted back to its original state (Fig. 4d). In marked contrast to its lack of effect on monocular deprivation, 1NM-PP1 treatment of TrkBF616A mice during the binocular vision prevented this recovery (Fig. 4b–d). Both the previously deprived- and open-eye responses failed to recover from the effect of monocular deprivation, resulting in a lack of ODI restoration (Fig. 4d).
Figure 4.

TrkB inactivation blocks the recovery of cortical responses to the previously deprived eye. (a) Experimental schedule and examples of response amplitude maps recorded with repeated imaging in vehicle- and 1NM-PP1–treated A/A mice before lid suture (Pre), after 5 d of MD and after 4 d of binocular vision (Rec). (b) Average response amplitude (± s.e.m.) to contralateral and ipsilateral eye stimulation in A/A mice treated with vehicle solution (n = 5) or 1NM-PP1 (n = 5) and in WT mice treated with 1NM-PP1 (n = 4). (c) Recovery index was calculated as (recovery amplitude – MD amplitude) / (MD amplitude – pre amplitude) and presented as mean ± s.e.m.; a value of 1 indicates a complete reversal of MD-induced effects. (d) Individual ODIs computed in animals shown in b. Horizontal bars represent group averages. (e) Average response amplitudes (± s.e.m.) in WT mice treated with vehicle solution (n = 4) or K252a (n = 4) and in A/A mice treated with K252a (n = 4). (f) ODIs (mean ± s.e.m.) in animals whose response amplitudes are shown in e. *P < 0.05 and **P < 0.01 compared with pre-MD, repeated measure ANOVA (b,d–f); *P < 0.05 versus control groups, one-way ANOVA (c).
Consistent with the results of TrkB inactivation in mutant mice, administration of the broad-spectrum tyrosine kinase inhibitor K252a during re-established binocular vision in wild-type animals also blocked recovery. Impaired recovery was evident in deprived- and open-eye responses, as well as in ODI, whereas binocular responses in vehicle-treated animals were restored to pre–monocular deprivation levels (Fig. 4e,f). Notably, when K252a was administered to TrkBF616A mice during binocular vision, it did not produce detectable effects on recovery (Fig. 4e,f), suggesting that the mutant TrkB kinase is relatively insensitive to K252a, a suggestion that is consistent with previous results in vitro24. These findings further suggest that the effect of K252a that we observed is exerted primarily through its action on TrkB.
We also examined ocular dominance recovery using acute intrinsic-signal imaging (Fig. 5a) and single-unit extracellular recording (Fig. 5b,c). Consistent with the results that we obtained in our chronic imaging experiments, acute imaging showed that the mean ODI in 1NM-PP1–treated TrkBF616A mice after the re-establishment of binocular vision following monocular deprivation (−0.04 ± 0.06) differed significantly from those of all control recovery groups, but did not differ from those of wild-type and TrkBF616A mice after 5 d of monocular deprivation without recovery (one-way analysis of variance, ANOVA, with correction for multiple comparison). Even a much longer period of binocular vision (10–12 days) failed to restore normal ocular dominance under TrkB kinase inhibition (SC-L group in Fig. 5a; ODI, −0.02 ± 0.08). Single-unit recording confirmed that ocular dominance recovered almost fully in vehicle-treated TrkBF616A mice (Fisher exact test, P = 0.581 versus no monocular deprivation in Fig. 3b), but did not recover in 1NM-PP1–treated animals (Fisher exact test, P = 0.619 versus monocular deprivation in Fig. 3b, and P < 0.001 versus vehicle-recovery group) (Fig. 5b).
Figure 5.

Acute optical imaging and single-unit recording confirm the requirement of TrkB kinase activity for recovery. (a) Experimental schedule and ODI for all animals examined in acute intrinsic-signal imaging. For each group, a horizontal line indicates the mean. Either systemic (SC) or intracortical (CX) infusion of 1NM-PP1 in A/A mice substantially reduced the recovery, even after a longer period (10–12 d) of binocular vision (SC-L).
**P < 0.001 versus vehicle control groups.
(b) Distribution of ocular dominance (OD) scores in single-unit recording in A/A mice after 4 d of binocular vision following 5-d monocular deprivation was substantially different between SC-vehicle (69 cells, 3 mice) and SC–1NM-PP1 (72 cells, 3 mice) (P < 0.001, Fisher exact test) and between CX-vehicle (82 cells, 3 mice) and CX–1NM-PP1 (73 cells, 3 mice) (P < 0.001, Fisher exact test). (c) CBIs in individual animals (circles) and group averages (horizontal lines) computed from single-unit recording data. Data for nondeprived group (ND) were from Figure 3c.
**P < 0.01 versus control groups.
Cortical TrkB kinase activation is required for recovery
Because TrkB is expressed at multiple levels in the visual pathway, including the retina and dorsal lateral geniculate nucleus12,33, TrkB inhibition at those sites could have been responsible for the impaired ocular dominance recovery described above, where 1NM-PP1 administered subcutaneously could act systemically. Therefore, we probed the specific cortical contribution to this failure of ocular dominance recovery, taking advantage of the ability to inhibit TrkB activity locally by intracortical infusion of 1NM-PP1 in TrkBF616A mice (see Fig. 1c). Similar to systemic inhibition, local TrkB inactivation in the cortex prevented ocular dominance recovery (Vehicle-CX and 1NM-PP1–CX groups; Fig. 5a–c). These results indicate that TrkB kinase activity in the cortex is necessary for the ocular dominance recovery that is induced by binocular vision following a brief monocular deprivation.
DISCUSSION
Our findings, obtained using a chemical-genetic method, show contrasting roles for TrkB in plasticity of the mouse visual cortex; its kinase activity is dispensable for the ocular dominance shift caused by monocular deprivation, but is necessary for the subsequent recovery of deprived-eye responses that is induced by binocular vision. The present results establish that TrkB kinase signaling is simply not a part of the plasticity machinery responsible for the rapid loss of cortical responses in monocular deprivation, contrary to widely accepted models in which the reduction of deprived-eye activity leads to its inputs to the cortex being defeated in a competition with more active inputs from the open eye for limited quantities of BDNF. Instead, TrkB signaling in developing visual cortex has a conventional neurotrophic role in facilitating the growth or enhancement of connections: in this case, those serving an eye restored to normal levels of activity. Although our data in mutant mice do not exclude the possibility that BDNF acts as a retrograde signal during monocular deprivation through a mechanism other than TrkB kinase activity, such as the T1 isoform of TrkB or through unidentified receptors, the persistence of monocular deprivation effects after treatment with the specific ligand scavenger TrkB-IgG (Supplementary Fig. 2) argues against this possibility.
The effects of sensory deprivation and experience on BDNF expression in visual cortex parallel the roles for TrkB kinase activity that we demonstrate here in different forms of visual cortical plasticity. BDNF levels in the contralateral visual cortex decrease during monocular deprivation11,13; it thus makes sense that TrkB inactivation does not affect the loss of dominant-eye responses and synaptic pathways, as receptor activation is already low because of the scarcity of the ligand. On removal of the lid suture to initiate the recovery process, BDNF expression and secretion would be expected to return to the normal level rapidly, as they do after brief light exposure following a period of darkness10. This increase is consistent with the requirement for TrkB activation for the recovery of responses. Thus, BDNF-TrkB signaling appears to be unnecessary for the loss, but is necessary for the enhancement or growth of synaptic pathways that mediate deprived contralateral-eye responses. In this hypothesis, the fact that the absolute increase of response to the open eye following monocular deprivation does not depend on TrkB kinase function requires a different explanation. This increase, which took place only subsequent to the decrease in deprived-eye response (Fig. 3f), may be viewed as a homeostatic response to maintain the firing rates of cortical cells using the only input that remains active; in culture, inhibiting TrkB activation promotes synaptic upscaling34.
Differential effects of various experimental manipulations on the loss and recovery of binocular responses have suggested that distinct cellular processes, such as synapse removal or formation, branch retraction or growth, or long-term depression/potentiation, may underlie the two phenomena31,32. In particular, the requirement that we have found for TrkB kinase activation specifically in recovery, together with reports of a role for BDNF-TrkB signaling in the growth of new synapses and the potentiation of synaptic transmission8,9,35,36, suggests that these cellular processes may underlie recovery.
Several distinct signaling pathways are engaged by TrkB activation. Inactivation of TrkBF616A by 1NM-PP1 presumably prevents phosphorylation of cytoplasmic tyrosine residues, which would inhibit the majority of signaling pathways that start with the binding of adaptors and enzymes to docking sites served by phosphotyrosines, including Y515 (Ras-MAP kinase and PI3K-Akt pathways) and Y816 (phospholipase Cγ (PLCγ) pathway)37. The present results do not reveal which one or which combination of these signaling pathways is essential for recovery. Several observations, however, are relevant. First, PLCγ signaling has a major role in hippocampal long-term potentiation38, whereas the Shc pathway is less involved39. Second, TrkB activation by BDNF increases neurotransmitter release presynaptically40–42, which is mediated by the internal calcium stores and IP3 (refs. 43,44), again pointing toward the PLCγ pathway. Third, previous studies have demonstrated that neither protein synthesis32 nor CREB activation31 are required for recovery of deprived-eye responses in juvenile ferrets, although these findings have not yet been confirmed in mice. These two are well-characterized downstream targets/effectors of TrkB signaling. Fourth, BDNF exerts rapid effects on synaptic transmission by modulating ion channel properties through TrkB45, many of which are mediated by post-translational modifications and do not require new protein synthesis.
Relatively little is known about the molecular mechanisms underlying the restoration of cortical responses after deprivation is ended. Our results provide new insights into a potential molecular cascade that regulates the recovery of cortical function in response to sensory experience. In light of the potential clinical importance of these findings for patients with amblyopia, it will be necessary to elucidate the signaling pathways downstream of TrkB that are required for recovery.
METHODS
Animals
Generation of the mouse line carrying a knock-in mutation with a substitution of phenylalanine (F) to alanine (A) at the amino acid position 616 of the Trkb gene (TrkBF616A) was described previously24. 1NM-PP1 was synthesized as described previously25. Heterozygous mutant mice were maintained and experiments were carried out on homozygous and wild-type littermates from the cross of heterozygous mice unless otherwise noted. All experimental procedures were approved by the University of California San Francisco Institutional Animal Care and Use Committee.
Monocular deprivation and osmotic minipump implantation
To introduce monocular deprivation, the lids were sutured shut in the right eye, contralateral to the cerebral hemisphere examined, at P25–26 as described46. To examine the ocular dominance recovery following monocular deprivation, lids were sutured at P25 and opened at P30, followed by binocular vision for 4 d. Animals were checked daily to ensure that the eyes remained closed during monocular deprivation and open during the recovery period. We administered 1NM-PP1 or vehicle solution (4% (vol/vol) DMSO and 2% (vol/vol) Tween-20 in saline) using Alzet osmotic minipumps (Durect) at the rate of 0.5 nmol per g per h (subcutaneous) or 0.1 nmol h−1 (intracortical). The subcutaneous infusion rate was chosen on the basis of the previous report24. The intracortical infusion rate was determined in pilot experiments (data not shown). Cannula implantation for intracortical infusion was carried out as described47. The minipump was implanted about 24–36 h before monocular deprivation or suture removal.
TrkB-IgG (Regeneron Pharmaceuticals) in phosphate-buffered saline or K252a (Alexis Biochemicals) in artificial cerebrospinal fluid were infused to the cerebral cortex via a cannula connected to an osmotic minipump at a rate of 0.5 μg h−1 or 40–80 pmol h−1, respectively, starting 2 d before the lid suture.
Biochemical assays
Because the baseline level of phosphorylated TrkB in the cortex was low (Fig. 1a), we enhanced its level by systemically injecting kainic acid as described48. For acute TrkB inhibition, animals received an intraperitoneal injection of 1NM-PP1 (16.6 ng per g) followed by kainite (15 mg per kg) 10 or 60 min later. To examine TrkB inactivation by continuous infusion, 1NM-PP1 or vehicle were administered via osmotic minipumps for 1 or 6 d before the assay. In all cases, animals were quickly anesthetized with 4–5% isoflurane (Abbott) 20 min after kainate injection and were killed by decapitation. The cortices were then dissected and frozen in liquid nitrogen. For local inhibition experiments, the cortical area corresponding to the middle half in the medial-lateral extent from the caudal half was dissected separately from the left (infused side) and right (control side) cortices. Tissue was homogenized in cold lysis buffer containing 50 mM Tris (pH 8.0), 150 mM NaCl, 1% (wt/vol) Triton X-114, 0.1% (wt/vol) SDS, 0.5% (wt/vol) deoxycholic acid, protease inhibitor cocktail (Sigma) and 1 mM sodium orthovanadate. We incubated 1 mg of the extracted protein with 2 μg of rabbit antibody to mouse TrkB (gift from L. Reichardt, University of California San Francisco) for 1 h and then with protein A–Sepharose for 2 h. The precipitated protein was separated in SDS-PAGE and transferred to PVDF membrane. Phosphotyrosine was detected with antibody to phospho-Trk (mouse monoclonal IgG, Santa Cruz Biotechnology) using enhanced chemiluminescence and X-ray films. The membranes were then stripped using Restore Western Blot Stripping Buffer (Pierce) for 2–6 h at 20–22 °C (Supplementary Fig. 5 online) and reprobed with antibody to TrkB. The exposed films were digitized, and densitometric quantification was carried out using ImageJ (US National Institutes of Health). The phospho-TrkB levels were normalized to corresponding total TrkB signals and the deduced ratios were further normalized to that of the control wild-type mouse for statistical analyses with ANOVA followed by Bonferroni’s post hoc test. All chemicals were purchased from Sigma unless otherwise noted.
Optical imaging of intrinsic signals
Intrinsic-signal optical imaging and quantification of ocular dominance were carried out as described27. For repeated intrinsic-signal recording, we modified the acute preparation as follows. First, Nembutal was replaced with isoflurane (0.8%) in oxygen applied via a home-made nose mask, supplemented with a single intramuscular injection of 20–25 μg chlorprothixene. We monitored the concentration of isoflurane using an Ohmeda 5250 RGM (Datex-Ohmeda) throughout an imaging session. Second, we recorded transcranially; the scalp was suture closed at the end of the session and re-opened at the same location in the consequent session(s). Third, we shortened the duration of each run to 4 min, reducing the length of each imaging session to minimize the effects of anesthesia on ocular dominance plasticity. All repeated recordings were made in TrkBF616A mice. Typically, preparations took about 30 min, and animals stayed in a stable anesthetic level for 2–3 h. Animals regained normal awake behavior in 2–3 h after discontinuation of isoflurane and were kept in a regular housing condition between recordings.
For the initial assessment of retinotopy and response magnitude (as shown in Fig. 2 and Supplementary Fig. 1), we carried out intrinsic-signal imaging at P30–34 acutely under urethane anesthesia using full-screen bar stimulus as described49. The response area was calculated by selecting pixels with a response amplitude larger than 30% of the maximum value. Cortical magnification factor and map scatter were computed as described50.
Quantitative measurement of single-unit responses
To examine the effects of acute TrkB kinase inhibition on neuronal responsiveness, we recorded a total of 12 cells from three TrkBF616A homozygous mice at P32–36 using computer-driven stimulus presentation and spike collection with the System 3 workstation (Tucker-Davis Technologies). Single bright bars, 4° wide and 80° long at 80% contrast and drifting at 15° s−1, were used to drive cortical cells. Each trial consisted of the drifting bar in eight different directions and a period of blank screen in a random sequence. Recordings were made from the binocular area of V1 before and at several time points between 5 and 60 min after 1NM-PP1 injection (16.6 ng per g, intraperitoneal), with five trials at each time point. A peristimulus histogram for each stimulus direction was constructed using 100-ms bins. The response of the cell was defined as the sum of the spikes elicited by each stimulus direction at each time point. Visually evoked responses after 1NM-PP1 injection were compared with the preinjection response in individual animals, and changes were evaluated using repeated measure ANOVA. For statistical analysis of pooled data, the postinjection responses at each time point were normalized to the preinjection response.
The effects of longer-term TrkB kinase inactivation on response properties of visual cortical neurons were examined with single-unit recording in TrkBF616A homozygous mice after 6 d of continuous infusion of 1NM-PP1 or vehicle solution via subcutaneous osmotic minipump. The stimulus and recording settings were the same as those described above. Orientation selectivity index was calculated as (Rpref − Rnull)/(Rpref + Rnull), where Rpref is the highest number of spikes elicited among four different orientations and Rnull is the number of spikes elicited by the stimulus orthogonal to the preferred one. Receptive field size was computed as described50.
Statistical analysis
Statistical significance was determined by ANOVA, followed by multiple comparisons with Bonferroni’s correction. Repeated measure ANOVAs and paired t-tests were used for chronic imaging data. For ocular dominance scores in single-unit recording, we used the Fisher exact test.
Supplementary Material
Note: Supplementary information is available on the Nature Neuroscience website.
Acknowledgments
The TrkBF616A mice were provided by D. Ginty and with the permission of CGI and Princeton University. We thank members of the laboratory and P. McQuillen for discussions, J. Cang for help with analyses of optical imaging data, A. Shreiber, C.M. Beal and T. Tran for technical assistance, Regeneron Pharmaceuticals for TrkB-IgG, and L. Reichardt for TrkB antibody. This study was supported by grants from the US National Institutes of Health (P50-MH077972, R37-EY02874, and T32-EY07120).
Footnotes
AUTHOR CONTRIBUTIONS
M.K. carried out all of the biochemical analysis, optical imaging and single-unit recordings using TrkBF616A and their appropriate control mice, prepared all of the figures (except for Supplementary Fig. 3) and wrote the first draft of the manuscript. J.L.H. carried out experiments on K252a and TrkB-IgG mice. P.M.E. supplied mutant mice and 1NM-PP1. M.K., P.M.E. and M.P.S. designed the experiments and assisted with data analysis and revision of the manuscript.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions
References
- 1.Wiesel TN. Postnatal development of the visual cortex and the influence of environment. Nature. 1982;299:583–591. doi: 10.1038/299583a0. [DOI] [PubMed] [Google Scholar]
- 2.Reiter HO, Waitzman DM, Stryker MP. Cortical activity blockade prevents ocular dominance plasticity in the kitten visual cortex. Exp Brain Res. 1986;65:182–188. doi: 10.1007/BF00243841. [DOI] [PubMed] [Google Scholar]
- 3.Reiter HO, Stryker MP. Neural plasticity without postsynaptic action potentials: less-active inputs become dominant when kitten visual cortical cells are pharmacologically inhibited. Proc Natl Acad Sci USA. 1988;85:3623–3627. doi: 10.1073/pnas.85.10.3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hata Y, Stryker MP. Control of thalamocortical afferent rearrangement by post-synaptic activity in developing visual cortex. Science. 1994;265:1732–1735. doi: 10.1126/science.8085163. [DOI] [PubMed] [Google Scholar]
- 5.Hata Y, Tsumoto T, Stryker MP. Selective pruning of more active afferents when cat visual cortex is pharmacologically inhibited. Neuron. 1999;22:375–381. doi: 10.1016/s0896-6273(00)81097-1. [DOI] [PubMed] [Google Scholar]
- 6.Lessmann V, Gottmann K, Malcangio M. Neurotrophin secretion: current facts and future prospects. Prog Neurobiol. 2003;69:341–374. doi: 10.1016/s0301-0082(03)00019-4. [DOI] [PubMed] [Google Scholar]
- 7.Lu B. BDNF and activity-dependent synaptic modulation. Learn Mem. 2003;10:86–98. doi: 10.1101/lm.54603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annu Rev Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- 9.Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- 10.Castren E, Zafra F, Thoenen H, Lindholm D. Light regulates expression of brain-derived neurotrophic factor mRNA in rat visual cortex. Proc Natl Acad Sci USA. 1992;89:9444–9448. doi: 10.1073/pnas.89.20.9444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bozzi Y, et al. Monocular deprivation decreases the expression of messenger RNA for brain-derived neurotrophic factor in the rat visual cortex. Neuroscience. 1995;69:1133–1144. doi: 10.1016/0306-4522(95)00321-9. [DOI] [PubMed] [Google Scholar]
- 12.Schoups AA, Elliott RC, Friedman WJ, Black IB. NGF and BDNF are differentially modulated by visual experience in the developing geniculocortical pathway. Brain Res Dev Brain Res. 1995;86:326–334. doi: 10.1016/0165-3806(95)00043-d. [DOI] [PubMed] [Google Scholar]
- 13.Majdan M, Shatz CJ. Effects of visual experience on activity-dependent gene regulation in cortex. Nat Neurosci. 2006;9:650–659. doi: 10.1038/nn1674. [DOI] [PubMed] [Google Scholar]
- 14.Galuske RA, Kim DS, Castren E, Singer W. Differential effects of neurotrophins on ocular dominance plasticity in developing and adult cat visual cortex. Eur J Neurosci. 2000;12:3315–3330. doi: 10.1046/j.1460-9568.2000.00213.x. [DOI] [PubMed] [Google Scholar]
- 15.Gillespie DC, Crair MC, Stryker MP. Neurotrophin-4/5 alters responses and blocks the effect of monocular deprivation in cat visual cortex during the critical period. J Neurosci. 2000;20:9174–9186. doi: 10.1523/JNEUROSCI.20-24-09174.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lodovichi C, Berardi N, Pizzorusso T, Maffei L. Effects of neurotrophins on cortical plasticity: same or different? J Neurosci. 2000;20:2155–2165. doi: 10.1523/JNEUROSCI.20-06-02155.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shatz CJ. Neurotrophins and visual system plasticity. In: Cowan MW, Jessell TM, Zipursky SL, editors. Molecular and Cellular Approaches to Neural Development. Oxford University Press; New York: 1997. pp. 509–524. [Google Scholar]
- 18.Hanover JL, Huang ZJ, Tonegawa S, Stryker MP. Brain-derived neurotrophic factor overexpression induces precocious critical period in mouse visual cortex. J Neurosci. 1999;19:RC40. doi: 10.1523/JNEUROSCI.19-22-j0003.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang ZJ, et al. BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell. 1999;98:739–755. doi: 10.1016/s0092-8674(00)81509-3. [DOI] [PubMed] [Google Scholar]
- 20.Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270:593–598. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- 21.Bonhoeffer T. Neurotrophins and activity-dependent development of the neocortex. Curr Opin Neurobiol. 1996;6:119–126. doi: 10.1016/s0959-4388(96)80017-1. [DOI] [PubMed] [Google Scholar]
- 22.Hensch TK. Critical period plasticity in local cortical circuits. Nat Rev Neurosci. 2005;6:877–888. doi: 10.1038/nrn1787. [DOI] [PubMed] [Google Scholar]
- 23.Specht KM, Shokat KM. The emerging power of chemical genetics. Curr Opin Cell Biol. 2002;14:155–159. doi: 10.1016/s0955-0674(02)00317-4. [DOI] [PubMed] [Google Scholar]
- 24.Chen X, et al. A chemical-genetic approach to studying neurotrophin signaling. Neuron. 2005;46:13–21. doi: 10.1016/j.neuron.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 25.Wang H, et al. Inducible protein knockout reveals temporal requirement of CaMKII reactivation for memory consolidation in the brain. Proc Natl Acad Sci USA. 2003;100:4287–4292. doi: 10.1073/pnas.0636870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hensch TK, et al. Local GABA circuit control of experience-dependent plasticity in developing visual cortex. Science. 1998;282:1504–1508. doi: 10.1126/science.282.5393.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cang J, Kalatsky VA, Lowel S, Stryker MP. Optical imaging of the intrinsic signal as a measure of cortical plasticity in the mouse. Vis Neurosci. 2005;22:685–691. doi: 10.1017/S0952523805225178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mioche L, Singer W. Chronic recordings from single sites of kitten striate cortex during experience-dependent modifications of receptive-field properties. J Neurophysiol. 1989;62:185–197. doi: 10.1152/jn.1989.62.1.185. [DOI] [PubMed] [Google Scholar]
- 29.Frenkel MY, Bear MF. How monocular deprivation shifts ocular dominance in visual cortex of young mice. Neuron. 2004;44:917–923. doi: 10.1016/j.neuron.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 30.Bear MF, Kleinschmidt A, Gu QA, Singer W. Disruption of experience-dependent synaptic modifications in striate cortex by infusion of an NMDA receptor antagonist. J Neurosci. 1990;10:909–925. doi: 10.1523/JNEUROSCI.10-03-00909.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liao DS, Mower AF, Neve RL, Sato-Bigbee C, Ramoa AS. Different mechanisms for loss and recovery of binocularity in the visual cortex. J Neurosci. 2002;22:9015–9023. doi: 10.1523/JNEUROSCI.22-20-09015.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krahe TE, Medina AE, de Bittencourt-Navarrete RE, Colello RJ, Ramoa AS. Protein synthesis–independent plasticity mediates rapid and precise recovery of deprived eye responses. Neuron. 2005;48:329–343. doi: 10.1016/j.neuron.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 33.Ugolini G, Cremisi F, Maffei L. TrkA, TrkB and p75 mRNA expression is developmentally regulated in the rat retina. Brain Res. 1995;704:121–124. doi: 10.1016/0006-8993(95)01191-9. [DOI] [PubMed] [Google Scholar]
- 34.Rutherford LC, Nelson SB, Turrigiano GG. BDNF has opposite effects on the quantal amplitude of pyramidal neuron and interneuron excitatory synapses. Neuron. 1998;21:521–530. doi: 10.1016/s0896-6273(00)80563-2. [DOI] [PubMed] [Google Scholar]
- 35.Vicario-Abejon C, Owens D, McKay R, Segal M. Role of neurotrophins in central synapse formation and stabilization. Nat Rev Neurosci. 2002;3:965–974. doi: 10.1038/nrn988. [DOI] [PubMed] [Google Scholar]
- 36.Lu B, Pang PT, Woo NH. The yin and yang of neurotrophin action. Nat Rev Neurosci. 2005;6:603–614. doi: 10.1038/nrn1726. [DOI] [PubMed] [Google Scholar]
- 37.Reichardt LF. Neurotrophin-regulated signaling pathways. Phil Trans R Soc Lond B. 2006;361:1545–1564. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Minichiello L, et al. Mechanism of TrkB-mediated hippocampal long-term potentiation. Neuron. 2002;36:121–137. doi: 10.1016/s0896-6273(02)00942-x. [DOI] [PubMed] [Google Scholar]
- 39.Korte M, Minichiello L, Klein R, Bonhoeffer T. Shc-binding site in the TrkB receptor is not required for hippocampal long-term potentiation. Neuropharmacology. 2000;39:717–724. doi: 10.1016/s0028-3908(99)00273-7. [DOI] [PubMed] [Google Scholar]
- 40.Lessmann V, Heumann R. Modulation of unitary glutamatergic synapses by neurotrophin-4/5 or brain-derived neurotrophic factor in hippocampal microcultures: pre-synaptic enhancement depends on pre-established paired-pulse facilitation. Neuroscience. 1998;86:399–413. doi: 10.1016/s0306-4522(98)00035-9. [DOI] [PubMed] [Google Scholar]
- 41.Schinder AF, Berninger B, Poo M. Postsynaptic target specificity of neurotrophin-induced presynaptic potentiation. Neuron. 2000;25:151–163. doi: 10.1016/s0896-6273(00)80879-x. [DOI] [PubMed] [Google Scholar]
- 42.Tyler WJ, et al. BDNF increases release probability and the size of a rapidly recycling vesicle pool within rat hippocampal excitatory synapses. J Physiol (Lond) 2006;574:787–803. doi: 10.1113/jphysiol.2006.111310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li YX, Zhang Y, Lester HA, Schuman EM, Davidson N. Enhancement of neurotransmitter release induced by brain-derived neurotrophic factor in cultured hippocampal neurons. J Neurosci. 1998;18:10231–10240. doi: 10.1523/JNEUROSCI.18-24-10231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kang H, Schuman EM. Intracellular Ca2+ signaling is required for neurotrophin-induced potentiation in the adult rat hippocampus. Neurosci Lett. 2000;282:141–144. doi: 10.1016/s0304-3940(00)00893-4. [DOI] [PubMed] [Google Scholar]
- 45.Blum R, Konnerth A. Neurotrophin-mediated rapid signaling in the central nervous system: mechanisms and functions. Physiology (Bethesda) 2005;20:70–78. doi: 10.1152/physiol.00042.2004. [DOI] [PubMed] [Google Scholar]
- 46.Gordon JA, Stryker MP. Experience-dependent plasticity of binocular responses in the primary visual cortex of the mouse. J Neurosci. 1996;16:3274–3286. doi: 10.1523/JNEUROSCI.16-10-03274.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Taha S, Stryker MP. Rapid ocular dominance plasticity requires cortical but not geniculate protein synthesis. Neuron. 2002;34:425–436. doi: 10.1016/s0896-6273(02)00673-6. [DOI] [PubMed] [Google Scholar]
- 48.Aloyz R, Fawcett JP, Kaplan DR, Murphy RA, Miller FD. Activity-dependent activation of TrkB neurotrophin receptors in the adult CNS. Learn Mem. 1999;6:216–231. [PMC free article] [PubMed] [Google Scholar]
- 49.Kalatsky VA, Stryker MP. New paradigm for optical imaging: temporally encoded maps of intrinsic signal. Neuron. 2003;38:529–545. doi: 10.1016/s0896-6273(03)00286-1. [DOI] [PubMed] [Google Scholar]
- 50.Cang J, et al. Ephrin-as guide the formation of functional maps in the visual cortex. Neuron. 2005;48:577–589. doi: 10.1016/j.neuron.2005.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Note: Supplementary information is available on the Nature Neuroscience website.