

Abstract
The discovery of the RANKL/RANK/OPG system in the mid 1990s for the regulation of bone resorption has led to major advances in our understanding of how bone modeling and remodeling are regulated. It had been known for many years before this discovery that osteoblastic stromal cells regulated osteoclast formation, but it had not been anticipated that they would do this through expression of members of the TNF superfamily: receptor activator of NF-κB ligand (RANKL) and osteoprotegerin (OPG), or that these cytokines and signaling through receptor activator of NF-κB (RANK) would have extensive functions beyond regulation of bone remodeling. RANKL/RANK signaling regulates osteoclast formation, activation and survival in normal bone modeling and remodeling and in a variety of pathologic conditions characterized by increased bone turnover. OPG protects bone from excessive resorption by binding to RANKL and preventing it from binding to RANK. Thus, the relative concentration of RANKL and OPG in bone is a major determinant of bone mass and strength. Here, we review our current understanding of the role of the RANKL/RANK/OPG system in bone modeling and remodeling.
Keywords: Bone resorption, osteoclasts, RANKL, RANK, OPG
Normal bone modeling
With the exception of the bones of the calvaria, all bones in the mammalian skeleton are preformed in cartilage moulds from mesenchymal progenitors, which under appropriate stimuli also have the potential to differentiate into a variety of tissue types, including fibrous tissue, fat and muscle. Chondrocytes proliferate near the ends of the cartilage moulds to drive their longitudinal growth, while others in the centers of undergo hypertrophic differentiation. The hypertrophic chondrocytes at the periphery of the centers of these moulds are invaded by blood vessels and undergo apoptosis. Some of this hypertrophic cartilage survives as thin islands of cartilage in the centers of ossification in growing bones. Osteoblasts, which differentiate from progenitors in a collar of connective tissue around the middle of the bones where vascular invasion takes place, follow the endothelial cells and lay down bone matrix on the surfaces of these islands of cartilage to form bone struts or trabeculae (1). Osteoclast precursors (OCPs), derived from progenitors in the spleen and liver are attracted from blood in the invading blood vessels close to newly formed bone trabeculae. These osteoclast precursors fuse with one another to form multinucleated osteoclasts, which resorb most of the newly formed bone leaving only a few trabeculae to comprise the spongy bone of the secondary spongiosa. This secondary spongiosa comprises the metaphyseal bone between the epiphysis and the diaphysis of the bones. Osteoblasts lay down new bone on parts of the surfaces of these surviving trabeculae where there has been osteoclastic resorption and much of this new bone is subsequently resorbed by osteoclasts in a remodeling process that ensures that the volume of bone in the medullary cavity is limited and does not fill the space. This osteoclastic resorption of new bone provides space for the proliferation of hematopoietic cells, which like OCPs come from circulating precursors in the spleen and liver, and form the bone marrow. Osteoclastic resorption thus prevents the development of osteopetrosis, a congenital defect of endochondral ossification, which occurs if osteoclasts fail to form or have impaired activity (2, 3).
As the cartilaginous centers of the growing bones are removed and replaced by bone and marrow, condensations of proliferating and prehypertrophic chondrocytes form close to the ends of long bones where along with a layer of hypertrophic chondrocytes they constitute the epiphyseal growth plates. This process, called endochondral ossification, requires expression by mesenchymal osteoblast precursor cells of Runx2, the master transcription factor that regulates bone formation (4, 5) because in the absence of Runx2 vascular invasion of the cartilage moulds does not take place and no bone is formed (6, 7). Runx2 also mediates the osteoblast differentiating effects of BMP2 (8), which regulates chondrocyte differentiation as well as the formation of bone during endochondral ossification (9) in an NF-κB-dependent manner (10).
Hypertrophic and to a lesser extent prehypertrophic chondrocytes express RANKL, OPG and RANK. Mice deficient in RANKL, RANK and NF-κB p50 and p52 develop osteopetrosis because they do not form osteoclasts (see below) and have thickened hypertrophic cartilage zones in their growth plates. This defect is rectified spontaneously between 2 and 3 weeks of age in the RANKL−/− and RANK−/− mice and 2–3 weeks later in the NF-κB p50 and p52 double knockout mice (our unpublished observations). The precise role of RANKL/RANK/NF-κB signaling in chondrocytes during endochondral ossification remains poorly understood, but these findings suggest that it may regulate the lifespan of hypertrophic chondrocytes through NF-κB p50 and p52-regulated genes at least temporarily, similar to MMP-9 (11), which is regulated by NF-κB (12). All of these knockout mice are dwarfed, suggesting that RANKL/RANK/NF-κB signaling during the first 2–3 weeks of life is essential for attainment of full skeletal growth, but it is not known if this requirement is in cells of the osteoclast, osteoblast, chondrocyte, or endothelium lineages.
Recent studies suggest that hypertrophic chondrocytes are involved in the differentiation of osteoclast precursors at growth plates. Their expression of RANKL is regulated positively by Vitamin D3 (13) and BMP2 (14) in a dose-dependent manner in vitro. BMP2 is expressed by prehypertrophic chondrocytes in vivo and it induces higher expression of RANKL from chick upper sternal chondrocytes than from lower sternal chondrocytes (14), which consist predominantly of prehypertrophic cells. Upper sternal chondrocytes, in contrast are predominantly hypertrophic cells and these, like osteoblasts, also express OPG. However, these chondrocytes do not express OPG to the same degree as RANKL in response to BMP2 and thus the RANKL/OPG ratio changes in favor of osteoclast differentiation in response to BMP2. Chondrocytes express RANKL constitutively in vitro and surprisingly can support osteoclast differentiation from precursors when both cell types are cultured in a transwell assay system (14). Bone growth during embryonic development and in the first 2 weeks after birth occurs at a very high rate. Thus, it is essential that osteoclast formation is induced at the growth plate to mediate the rapid removal of calcified matrix and the formation of the marrow cavity. Hypertrophic chondrocytes are ideally situated in growth plates close to the blood vessels that bring osteoclast precursors from the circulation into developing bones to mediate this process. They secrete VEGF (11) and other factors that regulate vascular in-growth and could also promote egression of OCPs from these newly-formed blood vessels through RANKL expression. RANKL has a chemo-attractant effect on peripheral blood monocytes (15, 16). Therefore, local release of RANKL by chondrocytes and induction of its expression by BMP2 could mediate this OCP egression. Interestingly, osteoclasts are not required for the removal of the cartilage that forms during endochondral ossification since cartilage is resorbed and bone forms in the absence of osteoclasts, such as in RANKL and RANK knockout mice (17, 18). Presumably the cartilage is removed by chondroclasts, but these remain poorly defined cells, which may be in the monocyte lineage (11).
Bone remodeling
Bone has multiple functions in vertebrates, including protection of vital organs and hematopoietic marrow, structural support for muscles, and storage and release of vital ions, such as calcium, and of growth factors stored in the matrix. Bone in the adult skeleton is renewed continuously in response to a variety of stimuli by the process of bone remodeling. This involves removal of trenches or tunnels of bone from the surfaces of trabecular and cortical bone, respectively, by osteoclasts (19). Osteoblasts subsequently fill in these trenches by laying down new bone matrix in them. This sequence is similar to that which occurs in the trabeculae formed during endochondral ossification, but unlike in the latter where resorption exceeds formation to allow formation of the medullary cavity in bones, bone formation matches resorption during normal bone remodeling.
The processes that drive bone remodeling are still not fully explained, but they include damage to parts of bones in response to normal wear and tear, changes in mechanical forces following alterations in body shape or weight or exercise, and local release of cytokines or growth factors due to alterations in levels of systemic hormones. It is a tightly regulated process in that formation follows resorption in a site-specific manner and there are more than 1 million of these microscopic foci of remodeling at any time in the adult skeleton.
Recent studies suggest that at least in response to mechanical forces, osteocytes regulate the recruitment of osteoclasts to sites of bone resorption by inducing the expression of RANKL by osteoblastic cells in the local micro-environment (20). Despite this advance, the precise molecular mechanisms that control the initiation, progression and cessation of remodeling at any given site remain poorly understood. Osteocytes are terminally differentiated osteoblasts, which became embedded in the osteoid matrix they were laying down. They communicate with one another and with osteoblastic cells on the surface of overlying bone and through them with osteoblastic cells in the bone marrow cavity and in this way are thought to regulate RANKL expression by these latter cells, although they do not appear to express RANKL themselves.
The rate of bone remodeling and the number of remodeling sites are increased in a variety of pathologic conditions affecting the skeleton, including postmenopausal osteoporosis, hyperparathyroidism, and rheumatoid arthritis, in which local and/or systemic alterations in the levels of hormones or pro-inflammatory cytokines stimulate bone resorption (19). Most of these factors induce bone resorption predominantly by an indirect mechanism that involves upregulation of the expression of M-CSF and RANKL by osteoblastic and other cells (21). Our understanding of the molecular mechanisms that regulate the formation and activity of osteoclasts has advanced rapidly following the identification in the mid-to-late 1990s of the RANKL/RANK/OPG signaling system, coupled with study of bones from genetically altered mice and from animal models of bone diseases. More recently, it has become increasingly clear that osteoclasts are not simply bone resorbing cells, but that they also regulate osteoblast functions positively and negatively (22, 23), mediate the egression of hematopoietic stems from the marrow into the blood (24), and function as immunomodulators in pathologic states (25).
Regulation of Osteoclast Formation and Activation by OPG, RANKL and RANK
Osteoclasts are derived from mononuclear precursors in the myeloid lineage of hematopoietic cells that also give rise to macrophages. Understanding of the molecular mechanisms that regulate osteoclast formation and activation has advanced rapidly in the last 12 years since the discovery of the RANKL/RANK signaling system. M-CSF expression by osteoblastic stromal cells is required for progenitor cells to differentiate into osteoclasts, but M-CSF on its own is unable to complete this process. This requirement for M-CSF was discovered by the observation that op/op mice, which do not express functional M-CSF, have osteopetrosis because they lack osteoclasts (26). Completion of OCP differentiation requires expression of RANKL by osteoblastic stromal cells and of RANK by OCPs.
The requirement of osteoblastic stromal cell expression of a factor(s) that mediated OCP differentiation had been recognized for many years before the discovery of RANKL, its decoy receptor, OPG and its receptor, RANK. Many groups had attempted unsuccessfully to purify what turned out to be RANKL from bone cells for many years before its discovery by four groups working independently and using different approaches. Researchers at Amgen unexpectedly discovered the naturally-occurring inhibitor of RANKL, a molecule they named osteoprotegerin (OPG) because it protected against bone loss. They were making transgenic mice over-expressing various TNF receptor-related cDNAs in attempts to find molecules that could interfere with TNF signaling. They observed that mice over-expressing a particular cDNA developed osteopetrosis due to a lack of osteoclasts (27). Researchers at the Snow Brand Milk Products Co. in Japan discovered an identical molecule (28) by purifying a factor from ST-2 osteoblastic cells that inhibited osteoclast formation. Both groups used expression cloning and OPG as a probe and quickly identified its ligand, which they called OPG ligand (OPGL) and osteoclast differentiation factor (ODF), respectively (29, 30) This protein turned out to be identical to a member of the TNF super-family, which had been identified the year before and called receptor activator of nuclear factor-κB ligand (RANKL) (31) and TNF-related activation induced cytokine (TRANCE) (32). Researchers at Immunex had already discovered RANK while they were sequencing cDNAs from a human bone-marrow-derived myeloid dendritic-cell cDNA library (18) and it was soon identified as the receptor for OPGL/ODF. They had found that RANK was involved in the survival of dendritic cells and isolated RANKL by direct expression screening. They found that RANKL was expressed by T cells and that it increased proliferation and survival of dendritic cells. RANKL has come to be the generally accepted acronym for this cytokine following the recommendation of a nomenclature committee of the American Society for Bone and Mineral Research (33). RANKL/RANK/NF-κB signaling is also required for lymph node development and B cell maturation (18, 34). These discoveries that RANKL/RANK/NF-κB signaling is involved in osteoclast formation and immune responses and that T and B cells express RANKL have spawned the growing field of osteoimmunology.
OCP differentiation is regulated by a number of transcription factors and signaling pathways that are activated by RANKL/RANK interaction. The completion of OCP differentiation by RANKL requires the sequential expression of NF-κB, c-Fos and NFATc1 (35–37). Interestingly, TNF, which like RANKL can induce osteoclast differentiation directly from wt OCPs, also induces the sequential expression of NF-κB, c-Fos and NFATc1 (37). Importantly, TNF can also induce osteoclast formation from OCPs from RANKL−/− and RANK−/− mice in vitro (38) and thus could augment the osteoclast formation induced indirectly by itself and other factors through induction of osteoblastic cell expression of RANKL. c-Fos or NFATc1 can substitute for NF-κB in OCPs in this sequential activation program by RANKL and TNF (37), and when either of these transcription factors is over-expressed in OCPs IL-1 can induce osteoclast formation directly from them also (39). RANKL and TNF induce c-Fos expression in OCPs (37, 39). This may be an important role for these cytokines at sites of inflammation in bone where IL-1 concentrations are increased because this could facilitate direct induction of osteoclast formation by IL-1 and thus augment the effects of these cytokines on osteoclastogenesis.
To resorb bone effectively, osteoclasts must attach themselves firmly to the bone surface using specialized actin-rich podosomes. By means of these podosomes, they form tight seals with the underlying bone matrix in roughly circular extensions of their cytoplasm and within these sealed zones they form ruffled border membranes. This ruffling of the cytoplasmic membrane increases the area of the cell surface for secretion of the proteolytic enzyme, cathepsin K, and hydrochloric acid onto the bone surface (40). By this sealing and secretory mechanism, they simultaneously degrade the matrix and dissolve the mineral of bone, while protecting neighboring cells from the harmful effects of HCl. RANKL and beta integrin-mediated signaling from bone matrix activate osteoclasts (41). Osteoclast precursors fuse with one another and become multinucleated under the influence of RANKL. This fusion requires expression by OCPs of DC-STAMP (42) and of Atp6v0d2, a subunit of v-ATPase, a component of the V-type H+ ATP6i proton pump complex that secretes H+ from osteoclasts (23). RANKL also induces expression of tartrate-resistant acid phosphatase and cathepsin K through NFATc1 (35).
RANKL
RANKL exists as a homotrimeric protein and is typically membrane-bound on osteoblastic and activated T cells or is secreted by some cells, such as activated T cells (43–45). The secreted protein is derived from the membrane form as a result of either proteolytic cleavage or alternative splicing (46). The proteolytic cleavage of RANKL is carried out by matrix metalloproteases (MMP3 or 7) (47) or ADAM (a disintegrin and metalloprotease domain) (48). Most of the factors known to stimulate osteoclast formation and activity induce RANKL expression by osteoblastic stromal cells. However, RANKL is also highly expressed in lymph nodes, thymus, mammary glands and lung and at low levels in a variety of other tissues, including spleen and bone marrow (43). It is expressed by synovial cells and activated T cells in joints of patients with inflammatory arthritis to contribute at least in part to the joint destruction seen in patients with rheumatoid arthritis.
This joint destruction in RA is also mediated by TNF by a number of mechanisms: it increases the proliferation of OCPs in the bone marrow and the number of them circulating systemically by promoting their egression from the bone marrow (49); it also promotes the egression of OCPs from the blood to inflamed joints where along with RANKL and IL-1 it promotes fusion of these cells into osteoclasts (49). RANKL also stimulates the release of OCPs into the circulation; and recent studies using PTP -knockout mice suggest that osteoclasts themselves regulate the egression of hematopoietic stem cells (HSCs) from niches within the marrow under the control of RANKL. Osteoclasts from PTP -knockout mice have defective adhesion to bone and impaired resorption and RANKL did not induce HSC mobilization in these mice (24). Thus, RANKL-induced osteoclast activation appears to regulate HSC mobilization as part of homeostasis and host defense mechanisms, linking bone remodeling with the regulation of hematopoiesis.
RANKL is also expressed in epithelial cells in mammary gland lobules during pregnancy and is required for hyperplasia of these cell during lactation and thus milk production in mice (50). It is expressed by some malignant tumor cells which also express RANK, and thus RANKL signaling may regulate tumor cell proliferation (51) by either an autocrine or a paracrine mechanism when it is produced by accessory cells.
More recent studies have identified a role for RANKL signaling in tumor cell migration and bone metastasis (52). T cell production of RANKL also induces expression of INFβ by activated osteoclasts through c-Fos signaling to negatively regulate their formation (53), a mechanism that can be enhanced by T cell produced INFγ which degrades TRAF6, an essential adapter protein recruited to RANK to mediate RANK signaling (vide infra) (54). IFN-γ is a cytokine secreted primarily by activated T cells and NK cells and was originally characterized as a powerful macrophage activator that upregulated nitric oxide production and MHC class II expression in macrophages (55). It has been reported to be a strong suppressor of osteoclastogenesis in vitro through inhibition of RANKL signaling (54, 56). However, its effects on osteoclast formation are controversial. IFN-γ also enhances osteoclast generation in cultures of peripheral blood from osteopetrotic patients, in part by normalizing superoxide production (57) and has been shown to be efficacious in the treatment of osteoporosis in humans (58). A recent study analyzed the in vivo effects of IFN-γ in 3 mouse models of bone loss, including ovariectomy, LPS injection, and inflammation induced by silencing TGF-β signaling in T cells. It demonstrated that the net effect of IFN-γ in these conditions was bone loss due to increased osteoclast formation through the following mechanism: IFN-γ increased the antigen presenting function of bone marrow cells by up-regulating their expression of MHC class II molecules and thus stimulating T lymphocytes to produce RANKL, TNF and IFN-γ (59). It is likely that the effects of IFN-γ on osteoclast formation are complex and will depend on specific conditions in the bone microenvironment and relative concentrations of other cytokines that can affect the differentiation of OCPs. c-Fos/NFATc1-induced OCP differentiation can also be inhibited by reverse signaling through the OCP-expressed ligand, ephrin B2 (22). Interaction between ephrin B2 and its receptor, Eph 4 on osteoblast precursors prevents c-Fos activation of NFATc1 to inhibit OCP differentiation. Intriguingly, forward signaling through Eph 4 in osteoblast precursors promotes their differentiation (22), providing further evidence that osteoclastic cells have functions in bone other than bone resorption. It is likely that other mechanisms that limit osteoclast formation and activation through RANKL or TNF signaling will be identified.
Failure of osteoclast formation or activation leads to the development of osteopetrosis in mice, humans and other mammals (2). To date, most cases of osteopetrosis in humans have been attributed to mutations in genes, such as ClCN7, which encodes the chloride channel through which Cl flows from osteoclasts, OSTM1, which has a closely related function, TCIRG1, which encodes the a3 subunit of the H+ATPase of the proton pump, carbonic anhydrase II, which catalyzes the hydration of CO2 to H2CO3 to provide a source of H+, cathepsin K, which degrades the collagenous matrix, and Plekhm1, which encodes for a vesicle-associated protein linked to small GTPase signaling (2, 3, 60). Recently, the first report of a mutation in the RANKL gene was described in a kindred of First Nations individuals in Canada (61). The affected individuals had osteopetrosis, but they did not have obvious defect in immunologic parameters such as the number of B and T lymphocytes and frequency of infection. It is also not known if they have some of the other phenotypic features of RANKL−/− mice, such as absence of lymph nodes or failure of lactational hyperplasia. Aparent absence of anticipated immunologic features of RANKL deficiency in affected patients could reflect species-specific differences, or the possibility that these individuals make a mutant RANKL proteins that has some biological activity that mediates immune function, or that these individuals have immune cell deficiencies that have not yet been detected.
RANK
RANK is a homotrimeric transmembrane protein member of the TNF receptor superfamily. It appears to be expressed in fewer tissues than RANKL at the protein level, but in addition to OCPs, mature osteoclasts and dendritic cells, it is expressed in mammary glands (50) and some cancer cells, including breast and prostate cancers (51, 62), two tumors with high bone metastatic potential. No humans with osteopetrosis have been identified to date with mutations in rank. However, a deletion mutation that occurred spontaneously in rank was reported in transgenic mice. These mice had all of the features of mice with targeted deletion of RANK, confirming the importance of RANK for osteoclastogenesis (63). In contrast, activating mutations in exon 1 of rank have been reported in humans to account for the increased osteoclast formation, activity and osteolysis seen in some patients with familial Paget’s disease, confirming the importance of this system in humans (64). A potential role for RANK in tumor cell proliferation (51) is being investigated and if proven could be a future target for anti-tumor therapy.
OPG
OPG is secreted by many cell types in addition to osteoblasts, including those in the heart, kidney, liver, and spleen. A recent study reports that B cells may be responsible for 64% of total bone marrow OPG production and B cell-deficient mice are consistently osteoporotic, consistent with B cells being a major source of OPG in the bone marrow of normal mice (65). Most of the factors that induce RANKL expression by osteoblasts also regulate OPG expression (66). Although there some are contradictory data, in general when RANKL expression is up-regulated, OPG expression is down-regulated or not induced to the same degree as RANKL, such that the RANKL/OPG ratio changes in favor of osteoclastogenesis (43, 67). Osteoclast numbers and activity can increase if there is a change in the RANKL/OPG ratio due to either an increase in the former or a decrease in the latter or a change in both that leads to a change in the ratio in favor of RANKL.
OPG’s osteo-protective role in humans has been supported by homozygous partial deletions of opg in patients with juvenile Paget’s disease, an autosomal recessive disorder in which affected individuals have increased bone remodeling, osteopenia, and fractures (68); in addition, some patients with idiopathic hyperphosphatasia, an autosomal recessive bone disease in which affected children have increased bone turnover associated with deformities of long bones, kyphosis and acetabular protrusion have an inactivating deletion in exon 3 of OPG (69).
OPG expression is regulated in osteoblasts not only by a variety of cytokines, hormones and growth factors (67), but also by Wnt/β-catenin (70, 71). The Wnt/β-catenin pathway also regulates osteoblastic bone formation and the commitment of mesenchymal cells to the osteoblast lineage (72). Jagged1/Notch1 signaling negatively regulates osteoclast formation both directly in osteoclast precursors and indirectly by affecting the OPG/RANKL expression ratio in stromal cells (73). Thus, bone mass is determined by many influences on osteoblasts and osteoclasts and is regulated by osteoblasts through three major signaling pathways: RANKL/RANK, Wnt/β-catenin and Jagged1/Notch1.
Based on the presence of renal and aortic calcification in OPG−/− mice, OPG appears to protect large blood vessels from medial calcification (74). Calcification commonly complicates long-standing atherosclerosis, and the absence of OPG in OPG/apoE double knockout mice accelerates calcific atherosclerosis that develops in apoE−/− mice (75). These findings suggest that OPG could also limit calcification of atherosclerotic plaques (76). However, whether OPG or RANKL play important roles in cardiovascular disease remains to be determined and is controversial (77). For example, OPG serum levels are high in patients with high blood pressure and heart failure (78) and in patients with chronic renal failure, and cardiovascular disease is more common in osteoporotic patients (79). However, OPG does not appear to protect the skeleton against the increased bone resorption in patients with renal failure and associated secondary hyperparathyroidism mediated by PTH. Further studies will be required to determine the significance of high serum OPG levels in such patients because they raise questions about the importance of the RANKL/OPG ratio in serum samples being predictive of bone mass and bone resorption in these settings (80).
Transcription factor activation by RANKL/RANK in osteoclasts and OCPs
A key preliminary step in downstream signaling after RANKL ligation to RANK is the binding of TNF receptor-associated factors (TRAFs) to specific sites in the cytoplasmic domain of RANK (32, 81). RANK is a transmembrane protein, which like other TNF family receptors has no intrinsic protein kinase activating activity to mediate signaling. TRAFs 2, 5 and 6 all bind to RANK (81), but only TRAF6 appears to have essential functions in OCPs and osteoclasts, since deletion of only TRAF6 and no other TRAFs results in osteopetrosis (82, 83). TRAF6-deficient mice produced by 2 independent groups of investigators developed osteopetrosis (82, 83). Surprisingly, however, one set of mice has normal numbers of osteoclasts, but they are inactive (82), while the other has no osteoclasts (83). How inactivation of TRAF6 resulted in 2 different osteoclast phenotypes remains unexplained. Several signaling pathways are activated by RANK/TRAF-mediated protein kinase signaling; 4 directly mediate osteoclasts formation (inhibitor of NF-κB kinase (IKK)/NF-κB, c-Jun N-terminal kinase (JNK)/activator protein-1 (AP-1), c-myc, and calcineurin/NFATc1) and 3 mediate osteoclast activation (Src and MKK6/p38/MITF) and survival (Src and extracellular signal-regulated kinase) (84).
Several adapter molecules bind to the intracytoplasmic domain of RANK along with TRAFs to mediate signaling. These include Grb-2-associated binder (Gab) protein 2, a member of a family of proteins that are phosphorylated at tyrosine residues and recruit signaling molecules that contain Src homology-2 domains. Gab2-deficient mice have reduced RANKL-induced osteoclast differentiation, decreased bone resorption and mild osteopetrosis (85). The development of mild, rather than marked osteopetrosis in these mice suggests that Gab2 plays a significant, but not essential role in RANKL-induced osteoclast formation.
Essential roles for NF-κB, AP-1 and NFATc1 signaling in osteoclast formation was discovered after the generation of mice with targeted deletion of the genes encoding the precursor molecules of both p50 and p52 sub-units of NF-κB (34, 86), c-Fos (87), and NFATc1 (36). Over-expression of c-Fos rescues the defect in osteoclast formation in M-CSF-treated NF-κB p50/p52 double knockout osteoclast precursors in the absence of RANKL (37), and expression of NFATc1 in Fos−/− OCPs rescues their defect in differentiation (35). Activation of c-Fos by RANKL signaling requires expression of NF-κB p50 and p52, indicating that c-Fos and NFATc1 (37) are downstream from NF-κB (Figure). On the basis of all these studies, NFATc1 has been described as the master regulator of osteoclast formation (36). It is activated transiently in OCPs within 60 minutes of treatment with RANKL when it interacts with NF-κB p65 (88) and again during the fusion phase around 50–60 hours later by calcium-dependent calcineurin dephosphorylation. Several additional factors that are activated by RANKL and also participate in NFATc1 up-regulation include c-Fos and RNA polymerase II (89). Cyclosporine A, a calcineurin inhibitor, inhibits NFATc1 activation; yet treatment of patients with this immunosuppressant is associated with bone loss (90). NFATc1 also positively regulates expression of osterix, a transcription factor that regulates osteoblast differentiation and function (91). Thus the likely reason why cyclosporine A induces bone loss in vivo is that it has a greater inhibitory effect on osteoblasts than on osteoclasts (92).
Figure. Signaling pathways involved osteoclastogenesis in disease states leading to activation of NFATc1.
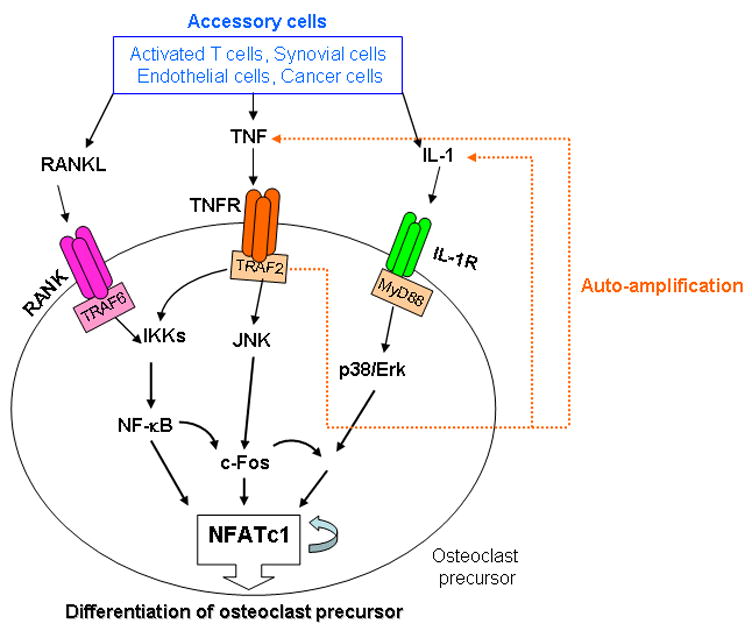
In inflammatory conditions, such as rheumatoid arthritis, the numbers of immune and accessory cells are increased in affected joints. Some of these cells produce RANKL in response to locally elevated levels of pro-inflammatory cytokines and other inflammatory mediators. RANKL binds to RANK on the surface of osteoclast precursors and recruits the adapter protein, TRAP6, leading to activation of NF-κB through phosphorylation and inactivation of inhibitory kappa kinases (IKKs) and NF-κB inhibitory kinase (not shown here). This induces activation of c-Fos. NF-κB and c-Fos interact with the NFATc1 promoter to trigger NFATc1 auto-amplification of NFATc1 and the transcription of genes, which mediate completion of the differentiation process. In addition to RANKL expression, these cells as well as macrophage/monocytes and osteoclasts themselves produce large amounts of TNF. TNF binds to the TNF receptor and this also activates c-Fos through both the NF-κB and JNK pathways in osteoclast precursors. TNF also stimulates its own expression and that of IL-1 by OCPs (orange lines). IL-1 does not activate c-Fos, but in OCPs in which c-Fos has been activated, for example by RANKL or TNF, IL-1 can induce osteoclastogenesis directly. This leads to more osteoclast formation using the same NFATc1 activated mechanism as RANKL. In this model, RANKL/RANK signaling is essential for OCP differentiation under both physiologic (through osteoblastic cells) and pathologic conditions (through accessory cells and OCP themselves), while TNF signaling appears to play a major role in inflammatory bone diseases.
Recently, signaling molecules other than the transcription factors mentioned above have been implicated in mediating the effect of RANKL/RANK in osteoclasts. Differential screening of a human osteoclastoma cDNA library demonstrated that the regulator of G-protein signaling 10 (RGS10) is specifically expressed in osteoclasts (93). The expression of RGS10 is induced by RANKL in OCPs, and ectopic expression of RGS10 dramatically increases the sensitivity of osteoclast differentiation to RANKL signaling. RGS10−/− mice exhibit severe osteopetrosis and impaired osteoclast differentiation. Deficiency of RGS10 resulted in the absence of [Ca2+]i oscillations and loss of NFATc1, which can be rescued by over-expression of NFATc1. RGS10 competitively interacts with Ca2+/calmodulin and PIP3 in a [Ca2+]I-dependent manner to mediate PLCγ activation and [Ca2+]i oscillations in osteoclasts (94). These studies indicate that RGS10 specifically regulates RANKL-evoked RGS10/calmodulin–[Ca2+]i oscillation–calcineurin–NFATc1 signaling in osteoclast differentiation and may be a potential therapeutic target for the treatment of bone diseases in which bone resorption is increased.
Immunoreceptors, Osteoimmunology and RANKL
Activation of calcium signaling during osteoclast formation appears to involve the Fc receptor common γ subunit (FcRγ) immunoreceptor expressed by osteoclasts and the adapter protein, DNAX-activating protein 12 (DAP12), which associates with an immunoreceptor tyrosine-based activation motif (ITAM) (95). DAP12/FcRγ double knockout mice are severely osteopetrotic due to impaired RANKL-induced NFATc1 activation and they do not form osteoclasts, while mice deficient in either gene have only mildly impaired osteoclast formation (96). Despite the essential functions of DAP12/FcRγ, this receptor-mediated signaling pathway cannot induce osteoclast formation on its own. Like M-CSF, it is necessary, but not sufficient for osteoclastogenesis. FcRγ-associating receptors include osteoclast-associated receptor (OSCAR) whose expression in OCPs is regulated by NFATc1 (97).
In inflammatory bone disorders, signaling through RANKL/RANK and these immunoreceptors provides an autocrine NFATc1-mediated self-amplifying mechanism to potentially increase osteoclast formation beyond that of RANKL alone. This mechanism for enhanced osteoclast formation in inflammatory bone diseases can also be augmented by TNF and other cytokines in a variety of ways. For example, TNF induces expression of c-fms, the receptor for M-CSF, by OCPs and increases OCP proliferation and survival (98). It also enhances OCP egression from the bone marrow into the bloodstream from where they can alight in increased numbers at sites of inflammation (49). These induced OCPs also increase their production of TNF in response to RANKL and TNF (our unpublished observation). As mentioned earlier, TNF also induces c-Fos expression in OCPs through NF-κB to induce osteoclast formation directly and to facilitate direct induction of osteoclastogenesis by IL-1 (Figure) (39, 99). By comparative microarray analysis of RNA from OCPs isolated from the blood of TNF transgenic (TNF-Tg) mice with established inflammatory erosive joint disease and from wild type littermates, we found that vascular endothelial growth factor (VEGF)-C expression is significantly increased in OCPs from the TNF-Tg mice (100). Interestingly, RANKL significantly increases the expression VEGF-C in osteoclasts and OCPs, and VEGF-C stimulates osteoclast bone resorption. Furthermore, blockade of the VEGF-C receptor signaling reduces RANKL-induced osteoclast bone resorption (101). Thus, VEGF-C is another RANKL target gene and affects osteoclast function through an autocrine mechanism.
By all of these mechanisms, cytokines can induce self-amplifying autocrine and paracrine cycles in OCPs and osteoclasts to induce increased osteoclast formation and activation and aggressive bone loss. It is likely that osteoclasts and their precursors secrete many more factors that interact with immune and other cells to affect bone volume and turnover in a variety of bone disorders.
Pharmacologic inhibition of RANKL/RANK signaling
Numerous preclinical in vivo studies using inhibitors or RANKL/RANK signaling have confirmed the important roles of this system in rodents and non-human primates. For example, OPG and RANK:Fc inhibited bone loss in models of sex-steroid deficiency and glucocorticoid-induced osteoporosis, rheumatoid arthritis, multiple myeloma, and metastatic bone disease (102–105). These studies were followed by phase 1 clinical trials of two forms of OPG: Fc-OPG and OPG-Fc, Single injections of these OPG constructs into normal volunteers resulted in prolonged dose-dependent reductions in biochemical markers of bone resorption (106, 107). Neither of these proteins was developed for further clinical trials, perhaps because of concerns about the possibility of immune responses to them and consequent unwanted adverse effects on the immune system. They were replaced by Denosumab, a fully human monoclonal antibody developed by injecting mice with human RANKL, which binds to and inactivates RANKL, similar to the action of OPG. Compared to OPG:Fc, Denosumab has a significantly longer circulating half-life and a more prolonged effect to reduce serum levels of markers of bone resorption and formation (108). There are several phase 2 and 3 clinical trails investigating the efficacy of Denosumab in patients with a variety of bone disorders, including postmenopausal osteoporosis, rheumatoid arthritis, multiple myeloma and metastatic bone disease (43, 109). To date, treatment of patients in these studies has resulted in significant inhibition of bone resorption without any obvious significant adverse effects.
Summary
Discovery of the RANKL/RANK/OPG system has been one of the most important advances in bone biology in the last decade. This signaling system is essential for skeletal homeostasis, and disruption of it leads to inhibition of bone resorption in vitro and in animal models of most bone diseases characterized by increased resorption. RANKL/RANK signaling plays important roles in tissues other than bone. Elucidation of the specific roles of RANKL/RANK in these various types of cells will likely link bone remodeling in normal and disease states with regulation of the function of other organ systems in health and disease. The discovery of RANKL, RANK and OPG has led to the development of specific inhibitors of RANKL, some of which, such as OPG and a monoclonal antibody to RANKL, have been tested in humans in clinical trials with successful inhibition of bone resorption. Thus, it will be important to determine if long-term inhibition of RANKL has any unwanted adverse effects in these tissues and on immune responses in particular.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Karsenty G, Wagner EF. Reaching a genetic and molecular understanding of skeletal development. Dev Cell. 2002;2:389–406. doi: 10.1016/s1534-5807(02)00157-0. [DOI] [PubMed] [Google Scholar]
- 2.Del Fattore A, Cappariello A, Teti A. Genetics, pathogenesis and complications of osteopetrosis. Bone. 2008;42:19–29. doi: 10.1016/j.bone.2007.08.029. [DOI] [PubMed] [Google Scholar]
- 3.Tolar J, Teitelbaum SL, Orchard PJ. Osteopetrosis. N Engl J Med. 2004;351:2839–2849. doi: 10.1056/NEJMra040952. [DOI] [PubMed] [Google Scholar]
- 4.Lian JB, Stein GS. Runx2/Cbfa1: a multifunctional regulator of bone formation. Curr Pharm Des. 2003;9:2677–2685. doi: 10.2174/1381612033453659. [DOI] [PubMed] [Google Scholar]
- 5.Karsenty G. Role of Cbfa1 in osteoblast differentiation and function. Semin Cell Dev Biol. 2000;11:343–346. doi: 10.1006/scdb.2000.0188. [DOI] [PubMed] [Google Scholar]
- 6.Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89:747–754. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- 7.Komori Y, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, et al. Targeted disruption of Cbfa 1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–764. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- 8.Hassan MQ, Tare RS, Lee SH, Mandeville M, Morasso MI, Javed A, van Wijnen AJ, Stein JL, Stein GS, Lian JB. BMP2 commitment to the osteogenic lineage involves activation of Runx2 by DLX3 and a homeodomain transcriptional network. J Biol Chem. 2006;281:40515–40526. doi: 10.1074/jbc.M604508200. [DOI] [PubMed] [Google Scholar]
- 9.Drissi MH, Li X, Sheu TJ, Zuscik MJ, Schwarz EM, Puzas JE, Rosier RN, O’Keefe RJ. Runx2/Cbfa1 stimulation by retinoic acid is potentiated by BMP2 signaling through interaction with Smad1 on the collagen X promoter in chondrocytes. J Cell Biochem. 2003;90:1287–1298. doi: 10.1002/jcb.10677. [DOI] [PubMed] [Google Scholar]
- 10.Feng JQ, Xing L, Zhang JH, Zhao M, Horn D, Chan J, Boyce BF, Harris SE, Mundy GR, Chen D. NF-kappaB specifically activates BMP-2 gene expression in growth plate chondrocytes in vivo and in a chondrocyte cell line in vitro. J Biol Chem. 2003;278:29130–29135. doi: 10.1074/jbc.M212296200. [DOI] [PubMed] [Google Scholar]
- 11.Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D, Shapiro SD, Senior RM, Werb Z. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rhee JW, Lee KW, Sohn WJ, Lee Y, Jeon OH, Kwon HJ, Kim DS. Regulation of matrix metalloproteinase-9 gene expression and cell migration by NF-kappa B in response to CpG-oligodeoxynucleotides in RAW 264.7 cells. Mol Immunol. 2007;44:1393–1400. doi: 10.1016/j.molimm.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 13.Masuyama R, Stockmans I, Torrekens S, Van Looveren R, Maes C, Carmeliet P, Bouillon R, Carmeliet G. Vitamin D receptor in chondrocytes promotes osteoclastogenesis and regulates FGF23 production in osteoblasts. J Clin Invest. 2006;116:3150–3159. doi: 10.1172/JCI29463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Usui M, Xing L, Drissi H, Zuscik M, O’Keefe R, Chen D, Boyce BF. Murine and Chicken Chondrocytes Regulate Osteoclastogenesis by Producing RANKL in Response to BMP2. J Bone Miner Res. 2007 doi: 10.1359/JBMR.071025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Breuil V, Schmid-Antomarchi H, Schmid-Alliana A, Rezzonico R, Euller-Ziegler L, Rossi B. The receptor activator of nuclear factor (NF)-kappaB ligand (RANKL) is a new chemotactic factor for human monocytes. Faseb J. 2003;17:1751–1753. doi: 10.1096/fj.02-1188fje. [DOI] [PubMed] [Google Scholar]
- 16.Mosheimer BA, Kaneider NC, Feistritzer C, Sturn DH, Wiedermann CJ. Expression and function of RANK in human monocyte chemotaxis. Arthritis Rheum. 2004;50:2309–2316. doi: 10.1002/art.20352. [DOI] [PubMed] [Google Scholar]
- 17.Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, Morony S, Oliveira-dos-Santos AJ, Van G, Itie A, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315–323. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- 18.Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, De Smedt T, Daro E, Smith J, Tometsko ME, Maliszewski CR, et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999;13:2412–2424. doi: 10.1101/gad.13.18.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boyce BF, Xing L, Shakespeare W, Wang Y, Dalgarno D, Iuliucci J, Sawyer T. Regulation of bone remodeling and emerging breakthrough drugs for osteoporosis and osteolytic bone metastases. Kidney Int Suppl. 2003:2–5. doi: 10.1046/j.1523-1755.63.s85.2.x. [DOI] [PubMed] [Google Scholar]
- 20.Tatsumi S, Ishii K, Amizuka N, Li M, Kobayashi T, Kohno K, Ito M, Takeshita S, Ikeda K. Targeted Ablation of Osteocytes Induces Osteoporosis with Defective Mechanotransduction. Cell Metabolism. 2007;5:464–475. doi: 10.1016/j.cmet.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 21.Boyce BF, Xing L. Biology of RANK, RANKL, and osteoprotegerin. Arthritis Res Ther. 2007;9 Suppl 1:S1. doi: 10.1186/ar2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao C, Irie N, Takada Y, Shimoda K, Miyamoto T, Nishiwaki T, Suda T, Matsuo K. Bidirectional ephrinB2-EphB4 signaling controls bone homeostasis. Cell Metabolism. 2006;4:111–121. doi: 10.1016/j.cmet.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 23.Lee SH, Rho J, Jeong D, Sul JY, Kim T, Kim N, Kang JS, Miyamoto T, Suda T, Lee SK, et al. v-ATPase V0 subunit d2-deficient mice exhibit impaired osteoclast fusion and increased bone formation. Nat Med. 2006;12:1403–1409. doi: 10.1038/nm1514. [DOI] [PubMed] [Google Scholar]
- 24.Kollet O, Dar A, Shivtiel S, Kalinkovich A, Lapid K, Sztainberg Y, Tesio M, Samstein RM, Goichberg P, Spiegel A, et al. Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat Med. 2006;12:657–664. doi: 10.1038/nm1417. [DOI] [PubMed] [Google Scholar]
- 25.Xing L, Schwarz EM, Boyce BF. Osteoclast precursors, RANKL/RANK, and immunology. Immunol Rev. 2005;208:19–29. doi: 10.1111/j.0105-2896.2005.00336.x. [DOI] [PubMed] [Google Scholar]
- 26.Yoshida H, Hayashi S, Kunisada T, Ogawa M, Nishikawa S, Okamura H, Sudo T, Shultz LD. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature. 1990;345:442–444. doi: 10.1038/345442a0. [DOI] [PubMed] [Google Scholar]
- 27.Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–319. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- 28.Yasuda H, Shima N, Nakagawa N, Mochizuki SI, Yano K, Fujise N, Sato Y, Goto M, Yamaguchi K, Kuriyama M, et al. Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG): a mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro. Endocrinology. 1998;139:1329–1337. doi: 10.1210/endo.139.3.5837. [DOI] [PubMed] [Google Scholar]
- 29.Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, Tomoyasu A, Yano K, Goto M, Murakami A, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A. 1998;95:3597–3602. doi: 10.1073/pnas.95.7.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- 31.Anderson DM, Maraskovsky E, Billingsley WL, Dougall WC, Tometsko ME, Roux ER, Teepe MC, DuBose RF, Cosman D, Galibert L. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature. 1997;390:175–179. doi: 10.1038/36593. [DOI] [PubMed] [Google Scholar]
- 32.Wong BR, Rho J, Arron J, Robinson E, Orlinick J, Chao M, Kalachikov S, Cayani E, Bartlett FS, III, Frankel WN, et al. TRANCE is a novel ligand of the tumor necrosis factor receptor family that activates c-Jun N-terminal kinase in T cells. J Biol Chem. 1997;272:25190–25194. doi: 10.1074/jbc.272.40.25190. [DOI] [PubMed] [Google Scholar]
- 33.Proposed standard nomenclature for new tumor necrosis factor family members involved in the regulation of bone resorption. The American Society for Bone and Mineral Research President’s Committee on Nomenclature. J Bone Miner Res. 2000;15:2293–2296. doi: 10.1359/jbmr.2000.15.12.2293. [DOI] [PubMed] [Google Scholar]
- 34.Franzoso G, Carlson L, Xing L, Poljak L, Shores EW, Brown KD, Leonardi A, Tran T, Boyce BF, Siebenlist U. Requirement for NF-kappaB in osteoclast and B-cell development. Genes Dev. 1997;11:3482–3496. doi: 10.1101/gad.11.24.3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsuo K, Galson DL, Zhao C, Peng L, Laplace C, Wang KZ, Bachler MA, Amano H, Aburatani H, Ishikawa H, et al. Nuclear factor of activated T-cells (NFAT) rescues osteoclastogenesis in precursors lacking c-Fos. J Biol Chem. 2004;279:26475–26480. doi: 10.1074/jbc.M313973200. [DOI] [PubMed] [Google Scholar]
- 36.Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, Saiura A, Isobe M, Yokochi T, Inoue J, et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3:889–901. doi: 10.1016/s1534-5807(02)00369-6. [DOI] [PubMed] [Google Scholar]
- 37.Yamashita T, Yao Z, Li F, Zhang Q, Badell IR, Schwarz EM, Takeshita S, Wagner EF, Noda M, Matsuo K, et al. NF-kappaB p50 and p52 regulate receptor activator of NF-kappaB ligand (RANKL) and tumor necrosis factor-induced osteoclast precursor differentiation by activating c-Fos and NFATc1. J Biol Chem. 2007;282:18245–18253. doi: 10.1074/jbc.M610701200. [DOI] [PubMed] [Google Scholar]
- 38.Kim N, Kadono Y, Takami M, Lee J, Lee SH, Okada F, Kim JH, Kobayashi T, Odgren PR, Nakano H, et al. Osteoclast differentiation independent of the TRANCE-RANK-TRAF6 axis. J Exp Med. 2005;202:589–595. doi: 10.1084/jem.20050978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yao Z, Xing L, Qin C, Schwarz EM, Boyce BF. Osteoclast precursor interaction with bone matrix induces osteoclast formation directly by an IL-1-mediated autocrine mechanism. J Biol Chem. 2008 doi: 10.1074/jbc.M706415200. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Teitelbaum SL, Ross FP. Genetic regulation of osteoclast development and function. Nature Reviews Genetics. 2003;4:638–649. doi: 10.1038/nrg1122. [DOI] [PubMed] [Google Scholar]
- 41.Teitelbaum SL. Osteoclasts and integrins. Ann N Y Acad Sci. 2006;1068:95–99. doi: 10.1196/annals.1346.017. [DOI] [PubMed] [Google Scholar]
- 42.Yagi M, Miyamoto T, Sawatani Y, Iwamoto K, Hosogane N, Fujita N, Morita K, Ninomiya K, Suzuki T, Miyamoto K, et al. DC-STAMP is essential for cell-cell fusion in osteoclasts and foreign body giant cells. J Exp Med. 2005;202:345–351. doi: 10.1084/jem.20050645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kearns AE, Khosla S, Kostenuik P. RANKL and OPG Regulation of Bone Remodeling in Health and Disease. Endocr Rev. 2007 doi: 10.1210/er.2007-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wada T, Nakashima T, Hiroshi N, Penninger JM. RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends Mol Med. 2006;12:17–25. doi: 10.1016/j.molmed.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 45.Takayanagi H. Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol. 2007;7:292–304. doi: 10.1038/nri2062. [DOI] [PubMed] [Google Scholar]
- 46.Ikeda T, Kasai M, Utsuyama M, Hirokawa K. Determination of three isoforms of the receptor activator of nuclear factor-[kappa]B ligand and their differential expression in bone and thymus. Endocrinology. 2001;142:1419–1426. doi: 10.1210/endo.142.4.8070. [DOI] [PubMed] [Google Scholar]
- 47.Lynch CC, Hikosaka A, Acuff HB, Martin MD, Kawai N, Singh RK, Vargo-Gogola TC, Begtrup JL, Peterson TE, Fingleton B, et al. MMP-7 promotes prostate cancer-induced osteolysis via the solubilization of RANKL. Cancer Cell. 2005;7:485–496. doi: 10.1016/j.ccr.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 48.Hikita A, Yana I, Wakeyama H, Nakamura M, Kadono Y, Oshima Y, Nakamura K, Seiki M, Tanaka S. Negative regulation of osteoclastogenesis by ectodomain shedding of receptor activator of NF-kappa B ligand. J Biol Chem. 2006 doi: 10.1074/jbc.M606656200. [DOI] [PubMed] [Google Scholar]
- 49.Li P, Schwarz EM, O’Keefe RJ, Ma L, Looney RJ, Ritchlin CT, Boyce BF, Xing L. Systemic tumor necrosis factor alpha mediates an increase in peripheral CD11bhigh osteoclast precursors in tumor necrosis factor alpha-transgenic mice. Arthritis Rheum. 2004;50:265–276. doi: 10.1002/art.11419. [DOI] [PubMed] [Google Scholar]
- 50.Fata JE, Kong YY, Li J, Sasaki T, Irie-Sasaki J, Moorehead RA, Elliott R, Scully S, Voura EB, Lacey DL. The Osteoclast Differentiation Factor Osteoprotegerin-Ligand Is Essential for Mammary Gland Development. Cell. 2000;103:41–50. doi: 10.1016/s0092-8674(00)00103-3. [DOI] [PubMed] [Google Scholar]
- 51.Kim NS, Kim HJ, Koo BK, Kwon MC, Kim YW, Cho Y, Yokota Y, Penninger JM, Kong YY. Receptor activator of NF-kappaB ligand regulates the proliferation of mammary epithelial cells via Id2. Mol Cell Biol. 2006;26:1002–1013. doi: 10.1128/MCB.26.3.1002-1013.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jones DH, Nakashima T, Sanchez OH, Kozieradzki I, Komarova SV, Sarosi I, Morony S, Rubin E, Sarao R, Hojilla CV, et al. Regulation of cancer cell migration and bone metastasis by RANKL. Nature. 2006;440:692–696. doi: 10.1038/nature04524. [DOI] [PubMed] [Google Scholar]
- 53.Takayanagi H, Kim S, Matsuo K, Suzuki H, Suzuki T, Sato K, Yokochi T, Oda H, Nakamura K, Ida N, et al. RANKL maintains bone homeostasis through c-Fos-dependent induction of interferon-beta.[see comment] Nature. 2002;416:744–749. doi: 10.1038/416744a. [DOI] [PubMed] [Google Scholar]
- 54.Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K, Takaoka A, Yokochi T, Oda H, Tanaka K, et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature. 2000;408:600–605. doi: 10.1038/35046102. [DOI] [PubMed] [Google Scholar]
- 55.Chen H, Gilbert CA, Hudson JA, Bolick SC, Wright KL, Piskurich JF. Positive regulatory domain I-binding factor 1 mediates repression of the MHC class II transactivator (CIITA) type IV promoter. Mol Immunol. 2007;44:1461–1470. doi: 10.1016/j.molimm.2006.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Takayanagi H, Kim S, Taniguchi T. Signaling crosstalk between RANKL and interferons in osteoclast differentiation. Arthritis Res. 2002;4 Suppl 3:S227–232. doi: 10.1186/ar581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Madyastha PR, Yang S, Ries WL, Key LL., Jr IFN-gamma enhances osteoclast generation in cultures of peripheral blood from osteopetrotic patients and normalizes superoxide production. J Interferon Cytokine Res. 2000;20:645–652. doi: 10.1089/107999000414826. [DOI] [PubMed] [Google Scholar]
- 58.Key LL, Jr, Rodriguiz RM, Willi SM, Wright NM, Hatcher HC, Eyre DR, Cure JK, Griffin PP, Ries WL. Long-term treatment of osteopetrosis with recombinant human interferon gamma. N Engl J Med. 1995;332:1594–1599. doi: 10.1056/NEJM199506153322402. [DOI] [PubMed] [Google Scholar]
- 59.Gao Y, Grassi F, Ryan MR, Terauchi M, Page K, Yang X, Weitzmann MN, Pacifici R. IFN-gamma stimulates osteoclast formation and bone loss in vivo via antigen-driven T cell activation. J Clin Invest. 2007;117:122–132. doi: 10.1172/JCI30074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Van Wesenbeeck L, Odgren PR, Coxon FP, Frattini A, Moens P, Perdu B, MacKay CA, Van Hul E, Timmermans JP, Vanhoenacker F, et al. Involvement of PLEKHM1 in osteoclastic vesicular transport and osteopetrosis in incisors absent rats and humans. J Clin Invest. 2007;117:919–930. doi: 10.1172/JCI30328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sobacchi C, Frattini A, Guerrini MM, Abinun M, Pangrazio A, Susani L, Bredius R, Mancini G, Cant A, Bishop N, et al. Osteoclast-poor human osteopetrosis due to mutations in the gene encoding RANKL. Nat Genet. 2007;39:960–962. doi: 10.1038/ng2076. [DOI] [PubMed] [Google Scholar]
- 62.Chen G, Sircar K, Aprikian A, Potti A, Goltzman D, Rabbani SA. Expression of RANKL/RANK/OPG in primary and metastatic human prostate cancer as markers of disease stage and functional regulation. Cancer. 2006;107:289–298. doi: 10.1002/cncr.21978. [DOI] [PubMed] [Google Scholar]
- 63.Kapur RP, Yao Z, Iida MH, Clarke CM, Doggett B, Xing L, Boyce BF. Malignant autosomal recessive osteopetrosis caused by spontaneous mutation of murine Rank. J Bone Miner Res. 2004;19:1689–1697. doi: 10.1359/JBMR.040713. [DOI] [PubMed] [Google Scholar]
- 64.Hughes AE, Ralston SH, Marken J, Bell C, MacPherson H, Wallace RG, van Hul W, Whyte MP, Nakatsuka K, Hovy L, et al. Mutations in TNFRSF11A, affecting the signal peptide of RANK, cause familial expansile osteolysis. Nature Genetics. 2000;24:45–48. doi: 10.1038/71667. [DOI] [PubMed] [Google Scholar]
- 65.Li Y, Toraldo G, Li A, Yang X, Zhang H, Qian WP, Weitzmann MN. B cells and T cells are critical for the preservation of bone homeostasis and attainment of peak bone mass in vivo. Blood. 2007;109:3839–3848. doi: 10.1182/blood-2006-07-037994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hofbauer LC, Schoppet M. Clinical implications of the osteoprotegerin/RANKL/RANK system for bone and vascular diseases. Jama. 2004;292:490–495. doi: 10.1001/jama.292.4.490. [DOI] [PubMed] [Google Scholar]
- 67.Theoleyre S, Wittrant Y, Tat SK, Fortun Y, Redini F, Heymann D. The molecular triad OPG/RANK/RANKL: involvement in the orchestration of pathophysiological bone remodeling. Cytokine Growth Factor Rev. 2004;15:457–475. doi: 10.1016/j.cytogfr.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 68.Whyte MP, Obrecht SE, Finnegan PM, Jones JL, Podgornik MN, McAlister WH, Mumm S. Osteoprotegerin deficiency and juvenile Paget’s disease. N Engl J Med. 2002;347:175–184. doi: 10.1056/NEJMoa013096. [DOI] [PubMed] [Google Scholar]
- 69.Cundy T, Hegde M, Naot D, Chong B, King A, Wallace R, Mulley J, Love DR, Seidel J, Fawkner M, et al. A mutation in the gene TNFRSF11B encoding osteoprotegerin causes an idiopathic hyperphosphatasia phenotype. Hum Mol Genet. 2002;11:2119–2127. doi: 10.1093/hmg/11.18.2119. [DOI] [PubMed] [Google Scholar]
- 70.Glass DA, 2nd, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H, Taketo MM, Long F, McMahon AP, Lang RA, et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell. 2005;8:751–764. doi: 10.1016/j.devcel.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 71.Kieslinger M, Folberth S, Dobreva G, Dorn T, Croci L, Erben R, Consalez GGRG. EBF2 regulates osteoblast-dependent differentiation of osteoclasts. Cell Metabolism. 2005 doi: 10.1016/j.devcel.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 72.Hill TP, Spater D, Taketo MM, Birchmeier W, Hartmann C. Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev Cell. 2005;8:727–738. doi: 10.1016/j.devcel.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 73.Bai S, Kopan R, Zou W, Hilton MJ, Ong CT, Long F, Ross FP, Teitelbaum SL. Notch1 regulates osteoclastogenesis directly in osteoclast precursors and indirectly via osteoblast lineage cells. J Biol Chem. 2007 doi: 10.1074/jbc.M707000200. [DOI] [PubMed] [Google Scholar]
- 74.Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, Scully S, Tan HL, Xu W, Lacey DL, et al. osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998;12:1260–1268. doi: 10.1101/gad.12.9.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bennett BJ, Scatena M, Kirk EA, Rattazzi M, Varon RM, Averill M, Schwartz SM, Giachelli CM, Rosenfeld ME. Osteoprotegerin inactivation accelerates advanced atherosclerotic lesion progression and calcification in older ApoE−/− mice. Arterioscler Thromb Vasc Biol. 2006;26:2117–2124. doi: 10.1161/01.ATV.0000236428.91125.e6. [DOI] [PubMed] [Google Scholar]
- 76.Morony S, Tintut Y, Zhang Z, Cattley RC, Van G, Dwyer D, Stolina M, Kostenuik PJ, Demer LL. Osteoprotegerin inhibits vascular calcification without affecting atherosclerosis in ldlr(−/−) mice. Circulation. 2008;117:411–420. doi: 10.1161/CIRCULATIONAHA.107.707380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Collin-Osdoby P. Regulation of vascular calcification by osteoclast regulatory factors RANKL and osteoprotegerin. Circ Res. 2004;95:1046–1057. doi: 10.1161/01.RES.0000149165.99974.12. [DOI] [PubMed] [Google Scholar]
- 78.Ueland T, Yndestad A, Oie E, Florholmen G, Halvorsen B, Froland SS, Simonsen S, Christensen G, Gullestad L, Aukrust P. Dysregulated osteoprotegerin/RANK ligand/RANK axis in clinical and experimental heart failure. Circulation. 2005;111:2461–2468. doi: 10.1161/01.CIR.0000165119.62099.14. [DOI] [PubMed] [Google Scholar]
- 79.Sammartino A, Cirillo D, Mandato VD, Di Carlo C, Nappi C. Osteoporosis and cardiovascular disease: benefit-risk of hormone replacement therapy. J Endocrinol Invest. 2005;28:80–84. [PubMed] [Google Scholar]
- 80.Rogers A, Eastell R. Circulating osteoprotegerin and receptor activator for nuclear factor kappaB ligand: clinical utility in metabolic bone disease assessment. J Clin Endocrinol Metab. 2005;90:6323–6331. doi: 10.1210/jc.2005-0794. [DOI] [PubMed] [Google Scholar]
- 81.Kim HH, Lee DE, Shin JN, Lee YS, Jeon YM, Chung CH, Ni J, Kwon BS, Lee ZH. Receptor activator of NF-kappaB recruits multiple TRAF family adaptors and activates c-Jun N-terminal kinase. FEBS Lett. 1999;443:297–302. doi: 10.1016/s0014-5793(98)01731-1. [DOI] [PubMed] [Google Scholar]
- 82.Lomaga MA, Yeh WC, Sarosi I, Duncan GS, Furlonger C, Ho A, Morony S, Capparelli C, Van G, Kaufman S, et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 1999;13:1015–1024. doi: 10.1101/gad.13.8.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Naito A, Azuma S, Tanaka S, Miyazaki T, Takaki S, Takatsu K, Nakao K, Nakamura K, Katsuki M, Yamamoto T, et al. Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes Cells. 1999;4:353–362. doi: 10.1046/j.1365-2443.1999.00265.x. [DOI] [PubMed] [Google Scholar]
- 84.Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337–342. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- 85.Wada T, Nakashima T, Oliveira-dos-Santos AJ, Gasser J, Hara H, Schett G, Penninger JM. The molecular scaffold Gab2 is a crucial component of RANK signaling and osteoclastogenesis. Nat Med. 2005;11:394–399. doi: 10.1038/nm1203. [DOI] [PubMed] [Google Scholar]
- 86.Iotsova V, Caamano J, Loy J, Lewin A, Bravo R. Osteopetrosis in mice lacking NF-κB1 and NF-κB2. Nature Med. 1997;3:1285–1289. doi: 10.1038/nm1197-1285. [DOI] [PubMed] [Google Scholar]
- 87.Wang ZQ, OVitt C, Grigoriadis AE, Mohle-Steinlein U, Ruther U, Wagner EF. Bone and haematopoietic defects in mice lacking c-Fos. Nature. 1992;360:741–745. doi: 10.1038/360741a0. [DOI] [PubMed] [Google Scholar]
- 88.Asagiri M, Sato K, Usami T, Ochi S, Nishina H, Yoshida H, Morita I, Wagner EF, Mak TW, Serfling E, et al. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J Exp Med. 2005;202:1261–1269. doi: 10.1084/jem.20051150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fretz JA, Shevde NK, Singh S, Darnay BG, Pike JW. Rankl-Induced Nuclear Factor of Activated T Cells (C1) Autoregulates Its Own Expression in Osteoclasts and Mediates the Upregulation of Tartrate-Resistant Acid Phosphatase. Mol Endocrinol. 2007 doi: 10.1210/me.2007-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thiebaud D, Krieg MA, Gillard-Berguer D, Jacquet AF, Goy JJ, Burckhardt P. Cyclosporine induces high bone turnover and may contribute to bone loss after heart transplantation. Eur J Clin Invest. 1996;26:549–555. doi: 10.1046/j.1365-2362.1996.00170.x. [DOI] [PubMed] [Google Scholar]
- 91.Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, de Crombrugghe B. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108:17–29. doi: 10.1016/s0092-8674(01)00622-5. [DOI] [PubMed] [Google Scholar]
- 92.Koga T, Matsui Y, Asagiri M, Kodama T, de Crombrugghe B, Nakashima K, Takayanagi H. NFAT and Osterix cooperatively regulate bone formation. Nat Med. 2005;11:880–885. doi: 10.1038/nm1270. [DOI] [PubMed] [Google Scholar]
- 93.Yang S, Chen W, Stashenko P, Li YP. Specificity of RGS10A as a key component in the RANKL signaling mechanism for osteoclast differentiation. J Cell Sci. 2007;120:3362–3371. doi: 10.1242/jcs.008300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yang S, Li YP. RGS10-null mutation impairs osteoclast differentiation resulting from the loss of [Ca2+]i oscillation regulation. Genes Dev. 2007;21:1803–1816. doi: 10.1101/gad.1544107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Koga T, Inui M, Inoue K, Kim S, Suematsu A, Kobayashi E, Iwata T, Ohnishi H, Matozaki T, Kodama T, et al. Costimulatory signals mediated by the ITAM motif cooperate with RANKL for bone homeostasis. Nature. 2004;428:758–763. doi: 10.1038/nature02444. [DOI] [PubMed] [Google Scholar]
- 96.Takayanagi H. Mechanistic insight into osteoclast differentiation in osteoimmunology. J Mol Med. 2005;83:170–179. doi: 10.1007/s00109-004-0612-6. [DOI] [PubMed] [Google Scholar]
- 97.Kim Y, Sato K, Asagiri M, Morita I, Soma K, Takayanagi H. Contribution of nuclear factor of activated T cells c1 to the transcriptional control of immunoreceptor osteoclast-associated receptor but not triggering receptor expressed by myeloid cells-2 during osteoclastogenesis. J Biol Chem. 2005;280:32905–32913. doi: 10.1074/jbc.M505820200. [DOI] [PubMed] [Google Scholar]
- 98.Yao Z, Li P, Zhang Q, Schwarz EM, Keng P, Arbini A, Boyce BF, Xing L. Tumor Necrosis Factor-{alpha} Increases Circulating Osteoclast Precursor Numbers by Promoting Their Proliferation and Differentiation in the Bone Marrow through Up-regulation of c-Fms Expression. J Biol Chem. 2006;281:11846–11855. doi: 10.1074/jbc.M512624200. [DOI] [PubMed] [Google Scholar]
- 99.Yao Z, Xing L, Schwarz EM, Boyce BF. Osteoclast Precursors Induce their Differentiation to Osteoclasts by Interacting with Bone Matrix and Secreting Cytokines. JBMR. 2006;21:S262. [Google Scholar]
- 100.Zhang Q, Lu Y, Proulx S, Guo R, Yao Z, Schwarz EM, Boyce BF, Xing L. Increased lymphangiogenesis in joints of mice with inflammatory arthritis. Arthritis Res Ther. 2007;9:R118. doi: 10.1186/ar2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Guo R, Zhang Q, Lu Y, Schwarz EM, Jin Z, Boyce BF, Xing L. VEGF-C, a Lymphatic Growth Factor, is a RANKL Target Gene in Osteoclasts that Enhances Osteoclastic Bone Resorption through an Autocrine Mechanism. JBMR. 2007;22:S30. doi: 10.1074/jbc.M708055200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schett G, Hayer S, Zwerina J, Redlich K, Smolen JS. Mechanisms of Disease: the link between RANKL and arthritic bone disease. Nat Clin Pract Rheumatol. 2005;1:47–54. doi: 10.1038/ncprheum0036. [DOI] [PubMed] [Google Scholar]
- 103.Humphrey EL, Williams JH, Davie MW, Marshall MJ. Effects of dissociated glucocorticoids on OPG and RANKL in osteoblastic cells. Bone. 2006;38:652–661. doi: 10.1016/j.bone.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 104.Blair JM, Zhou H, Seibel MJ, Dunstan CR. Mechanisms of disease: roles of OPG, RANKL and RANK in the pathophysiology of skeletal metastasis. Nat Clin Pract Oncol. 2006;3:41–49. doi: 10.1038/ncponc0381. [DOI] [PubMed] [Google Scholar]
- 105.Dougall WC, Chaisson M. The RANK/RANKL/OPG triad in cancer-induced bone diseases. Cancer Metastasis Rev. 2006;25:541–549. doi: 10.1007/s10555-006-9021-3. [DOI] [PubMed] [Google Scholar]
- 106.Bekker PJ, Holloway D, Nakanishi A, Arrighi M, Leese PT, Dunstan CR. The effect of a single dose of osteoprotegerin in postmenopausal women. J Bone Miner Res. 2001;16:348–360. doi: 10.1359/jbmr.2001.16.2.348. [DOI] [PubMed] [Google Scholar]
- 107.Body JJ, Greipp P, Coleman RE, Facon T, Geurs F, Fermand JP, Harousseau JL, Lipton A, Mariette X, Williams CD, et al. A phase I study of AMGN-0007, a recombinant osteoprotegerin construct, in patients with multiple myeloma or breast carcinoma related bone metastases. Cancer. 2003;97:887–892. doi: 10.1002/cncr.11138. [DOI] [PubMed] [Google Scholar]
- 108.Bekker PJ, Holloway DL, Rasmussen AS, Murphy R, Martin SW, Leese PT, Holmes GB, Dunstan CR, DePaoli AM. A single-dose placebo-controlled study of AMG 162, a fully human monoclonal antibody to RANKL, in postmenopausal women. J Bone Miner Res. 2004;19:1059–1066. doi: 10.1359/JBMR.040305. [DOI] [PubMed] [Google Scholar]
- 109.Lewiecki EM, Miller PD, McClung MR, Cohen SB, Bolognese MA, Liu Y, Wang A, Siddhanti S, Fitzpatrick LA. Two-year treatment with denosumab (AMG 162) in a randomized phase 2 study of postmenopausal women with low BMD. J Bone Miner Res. 2007;22:1832–1841. doi: 10.1359/jbmr.070809. [DOI] [PubMed] [Google Scholar]