

Summary
This report describes a phase I clinical trial using nonmyeloablative, lympho-depleting chemotherapy in combination with adoptive immunotherapy in patients with metastatic melanoma. The chemotherapy-conditioning schedule that induced transient lymphopenia consisted of cyclophosphamide (30 or 60 mg/kg per day for 2 days) followed by fludarabine (25 mg/m2 per day for 5 days). Immunotherapy for all patients consisted of in vitro expanded, tumor-reactive, autologous T-cell clones selected for high avidity recognition of melanoma antigens. Cohorts of three to six patients each received either no interleukin (IL)-2, low-dose IL-2 (72,000 IU/kg intravenously three times a day to a maximum of 15 doses), or high-dose IL-2 (720,000 IU/kg intravenously three times a day for a maximum of 12 doses). The toxicities associated with this treatment were transient and included neutropenia and thrombocytopenia that resolved in all patients. High dose intravenous IL-2 was better tolerated by patients after chemotherapy than during previous immunotherapy cycles without chemotherapy. No patient exhibited an objective clinical response to treatment, although five patients demonstrated mixed responses or transient shrinkage of metastatic deposits. This study established a nonmyeloablative-conditioning regimen that could be safely administered in conjunction with adoptive T-cell transfer and IL-2 in patients with metastatic melanoma.
Keywords: Adoptive transfer therapy, Nonmyeloablative chemotherapy, T-cell clones, Interleukin, 2 therapy, Melanoma
Immunotherapeutic approaches to patients with metastatic melanoma are based on developing lymphocytes with the ability to recognize and destroy tumor cells. High-dose interleukin (IL)-2 therapy is capable of inducing durable, complete responses in a subset of patients with melanoma (1,2) and is thought to act through T lymphocytes to mediate tumor eradication. Direct evidence for the antitumor capacity of T cells in patients with melanoma comes from studies in which lymphocytes were isolated directly from resected tumor specimens, expanded with IL-2 in vitro, and administered to patients for therapy. These tumor-infiltrating lymphocytes (TIL), when administered together with high-dose IL-2, produced objective tumor regressions in 34% of treated patients including patients refractory to IL-2 therapy (3).
Many genes encoding tumor antigens that are recognized by T cells have been cloned, and the HLA-restricted peptide epitopes derived from them have been identified (4). Clinical evaluation of the immunogenicity and therapeutic efficacy of modified peptide epitopes from gp100 and other melanoma-melanocyte differentiation antigens (5,6) is currently underway. Administration of the anchor-modified gp100:209–217(210M) peptide (referred to as g209–2M) in incomplete Freund’s adjuvant in combination with IL-2 therapy resulted in objective clinical responses in 13 of 31 patients (42%) (7). The g209–2M peptide has been shown to be highly immunogenic, inducing specific T-cell responses that comprised up to 1% of peripheral blood CD8+ lymphocytes (8). Cloning and evaluation of lymphocytes from patients vaccinated with the g209–2M peptide showed that the induced T-cell response was heterogeneous and included T cells that were highly avid for the native g209 peptide as well as strongly reactive with HLA-A2 matched melanoma cell lines (9,10). These results have led to the further evaluation of g209–2M and other tumor antigens for the therapy of patients with melanoma and other cancers.
The ability to routinely immunize patients using peptide vaccination provided the rationale for re-examining T-cell transfer as a therapeutic modality, and in a previous study, we reported on the isolation and in vitro expansion of peptide-specific, tumor-reactive T-cell clones from immunized patients for adoptive therapy (11). Thirteen patients were treated with multiple cycles of highly avid T-cell clones, with or without concomitant high-dose IL-2. Although the treatment was safe and well tolerated, no objective clinical responses were noted. Significantly, analysis of peripheral blood samples of treated patients revealed that the transferred clones disappeared rapidly from the circulation and decreased to undetectable levels within 1 week of transfer. Similarly, radionuclide labeling of aliquots of infused cells provided no evidence for traffic of transferred clones to tumor deposits.
This study reports on the use of a nonmyeloablative chemotherapy regimen before the adoptive transfer of cloned T cells for the therapy of patients with metastatic melanoma. This study was initiated based on the indirect evidence from clinical trials and direct evidence from preclinical model systems that immune suppression may enhance the efficacy of lymphocyte transfer therapy. Fifteen patients with malignant melanoma were treated. The trial was designed as a phase I dose-escalation study to evaluate the toxicity of the multi-modality treatment that included immunosuppressive chemotherapy, T-cell transfer, and IL-2 administration. Hematologic effects of the combination therapy, as well as nonhematologic toxicities and clinical responses were evaluated.
MATERIALS AND METHODS
Patients and Protocol Design
Patients with metastatic melanoma who were HLA-A*0201 positive, HIV negative, hepatitis B and C negative, and who had progressive disease after a gp100-based therapy including high-dose IL-2 were eligible for this protocol. The protocol design called for treatment of sequential cohorts of three to six patients with escalation of the doses of chemotherapy, followed by escalation of the IL-2 dose at the maximum chemotherapy dose. Before initiation of nonmyeloablative therapy, all patients underwent 5 days of granulocyte colony-stimulating factor administration and peripheral stem cell harvest by cytapheresis, to bank stem cells against the possibility that irreversible hematologic deficiencies could develop during the course of treatment.
All patients were admitted to the Surgery Branch unit of the National Cancer Institute’s Clinical Center Hospital and treated as in-patients for the duration of each treatment cycle. Before the initiation of cyclophosphamide administration, patients received intravenous hydration overnight. Cohort 1 (three patients) received 30 mg/kg cyclophosphamide each day for 2 days (termed day –7 and day –6), followed by fludarabine at 25 mg/m2 each day for 5 days (day –5 through day –1). On the first day after the final dose of fludarabine, patients received the infusion of in vitro expanded T cells (day 0). Cohort 2 (three patients) received 60 mg/kg cyclophosphamide per day for 2 days, then the same schedule as Cohort 1 (i.e., fludarabine for 5 days followed by cells). Cohort 3 received 60 mg/kg cyclophosphamide each day for 2 days, followed by 5 days of fludarabine, adoptive cell transfer, then low-dose intravenous IL-2 (72,000 IU per kg three times a day for 5 days). Cohort 4 received 60 mg per kg cyclophosphamide per day for 2 days, then fludarabine, T-cell infusion, and then high-dose intravenous IL-2 (720,000 IU per kg three times a day to tolerance, with a maximum of 12 doses).
Cyclophosphamide was administered with granisetron, mesna, and furosemide, and urine output was monitored closely. In each cohort, peptide vaccination was given a few hours before cell transfer by intradermal injection into each thigh of 1 m g of g209–2M peptide emulsified in 1 m L of incomplete Freund’s adjuvant, only if the patient’s infused cloned cells were specific for the gp100:209–217 epitope (g209). Granulocyte colony-stimulating factor was administered to all patients starting on the day of cell transfer and continuing until the absolute neutrophil count exceeded 0.5 × 106 per mm3. Two to 4 weeks after the first administration of cells, most patients were given a second treatment cycle consisting of T cells, vaccination, and IL-2 according to their protocol cohort, but without further chemotherapy.
Patients’ hematologic parameters were monitored by obtaining complete and differential blood counts and by flow cytometric analysis of peripheral mononuclear cells. Patients were evaluated 1 month after completion of each course by standard radiographic studies and physical examination. A partial response was defined as a 50% or greater decrease in the sum of the products of perpendicular diameter of all measurable lesions for at least 1 month with no new lesions. A minor response was defined as shrinkage of all tumor sites by 25%–49% with no new lesions. A mixed response was defined as greater than 25% shrinkage of an individual lesion with growth of other lesions or appearance of new lesions. Patients demonstrating a partial, mixed, or minor response, or stable disease were eligible to undergo additional courses of treatment.
Generation of Cloned T Cells
All T cells used for adoptive therapy in this protocol were generated by a three-step strategy. First, a reactive “bulk” culture was established; second, clonal T-cell cultures were established by limiting dilution cloning; and finally, clones were expanded to treatment levels using a modified version of the previously described rapid expansion protocol (12,13). Specific bulk cultures were established for each patient by one of two methods: in vitro sensitization of peripheral blood mononuclear cells (PBMC), or expansion in high-dose IL-2 of tumor infiltrating lymphocytes. For in vitro sensitization, PBMC were obtained from patients who had been previously vaccinated with g209–2M peptide and were highly immunized (7). In vitro sensitization was performed on thawed cells as previously described (10). Briefly, 3 ×106 cells per well were cultured in 24-well plates in complete medium (CM; RPMI 1640-based medium with 10% human serum) with 1 μg per mL g209–2M peptide (IMDQVPFSV) and 300 IU per mL IL-2 (Chiron, Emeryville, CA, U.S.A.) for 10–14 days. Bulk TIL cultures were generated from single-cell tumor digests by culture for 2–8 weeks in CM with 6,000 IU per mL of IL-2 as previously described (14).
T cells were cloned as previously described (9) from bulk peripheral blood lymphocyte or TIL cultures by limiting dilution at 0.6–2.0 cells per well in 96 well U-bottom plates in 200 microliters of CM, 30 ng/mL of OKT-3 (Ortho Biotech Inc., Raritan, NJ, U.S.A.), 300 IU per mL of IL-2, and 5 × 104 allogeneic irradiated (34 Gy) PBMC per well. After 2 weeks, growth positive wells were screened for specific recognition of g209 peptide (ITDQVPFSV) or HLA-A2+ melanoma cell lines by cytokine release assay. A fraction of the cells from each microwell was split into replicate plates for a cytokine release assay. T cells were cultured overnight with antigen presenting cells, including a control stimulator (T2 cells pulsed with an irrelevant peptide or an HLA-A2 negative melanoma cell line) and an antigen-expressing stimulator cell line (T2 cells pulsed with g209 peptide or an HLA-A2+ gp100-expressing tumor cell line). Quantitative enzyme-linked immunosorbent assay (Endogen, Wobern, MA, U.S.A.) determined IFN-γ release, and specific stimulation was calculated by subtracting the control stimulator value from the antigen-expressing stimulator value.
Antigen-reactive microcultures were expanded for further functional screening using a modified rapid expansion protocol (9) with allogeneic irradiated PBMC, IL-2, and OKT-3 in CM. Cell number was determined after a single round of expansion, and T-cell clones were analyzed by FACS for expression of CD4 and CD8. T-cell clones were also tested by cytokine release assay (using IFN-γ and IL-2 secretion) for recognition of nanomolar g209 peptide concentrations and reactivity against a panel of melanoma cell lines. A single T-cell clone that was capable of rapid in vitro expansion was selected for each patient with high apparent g209 peptide avidity and strong recognition of melanoma cell lines. Each clone was further expanded to large cell numbers for treatment using multiple rounds of the rapid expansion protocol.
RESULTS
Demographics and Selected Characteristics of Patients
All 15 patients treated on this protocol had metastatic melanoma with evidence of progressive disease refractory to high-dose IL-2 therapy and had received an immunization regimen containing the gp100:209–217(210M) peptide. Demographic characteristics of the patients enrolled in this trial are shown in Table 1. These patients had been extensively pretreated (three to five therapy modalities before), and most had multiple sites of metastatic disease.
TABLE 1.
Characteristics of patients enrolled in this study
| Cohort | Patient # | Sex | Age | PS* | Prior Rx† | Sites of disease‡ |
|---|---|---|---|---|---|---|
| 1 | 1 | F | 35 | 1 | RSCI | AX, BO, IL, LI, LU, SU |
| 1 | 2 | M | 37 | 0 | RSCHI | AX, LI, LU, SU |
| 1 | 3 | M | 52 | 0 | SI | AX, LI, RP |
| 2 | 4 | M | 57 | 0 | SCI | LU |
| 2 | 5 | F | 52 | 0 | SI | LI, LU |
| 2 | 6 | M | 48 | 0 | RSCI | AB, BN, LU, MO, NK |
| 3 | 7 | M | 39 | 0 | SCI | ME, SU |
| 3 | 8 | F | 57 | 0 | SCI | IL, IN, SK |
| 3 | 9 | M | 31 | 0 | SCI | ME, PL |
| 4 | 10 | M | 37 | 0 | SI | LU, SU |
| 4 | 11 | F | 32 | 0 | RSI | AX, CL, MT, PV |
| 4 | 12 | M | 18 | 0 | SCI | LI, SP |
| 4 | 13 | M | 53 | 0 | SCI | LU, SK |
| 4 | 14 | F | 30 | 1 | RSCI | SK, SU |
| 4 | 15 | F | 43 | 0 | SI | LU, SK, SU |
Performance status—Eastern Cooperative Oncology Group performance scale.
Prior treatments by category: R, radiation therapy; S, surgery; C, chemotherapy; H, hormone therapy; I, immunotherapy.
Sites of evaluable disease: AB, abdominal; AX, axillary nodes; BN, brain; BO, bone; CL, supraclavicular nodes; IL, iliac nodes; IN, inguinal nodes; LI, liver; LU, lungs; ME, mediastinal; MO, mouth; MT, mesenteric; NK, neck; PL, pleura; PV, pelvis; RP, retroperitoneal; SK, skin; SP, spleen; SU, subcutaneous.
Patients Were Treated With Highly Reactive Tumor Antigen-Specific T-Cell Cultures
On the day after completion of chemotherapy, each patient received an intravenous infusion of cloned tumor antigen-specific, highly active T cells (Table 2). Twelve patients received T-cell clones that were specific for the g209 peptide antigen, including nine derived from peripheral blood cells and three derived from TIL. Three patients received a T-cell clone with specificity for an antigen other than g209, including one PBMC clone and one TIL clone that recognized the MART-1:27–35 peptide, and one TIL clone that was stimulated exclusively by the patient’s autologous tumor-cell line (Table 2). During the first cycle of treatment, patients received an average of 10.4 × 109 cells (range 0.9 × 109 to 24.2 ×109) (Table 3). Most patients received a second cycle of treatment without additional chemotherapy 2–3 weeks after administration of the first cycle (3–5 weeks for patients in Cohort 4). The second treatment cycle consisted of cloned T cells, g209–2M vaccination emulsified in incomplete Freund’s adjuvant if appropriate, and the same IL-2 regimen they had received during the first treatment cycle.
TABLE 2.
Specificity and activity of T-cell clones
| T2 + peptide* |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| g209§ |
A2− Tumor line
|
A2+ Tumor line
|
||||||||
| m27† 1.0 | g280‡ 1.0 | 1.0 | 0.01 | 0.0001 | 888mel | 938mel | 526mel | 624mel | Auto|| | |
| Patient | ||||||||||
| 1 | 131 | 19770 | 11050 | >1000 | 143 | 179 | 2195 | 996 | ||
| 2 | 4 | 55150 | 43100 | 512 | 0 | 0 | 3335 | 2815 | ||
| 3 | 85 | 71000 | 59400 | 9300 | 90 | 87 | 5245 | 2130 | ||
| 4 | 20 | 30750 | 6400 | 70 | 25 | 24 | 825 | 567 | ||
| 5 | 1235 | 10 | 8 | 11 | > 29700 | 8270 | ||||
| 6 | 90 | 45400 | 31450 | 537 | 47 | 52 | 5810 | 3980 | ||
| 7 | 30 | > 128000 | 65400 | 299 | 36 | 11 | > 12000 | 5350 | ||
| 8 | 17 | 58900 | 35750 | 373 | 13 | 15 | > 14900 | 4625 | ||
| 9 | 27 | 15850 | 5160 | 248 | 18 | 24 | 4450 | 3690 | ||
| 10 | 47050 | 657 | 419 | 418 | 27150 | >9665 | ||||
| 11 | 1 | 1 | 1 | 2 | 2 | 19900 | ||||
| 12 | 17 | 18515 | 3874 | 265 | 12 | 12 | 778 | 56 | ||
| 13 | 32 | 21750 | 19750 | 2950 | 28 | 39 | 2385 | 2710 | ||
| 14 | 9 | 25900 | 8720 | 151 | 10 | 8 | 1840 | 3120 | ||
| 15 | 24 | 52150 | 34950 | > 16360 | 20 | 15 | > 11180 | 7995 | ||
Interferon-gamma secretion (pg/mL) by cloned T cells when stimulated with T2 cells pulsed with the indicated peptide concentration (micromolar) or with melanoma cell lines.
MART-1:27–35 peptide
gp100:280–288 peptide
gp100:209–217 peptide
Autologous tumor-cell line.
TABLE 3.
Treatment received by each patient
| Cloned cells administered (×10−9)
|
||||||
|---|---|---|---|---|---|---|
| Patient | Cy* | Flu† | IL-2‡ | Source | First cycle | Second cycle |
| 1 | 30 | 25 | — | TIL | 22.4 | 10.4 |
| 2 | 30 | 25 | — | PBL | 21.5 | 16.0 |
| 3 | 30 | 25 | — | PBL | 15.0 | 14.5 |
| 4 | 60 | 25 | — | TIL | 9.3 | 11.0 |
| 5 | 60 | 25 | — | TIL | 4.1 | 6.0 |
| 6 | 60 | 25 | — | PBL | 5.5 | nd§ |
| 7 | 60 | 25 | 72,000 | PBL | 11.0 | 11.0 |
| 8 | 60 | 25 | 72,000 | TIL | 6.8 | 7.3 |
| 9 | 60 | 25 | 72,000 | PBL | 3.2 | 1.8 |
| 10 | 60 | 25 | 720,000 | PBL | 2.8 | 5.6|| |
| 11 | 60 | 25 | 720,000 | TIL | 11.3 | 14.0 |
| 12 | 60 | 25 | 720,000 | PBL | 0.9 | nd§ |
| 13 | 60 | 25 | 720,000 | PBL | 4.9 | 6.7 |
| 14 | 60 | 25 | 720,000 | PBL | 12.6 | nd¶ |
| 15 | 60 | 25 | 720,000 | PBL | 24.2 | nd¶ |
Cytoxan (mg/kg)
Fludarabine (mg/m2)
Interleukin-2 (IU/kg)
Not done: Patient 6 and Patient 12 received only one infusion of cells due to rapid disease progression.
Patient 10 received a mixture of three CD8+ T-cell clones in the second infusion cycle with a total cell number of 5.6 × 109 cells.
Not done: Patient 14 and Patient 15 received only one infusion of cloned cells, then they were transferred to other treatment protocols.
Hematologic Effects of Treatment
Administration of cyclophosphamide and fludarabine with or without subsequent IL-2 caused a severe lymphopenia that lasted approximately 1 week after the last dose of fludarabine (Fig. 1 and Table 4). Absolute lymphocyte counts dropped from the normal range (460–4700 per mm3) to nadir values between 0 and 30 per mm3. The nadir values and time to recovery for absolute neutrophil count, absolute lymphocyte count, and platelet count are shown in Table 4. Patients enrolled in this protocol experienced nadir absolute neutrophil counts in the range of 0.001–0.303 × 103 per mm3. Similarly, although one patient in the first cohort receiving 30 mg/Kg cyclophosphamide maintained platelet counts in the normal range (154–345 × 103 per mm3), the platelet counts from the other 14 patients dropped to nadir values between 7 and 84 × 103 per mm3. Twelve patients received red blood cell transfusions, and eleven patients received platelet transfusions during the treatment cycle (Table 5). The variation in hematologic parameters between individual patients even within a single cohort probably reflected differences such as prior chemotherapy or the sequelae of advanced disease. Importantly, all hematologic values returned to safe levels in all patients without the need for infusion of autologous stem cells (Table 4).
FIG. 1.
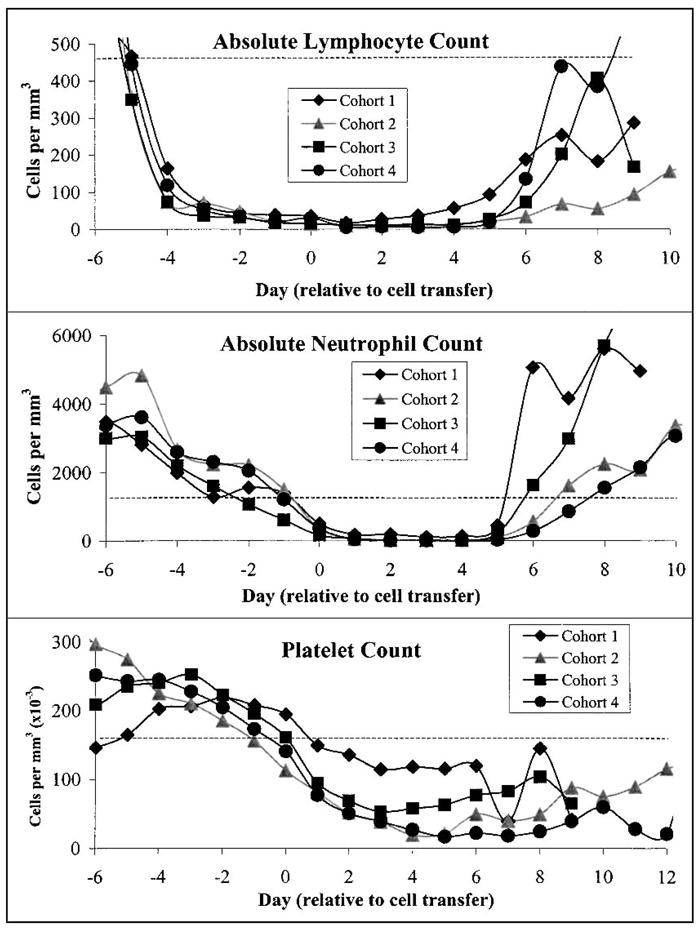
Patients who underwent treatment including nonmyelodepleting chemotherapy exhibited profound but transient lymphopenia, neutropenia, and thrombocytopenia. Average absolute lymphocyte counts (ALC), absolute neutrophil counts (ANC), and platelet counts from all patients in each cohort are plotted as a function of time. All patients received chemotherapy consisting of cyclophosphamide once per day on days –7 and –6 (Cohort 1, 30 mg/Kg; Cohorts 2, 3, and 4, 60 mg/Kg) and fludarabine once per day on days –5 through –1 (25 mg/m2). T-cell clones and peptide vaccinations were administered on day 0. Patients in Cohorts 3 and 4 received IL-2 starting on day 1. A dashed line indicates the lower limit of normal values. Immunosuppression was evident for about 5 days in all cohorts, and then counts recovered to safe levels within a few days. After day 7, values were not obtained for each patient everyday, and “average” counts may not include patients who were discharged as their counts normalized.
TABLE 4.
Hematologic toxicities associated with the treatment
| Nadir* (cells/mm3)
|
Time to recovery† (days)
|
|||||
|---|---|---|---|---|---|---|
| Patient | ANC | ALC | Plat (×10−3) | ANC >500 | ALC >200 | Plat >20 |
| 1 | 3 | 1 | 18 | 6 | 7 | 5 |
| 2 | 303 | 17 | 83 | 5 | 14 | —‡ |
| 3 | 20 | 30 | 220 | 6 | 6 | — |
| 4 | 3 | 3 | 14 | 6 | 15 | 5 |
| 5 | 1 | 5 | 11 | 8 | 11 | 6 |
| 6 | 7 | 13 | 18 | 6 | 9 | 6 |
| 7 | 3 | 4 | 45 | 6 | 8 | — |
| 8 | 8 | 9 | 74 | 5 | 7 | — |
| 9 | 13 | 11 | 8 | 7 | 7 | 7 |
| 10 | 2 | 5 | 14 | 6 | 7 | 10 |
| 11 | 5 | 0 | 12 | 9 | 8 | 10 |
| 12 | 6 | 5 | 19 | 8 | 9 | 8 |
| 13 | 1 | 1 | 7 | 11 | 12 | 12 |
| 14 | 9 | 2 | 18 | 6 | 6 | 8 |
| 15 | 7 | 3 | 9 | 7 | 8–50 | 6 |
Normal ranges for these parameters are ANC (1.32–7.5 × 103 per mm3), ALC (0.46–4.7 × 103 per mm3), and platelets (154–345 × 103 per mm3).
Values were obtained at least every other day unless a range is given.
Platelet count was never below 20 × 103 per mm3.
ALC, absolute lymphocyte counts; ANC, absolute neutrophil counts.
TABLE 5.
Grade 3–4 nonhematologic toxicities, transfusion requirements, and episodes of febrile neutropenia during the first cycle of treatment
| Number of occurrences* |
|||||
|---|---|---|---|---|---|
| Study group | Arm 1 | Arm 2 | Arm 3 | Arm 4 | Total |
| Toxicity | |||||
| Hypotension | 0 | 0 | 0 | 1 | 1 |
| Malaise | 0 | 0 | 0 | 0 | 0 |
| Nausea | 2 | 0 | 0 | 2 | 4 |
| Neurologic | 0 | 0 | 0 | 0 | 0 |
| Febrile neutropenia | 0 | 2 | 1 | 3 | 6 |
| RBC transfusion | 1 | 2 | 3 | 6 | 12 |
| Platelet transfusion | 1 | 3 | 1 | 6 | 11 |
Counted once for each patient at the highest grade.
RBC, red blood cell.
Hematologic recovery from chemotherapy was further analyzed in sequential samples of peripheral blood lymphocytes by flow cytometric quantitation of lymphocyte subsets. Data collected from patients who received the highest dose of cyclophosphamide (60 mg/Kg; Cohorts 2, 3, and 4) is shown graphically in Figure 2. B cells (CD20+), and natural killer cells (CD16+ or CD56+, and CD3−) had recovered to pretreatment levels 3–4 months after chemotherapy. T lymphocyte counts (CD3+) appeared to remain depressed throughout the duration of the study, reflecting very low CD4+ cell numbers. CD4 and CD8 lymphocyte subsets were highly depleted immediately after cyclophosphamide and fludarabine chemotherapy. Subsequently, CD8 cell counts increased over time and approached pre-ablation values 3–4 months after chemotherapy. However, CD4 counts remained relatively low for the duration of the study. Thus, most patients experienced an extended period of recovery during which their CD4/CD8 ratio was below normal.
FIG. 2.
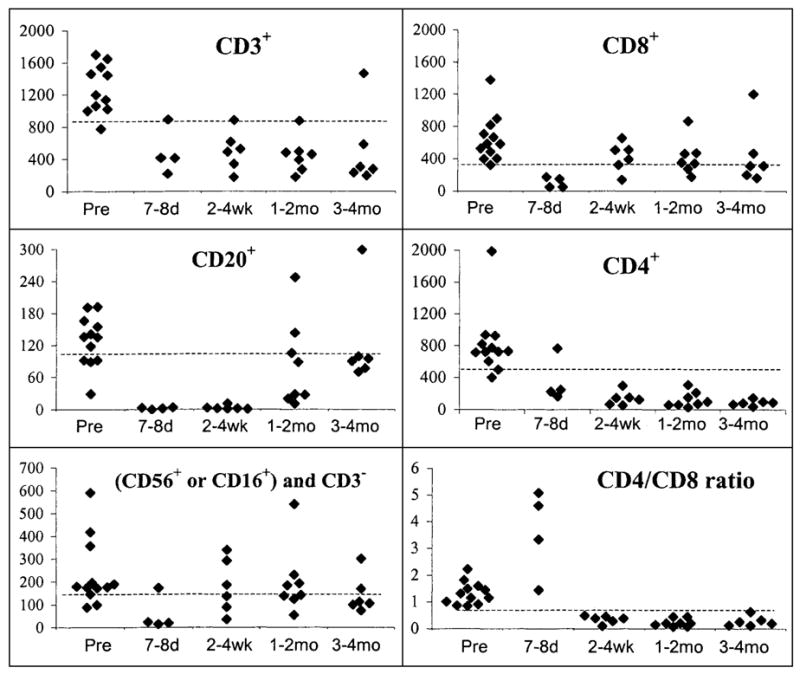
Recovery of lymphocyte subsets after treatment (absolute cell counts per mm3). Flow cytometric analysis of lymphocyte subsets was performed on peripheral blood samples within the indicated time ranges. All data from patients who received 60 mg/Kg cyclophosphamide (Cohorts 2, 3, and 4) was included. No patient is represented more than once in a single time range. Pre: values assayed before the start of chemotherapy. A dashed line represents the lower limit of normal values. B cells (CD20+), and NK cells (CD16+ or CD56+ and CD3−) and CD8+ T cells recovered to pre-ablation levels within a few months. CD4+ cell counts remained low for the duration of the study, resulting in low values for total T cells (CD3+), and a depressed CD4+/CD8+ cell ratio.
Clinical Findings: Nonhematologic Toxicities and Tumor Response
The nonhematologic effects of this combination of chemotherapy and adoptive immunotherapy were well tolerated. Patients on all arms of this protocol experienced a variety of toxicities in addition to hematologic depletion that are typically experienced by patients receiving this chemotherapy alone or high-dose IL-2 therapy alone (Table 5). Six patients experienced at least one episode of febrile neutropenia (a rise in axillary temperature to above 38.5°C for more than 1 hour while having an absolute neutrophil count (<0.5 × 103 per mm3). Nausea and (in Cohort 4) low-grade malaise were also common side effects of treatment. There were no treatment-related deaths.
This trial represents the first time a nonmyeloablative chemotherapy was administered together with high-dose IL-2. Interestingly, the patients in Cohort 4 tolerated high-dose IL-2 therapy on this adoptive transfer protocol extremely well. Before treatment in this protocol, all patients had received multiple cycles of IL-2 therapy and had tumors that were refractory to IL-2. Patients had tolerated an average of 9.5 doses of IL-2 during their first cycle of IL-2, and all patients tolerated fewer doses in subsequent treatment cycles (Table 6). Patients tolerated only an average of 5.3 doses of IL-2 in the treatment cycle immediately preceding chemotherapy. Surprisingly, in the treatment cycle directly after cyclophosphamide and fludarabine administration, patients tolerated an average of 11.2 doses of IL-2. In subsequent courses of IL-2 therapy after endogenous lymphocyte counts had recovered, patients tolerated an average of 6.4 doses.
TABLE 6.
Lymphodepletion increased patient tolerance to high dose IL-2 administration
| Doses of IL-2 tolerated for each treatment cycle
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Treatment cycles before ablation
|
|||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | During ablation 1 | After recovery* 2 | |
| Pt# | |||||||||||
| 10 | 8 | 9 | 6 | 15† | 6 | ||||||
| 11 | 10 | 8 | 7 | 6 | 12 | 8 | |||||
| 12 | 8 | 6 | 5 | 8 | |||||||
| 13 | 12 | 12 | 6 | 6 | 1 | 4 | 4 | 4 | 5 | 12 | 8 |
| 14 | 10 | 9 | 8 | 9 | 9 | 7 | 6 | 12 | 6 | ||
| 15 | 9 | 7 | 7 | 8 | 5 | 6 | 4 | 8 | 4 | ||
| Average | 9.5 | 8.5 | 6.5 | 7.3 | 5 | 5.7 | 4.7 | 4 | 5 | 11.2 | 6.4 |
Patients treated subsequently were approved to receive up to 12 doses of IL-2.
Three to ten weeks after administration of chemotherapy, five patients received either a second cycle of cloned T cells or other immunotherapy, including high dose IL-2 therapy.
Patient 10 was treated with regulatory approval to receive up to 15 doses of IL-2.
No objective partial response was observed in any patient in this protocol. Of interest, five patients, including two patients receiving low-dose IL-2 and three patients receiving high-dose IL-2, demonstrated marked reduction at individual tumor sites. Patient 7 had a mixed response with 36% shrinkage of a hilar mass, stabilization of a mediastinal mass, and concurrent appearance of a subcutaneous mass. Patient 8 had minor regression of multiple sites of cutaneous disease and stability in others, but the response was transient and lesions recurred within 3 months. Patient 11 experienced a transient shrinkage of axillary and abdominal masses, with increase in a pelvic mass. Patient 13 experienced shrinkage of multiple cutaneous and subcutaneous lesions on his lower extremity with progression of several lesions on the same extremity. And patient 14 experienced a transient minor response of extensive facial and neck cutaneous and subcutaneous metastases.
DISCUSSION
The cyclophosphamide/fludarabine schedule used in this clinical trial effectively achieved the immunosuppression of patients before subsequent immunotherapy. All patients on the high dose (60 mg/kg) cyclophosphamide arms of this trial developed severe neutropenia including lymphopenia, maintaining average absolute lymphocyte counts below 20 per mm3 for 5 or more days (Table 4 and Fig. 1)
Several lines of evidence, direct and indirect, suggest that lymphoablation can enhance the efficacy of adoptive T-cell transfer. In preclinical studies using transplantable tumors in mice, it was shown that large hepatic and pulmonary tumors could be cured by the administration of adoptively transferred TIL and IL-2 only when combined with the administration of cyclophosphamide (15). This and other studies (16–18) suggest that immune ablation is an effective preconditioning regimen for adoptive transfer in these models. In our previous clinical studies, the administration of a single dose of cyclophosphamide before the administration of radionuclide-labeled TIL was correlated with enhanced trafficking of TIL to melanoma metastasis (19). Adoptive transfer of cloned T cells after bone marrow transplantation was successful in preventing CMV-specific and EBV-specific viral disease (20–22). Similarly, dramatic graft versus leukemia effects have been well documented (23,24), and a recent report extends the graft versus tumor effects to renal cell carcinoma in a “mini transplant” setting (25). In all of these clinical examples of adoptive transfer of allogeneic cells, the contributions of immunosuppression for the final disease outcome cannot be dissociated from the requirements of immunosuppression for donor engraftment.
Several models have been proposed to explain how immune suppression might improve adoptive transfer therapy in preclinical models. North et al. examined the hypothesis that endogenous lymphocytes acting as suppressor cells were specifically attenuating a lymphocyte mediated antitumor response (18). Recent investigations have rekindled an interest in the activity of suppressor cells and have defined a population of CD4+CD25+ T lymphocytes in peripheral blood that can mediate potent immunosuppressive effects in vitro (26–29). Eradication of these or other endogenous immune regulatory cells could be required for the establishment and propagation of an inflammatory antitumor immune response. An alternate mechanism could operate through the creation of a “niche” in the immune system into which transferred cells could colonize. Potent homeostatic mechanisms control the number of peripheral blood T lymphocytes (30). For patients recovering from lympho-depleting chemotherapy, the ablation of endogenous lymphocytes could enable the persistence and/or activation of adoptively transferred T lymphocytes. Regardless of which potential mechanism might be relevant for increasing the efficacy of adoptive transfer, the cyclophosphamide/fludarabine combination used in this trial is clearly adequate for transient immunosuppression of patients before adoptive cell transfer.
Importantly, all of the patients treated on this study recovered rapidly from the hematologic and nonhematologic toxicities of treatment (Table 4). The most acute hematologic consequences of the chemotherapy, neutropenia, and thrombocytopenia were managed by standard medical treatments. Although all patients underwent granulocyte colony-stimulating factor administration and stem cell harvest before ablation, no patient needed a stem cell transfusion to recover marrow function. Similarly, absolute lymphocyte counts and most lymphocyte subsets spontaneously returned to normal levels. In contrast, a prolonged depression of CD4+ cell counts was seen among patients receiving 60 mg/kg of cyclophosphamide (Fig. 2) as has been reported previously in patients recovering from intensive chemotherapy (30). T-cell subset imbalance after chemotherapy could reflect different mechanisms of CD4+ and CD8+ lymphocyte recovery in the context of reduced thymic function in older patients (31). Other (nonhematologic) toxicities of this therapy included known sequelae of the chemotherapy regimen (e.g., nausea) and the IL-2 regimen (e.g., malaise). No toxicities were ascribed to administration of the cloned lymphocytes, and no synergistic toxicities of the combined treatment modalities could be identified.
An interesting clinical observation revealed by this study was the reduced toxicity of intravenous high-dose IL-2 in the context of immune ablation. Although most patients undergoing IL-2 therapy demonstrate increased toxicity with repeated courses of therapy (32), patients who received lympho-depleting chemotherapy before cell transfer and IL-2 therapy tolerated more doses of IL-2 than they had tolerated when treated previously in our group on other immunotherapy protocols. This finding strongly suggested that most of the toxicity observed after administration of high-dose bolus IL-2 is mediated through secondary cytokines produced by endogenous lymphocytes. The CD8+ clones transferred immediately before IL-2 administration apparently were not capable of producing these secondary cytokines in sufficient amounts when exposed to IL-2 in vivo. The reduced number of IL-2 doses that patients tolerated in IL-2 courses after the recovery of endogenous lymphocytes (Table 5) bolsters this interpretation.
The therapeutic potential for the combination of nonmyelo-depleting chemotherapy, cloned T-cell infusion, and high-dose IL-2 administration remains undetermined. Although five patients exhibited a measurable impact on at least one site of metastatic disease, including mixed and minor responses, no patient experienced an objective clinical response or a lasting antitumor effect. It is unlikely that any tumor responded exclusively to IL-2 therapy, because all patients had disease that was refractory to prior extensive treatment with IL-2. The cyclophosphamide and fludarabine combination used in this study may have been responsible for some of the transient tumor reductions although these agents have very little, if any, reported activity against melanoma.
The lack of durable responses in this clinical trial suggests that additional manipulations will be required to achieve the optimal therapeutic benefit of T-cell transfer. It is possible that the CD8+ clones transferred in this study underwent apoptosis or simply failed to traffic to tumor sites in the absence of CD4+ “helper” T cells. Preliminary evaluation of gene-marked CD8+ T cell clones in two patients receiving the highest dose of chemotherapy and IL-2 indicated that the transferred clones were not detectable in the peripheral blood by 2 weeks after their administration. The important endpoints of T-cell persistence and trafficking in patients receiving nonmyeloablative chemotherapy and adoptively transferred T lymphocytes need to be further investigated. Alternately, transfer of monospecific T-cell clones may have resulted in the selection of resistant, antigen loss tumor variants. To address these issues, future adoptive transfer trials should include transfer of tumor-reactive CD4+ T cells, cells with multiple antigen specificities, or cells with heterogenous effector functions.
Acknowledgments
The authors wish to express their gratitude to the TIL lab technicians, Linda Parker, Tom Shelton, and Jenny Westwood, the clinical nursing staff on the 2E and 2J units of the Clinical Center, National Institutes of Health, and the clinical nurses and fellows in the Surgery Branch, National Cancer Institute for their valuable contributions to this study.
References
- 1.Atkins MB, Kunkel L, Sznol M, et al. High-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: long-term survival update. Cancer J Sci Am. 2000;6(Suppl 1):S11–4. [PubMed] [Google Scholar]
- 2.Rosenberg SA. Interleukin-2 and the development of immunotherapy for the treatment of patients with cancer. Cancer J Sci Am. 2000;6(Suppl 1):S2–7. [PubMed] [Google Scholar]
- 3.Rosenberg SA, Yannelli JR, Yang JC, et al. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J Natl Cancer Inst. 1994;86:1159–66. doi: 10.1093/jnci/86.15.1159. [DOI] [PubMed] [Google Scholar]
- 4.Renkvist N, Castelli C, Robbins PF, et al. A listing of human tumor antigens recognized by T cells. Cancer Immunol Immunother. 2001;50:3–15. doi: 10.1007/s002620000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawakami Y, Eliyahu S, Jennings C, et al. Recognition of multiple epitopes in the human melanoma antigen gp100 by tumor-infiltrating T lymphocytes associated with in vivo tumor regression. J Immunol. 1995;154:3961–8. [PubMed] [Google Scholar]
- 6.Kawakami Y, Eliyahu S, Delgado CH, et al. Cloning of the gene coding for a shared human melanoma antigen recognized by autologous T cells infiltrating into tumor. Proc Natl Acad Sci U S A. 1994;91:3515–9. doi: 10.1073/pnas.91.9.3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenberg SA, Yang JC, Schwartzentruber DJ, et al. Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat Med. 1998;4:321–7. doi: 10.1038/nm0398-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee KH, Wang E, Nielsen MB, et al. Increased vaccine-specific T cell frequency after peptide-based vaccination correlates with increased susceptibility to in vitro stimulation but does not lead to tumor regression. J Immunol. 1999;163:6292–6300. [PubMed] [Google Scholar]
- 9.Dudley ME, Ngo LT, Westwood J, et al. T-cell clones from melanoma patients immunized against an anchor-modified gp100 peptide display discordant effector phenotypes. Cancer J Sci Am. 2000;6:69–77. [PubMed] [Google Scholar]
- 10.Dudley ME, Nishimura MI, Holt AK, et al. Antitumor immunization with a minimal peptide epitope (G9–209–2M) leads to a functionally heterogeneous CTL response. J Immunother. 1999;22:288–98. doi: 10.1097/00002371-199907000-00002. [DOI] [PubMed] [Google Scholar]
- 11.Dudley ME, Wunderlich J, Nishimura MI, et al. Adoptive transfer of cloned melanoma-reactive T lymphocytes for the treatment of patients with metastatic melanoma. J Immunother. 2001;24:363–73. doi: 10.1097/00002371-200107000-00012. [DOI] [PubMed] [Google Scholar]
- 12.Yee C, Gilbert MJ, Riddell SR, et al. Isolation of tyrosinase-specific CD8+ and CD4+ T cell clones from the peripheral blood of melanoma patients after in vitro stimulation with recombinant vaccinia virus. J Immunol. 1996;157:4079–86. [PubMed] [Google Scholar]
- 13.Riddell SR, Greenberg PD. The use of anti-CD3 and anti-CD28 monoclonal antibodies to clone and expand human antigen-specific T cells. J Immunol Methods. 1990;128:189–201. doi: 10.1016/0022-1759(90)90210-m. [DOI] [PubMed] [Google Scholar]
- 14.Topalian SL, Muul LM, Solomon D, et al. Expansion of human tumor infiltrating lymphocytes for use in immunotherapy trials. J Immunol Methods. 1987;102:127–41. doi: 10.1016/s0022-1759(87)80018-2. [DOI] [PubMed] [Google Scholar]
- 15.Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science. 1986;233:1318–21. doi: 10.1126/science.3489291. [DOI] [PubMed] [Google Scholar]
- 16.Berenson JR, Einstein AB, Jr, Fefer A. Syngeneic adoptive immunotherapy and chemoimmunotherapy of a Friend leukemia: requirement for T cells. J Immunol. 1975;115:234–8. [PubMed] [Google Scholar]
- 17.Cameron RB, Spiess PJ, Rosenberg SA. Synergistic antitumor activity of tumor-infiltrating lymphocytes, interleukin 2, and local tumor irradiation. Studies on the mechanism of action. J Exp Med. 1990;171:249–63. doi: 10.1084/jem.171.1.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.North RJ. Cyclophosphamide-facilitated adoptive immunotherapy of an established tumor depends on elimination of tumor-induced suppressor T cells. J Exp Med. 1982;155:1063–74. doi: 10.1084/jem.155.4.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pockaj BA, Sherry RM, Wei JP, et al. Localization of 11 indium-labeled tumor infiltrating lymphocytes to tumor in patients receiving adoptive immunotherapy. Augmentation with cyclophosphamide and correlation with response. Cancer. 1994;73:1731–7. doi: 10.1002/1097-0142(19940315)73:6<1731::aid-cncr2820730630>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 20.Walter EA, Greenberg PD, Gilbert MJ, et al. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N Engl J Med. 1995;333:1038–44. doi: 10.1056/NEJM199510193331603. [DOI] [PubMed] [Google Scholar]
- 21.Rooney CM, Smith CA, Ng CY, et al. Infusion of cytotoxic T cells for the prevention and treatment of Epstein-Barr virus-induced lymphoma in allogeneic transplant recipients. Blood. 1998;92:1549–55. [PubMed] [Google Scholar]
- 22.Roskrow MA, Suzuki N, Gan Y, et al. Epstein-Barr virus (EBV)-specific cytotoxic T lymphocytes for the treatment of patients with EBV-positive relapsed Hodgkin’s disease. Blood. 1998;91:2925–34. [PubMed] [Google Scholar]
- 23.Weiden PL, Flournoy N, Thomas ED, et al. Antileukemic effect of graft-versus-host disease in human recipients of allogeneic-marrow grafts. N Engl J Med. 1979;300:1068–73. doi: 10.1056/NEJM197905103001902. [DOI] [PubMed] [Google Scholar]
- 24.Nash RA, Storb R. Graft-versus-host effect after allogeneic hematopoietic stem cell transplantation: GVHD and GVL. Curr Opin Immunol. 1996;8:674–80. doi: 10.1016/s0952-7915(96)80085-9. [DOI] [PubMed] [Google Scholar]
- 25.Childs R, Chernoff A, Contentin N, et al. Regression of metastatic renal-cell carcinoma after nonmyeloablative allogeneic peripheral-blood stem-cell transplantation. N Engl J Med. 2000;343:750–8. doi: 10.1056/NEJM200009143431101. [DOI] [PubMed] [Google Scholar]
- 26.Levings MK, Sangregorio R, Roncarolo MG. Human CD25(+)CD4(+) T regulatory cells suppress naive and memory T cell proliferation and can be expanded in vitro without loss of function. J Exp Med. 2001;193:1295–1302. doi: 10.1084/jem.193.11.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jonuleit H, Schmitt E, Stassen M, et al. Identification and functional characterization of human CD4(+)CD25(+) T cells with regulatory properties isolated from peripheral blood. J Exp Med. 2001;193:1285–94. doi: 10.1084/jem.193.11.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dieckmann D, Plottner H, Berchtold S, et al. Ex vivo isolation and characterization of CD4(+)CD25(+) T cells with regulatory properties from human blood. J Exp Med. 2001;193:1303–10. doi: 10.1084/jem.193.11.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ng WF, Duggan PJ, Ponchel F, et al. Human CD4(+)CD25(+) cells: a naturally occurring population of regulatory T cells. Blood. 2001;98:2736–44. doi: 10.1182/blood.v98.9.2736. [DOI] [PubMed] [Google Scholar]
- 30.Fry TJ, Mackall CL. Interleukin-7: master regulator of peripheral T-cell homeostasis? Trends Immunol. 2001;22:564–71. doi: 10.1016/s1471-4906(01)02028-2. [DOI] [PubMed] [Google Scholar]
- 31.Mackall CL, Gress RE. Thymic aging and T-cell regeneration. Immunol Rev. 1997;160:91–102. doi: 10.1111/j.1600-065x.1997.tb01030.x. [DOI] [PubMed] [Google Scholar]
- 32.Marroquin CE, White DE, Steinberg SM, et al. Decreased tolerance to interleukin-2 with repeated courses of therapy in patients with metastatic melanoma or renal cell cancer. J Immunother. 2000;23:387–92. doi: 10.1097/00002371-200005000-00012. [DOI] [PubMed] [Google Scholar]