

Azaspiracid-1, a toxin found in European shellfish, was first observed in the mid-1990s when several individuals became ill from eating mussels harvested off the coast of Western Ireland. Considerable synthetic attention has been directed toward this compound by our group[1] and others.[2] Additional excitement was generated by the revelation that the initial structure of azaspiracid-1 (1) had been misassigned (Figure 1).[3] The genesis of the error was believed to be in the ABCDE northern portion of the molecule. Recently, the revised structure 2 was determined in an impressive series of publications by Nicolaou et al.[4] Independently and concurrently, we also converged on the same stereochemical conclusion.[5] Herein, we disclose our successful synthesis of the C1–C26 northern portion of the revised structure of azaspiracid-1 (2).
Figure 1.
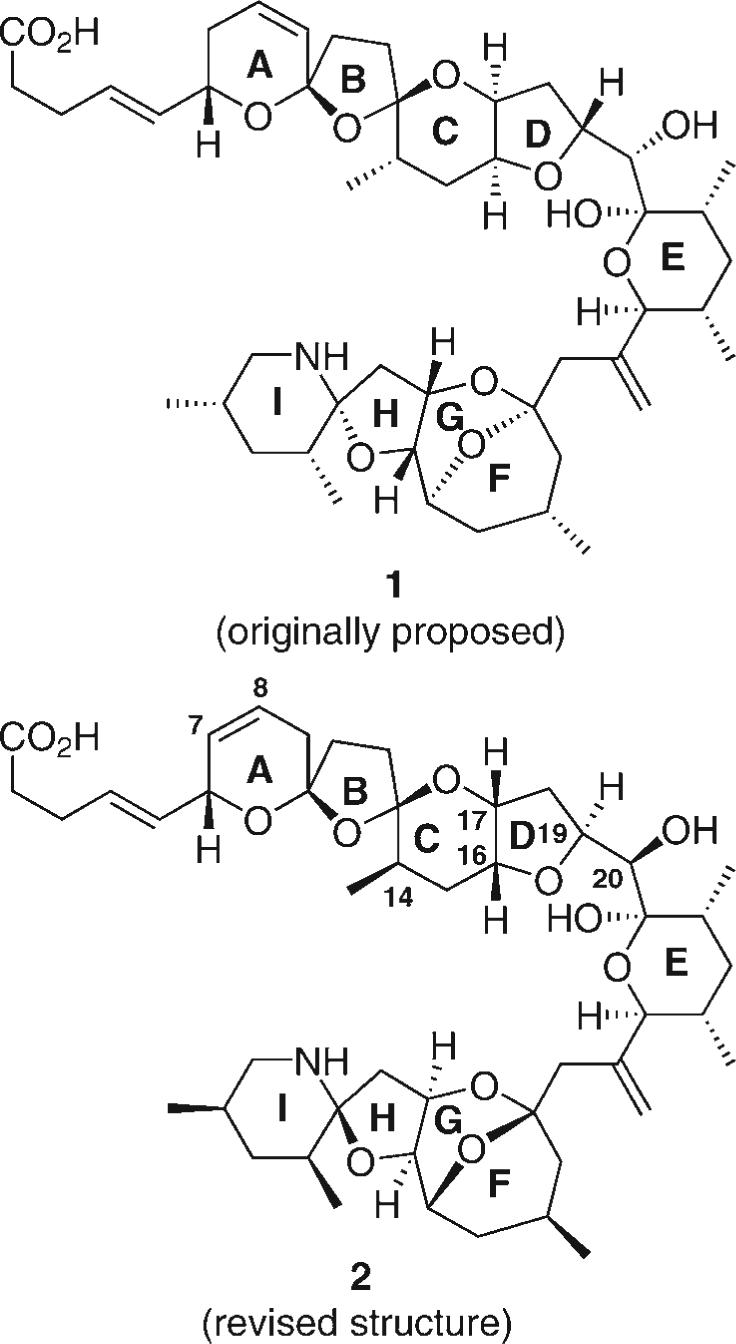
Original (1) and revised (2) structures of azaspiracid-1.
Our retrosynthetic analysis, as shown in Scheme 1, involved disconnection of 3 at the C19−20 linkage to provide the tetracycle 5 and the previously reported keto-phosphonate 6.[5] The bis-spiroketal could be accessed from the ketone 7, which would, in turn, be available from the sulfone 8 and the known aldehyde 9.[5] It is important to note that the revised structure of azaspiracid 2 contains a new challenge in its synthetic architecture: the bisallylic carbon–oxygen bond at C6. We hypothesized that attempted formation of the bisspiroketal in the presence of this labile functionality might prove problematic, particularly under acidic conditions. On the basis of this assumption, care was taken not to incorporate both alkenes in this region until the bis-spiroketal had been formed.
Scheme 1.
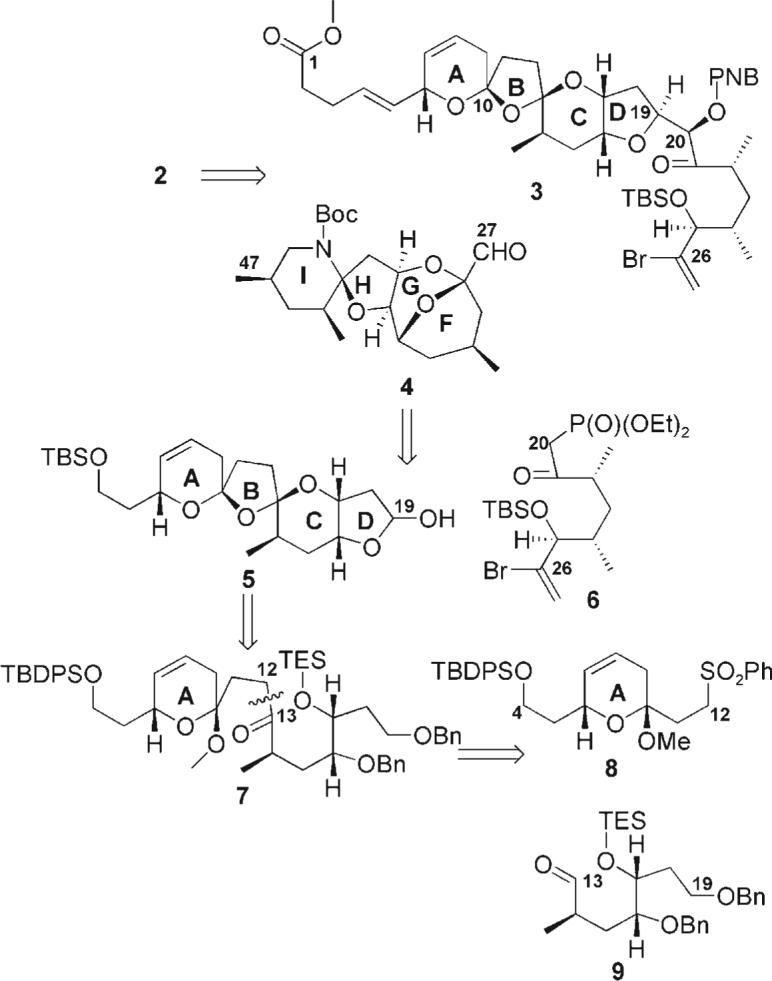
Retrosynthetic analysis of azaspiracid-1 (2). PNB = paranitrobenzoate; TBS = tert-butyldimethylsilyl; Boc = tert-butyloxycarbonyl; TBDPS = tert-butyldiphenylsilyl; TES = triethylsilyl; Bn = benzyl.
Synthesis of the sulfone fragment commenced with the commercially available maleic acid 10 (Scheme 2). Following a known four-step protocol,[6] the cis-unsaturated ester 11 was constructed. Subsequent reduction of the carbonyl moiety followed by conversion into the allylic bromide provided 12. Coupling of this electrophile with the readily available sulfone 14 (synthesized from the commercially available sodium salt of benzenesulfinic acid, thiophenol, and 1,3-dibromopropane (13)) and deprotection of the acetal gave the diol 15 as a mixture of stereoisomers at C10. Sequential protection at C4 and C6 provided the bis-silyl species 16. After careful optimization, we found that the sulfone 16 could be cleanly converted into the β,γ-unsaturated ketone 17 using sodium hexamethyldisilizane and bis(trimethylsilyl) peroxide.[7] Despite our fears of the possible instability of 17, this compound proved to be remarkably robust (stable to chromatography and prolonged storage in the freezer). Subsequent conversion into the methoxy ketal followed by oxidation of the sulfone[8] using Ley's TPAP reagent[9] yielded the coupling partner 8.
Scheme 2.
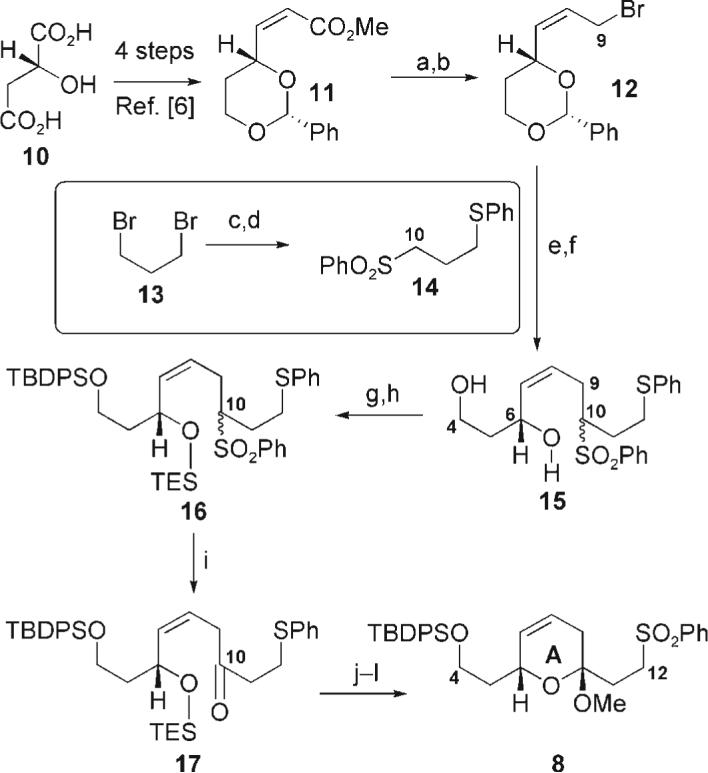
Synthesis of the sulfone. Reagents and conditions: a) DIBAL-H, CH2Cl2, 78°C, 98%; b) Ph3P, CBr4, MeCN, 92%; c) NaH, PhSH, DMF, 0°C, 73%; d) NaSO2Ph, DMF, 86%; e) nBuLi, 14 (2.6 equiv), THF, 78°C; f) 2 n HCl, MeCN, 58% (over two steps); g) TBDPSCl, imid., CH2Cl2, 92%; h) TESOTf, 2,6-lutidine, CH2Cl2, 95%; i) NaHMDS, THF then (TMSO)2, 72% (BORSM); j) NH4F, MeOH/CH2Cl2 (4:1); k) PPTS, MeOH, 68% (over two steps, BORSM); l) TPAP, NMO, MeCN, 40°C, 88%. DIBAL-H = diisobutylaluminum hydride; DMF = N,N-dimethylformamide; imid = imidazole; TESOTf = triethylsilyl triflate; HMDS = hexamethyldisilazide; TMS = trimethylsilyl; PPTS = pyridinium p-toluenesulfonate; TPAP = tetra-n-propylammonium perruthenate; NMO = N-methylmorpholine-N-oxide. BORSM = based on recovered starting material.
With the sulfone subunit 8 in hand, we embarked on the combination of the subunits 8 and 9 (Scheme 3). Julia coupling of 8 and 9 with LDA proved problematic; however, use of the more hindered lithium base derived from 2,2,6,6-tetramethylpiperidine nicely addressed this issue. Subsequent oxidation with TPAP provided the ketosulfone species 18 in excellent yield (92%) over the two steps. Desulfonylation of 18 using Na/Hg amalgam followed by the formation of the bisspiroketal of 7 under our second-generation conditions (PPTS, THF/H2O)[5] led cleanly to the desired transoidal bisspiroketal 19 with the corresponding cisoidal bis-spiroketal 20 as the minor product (2.5:1 19/20). This result is in stark contrast to the C8−9 series in which ketone 23 gave solely the transoidal bis-spiroketal 24.[5] Interestingly, resubmission of the cisoidal bis-spiroketal 20 to the same reaction conditions did not lead to formation of any transoidal bis-spiroketal 19. On the basis of these results, the location of the alkene in the A ring at the C8−9 position (allylic to C10) appears to be a catalyst for the unraveling and subsequent equilibration at C10. Even more intriguing, treatment of the TBDPS-protected cisoidal bis-spiroketal 20 under our first-generation conditions (CSA, tBuOH/PhMe)[1d–f] led to complete equilibration to the transoidal bis-spiroketal 19. It would appear from these experiments that the use of PPTS in THF/H2O leads to formation of the bis-spiroketal under kinetic control, whereas with CSA in tBuOH/PhMe the reaction proceeds under thermodynamic control. Additional support for this conclusion can be found in the exclusive formation of transoidal bis-spiroketal 19 from 7 upon treatment with CSA in tBuOH/PhMe. Note that decomposition appears to be a significant contributor to the low yield (20−30%) in the formation of the spiroketal from 7 using the first-generation conditions. The second-generation conditions were optimal for the initial formation of the bis-spiroketal of 7, whereas the first-generation conditions were preferred for the equilibration of 20.
Scheme 3.

Synthesis and equilibration of bis-spiroketals. Reagents and conditions: a) 2,2,6,6-tetramethylpiperidine, nBuLi, THF then 9; b) TPAP, NMO, CH2Cl2, M.S., 92% over two steps; c) Na/Hg, Na2HPO4, THF, H2O, 86%; d) PPTS, THF/H2O (4:1), 18 h; e) CSA, tBuOH/PhMe (1:1), 18 h. M.S. = molecular sieves; CSA = camphorsulfonic acid.
Removal of the C4-silyl protecting group on 20 revealed additional information (Scheme 4). Treatment of 21 under the first-generation conditions once again leads to complete conversion into the transoidal bis-spiroketal 22. Interestingly, use of our second-generation conditions also led to slow formation of the transoidal bis-spiroketal 22 (1:1 ratio of bis-spiroketals 21/22 after 72 h). Decomposition became a competitive pathway upon extended reaction times, making complete conversion of 21 into 22 under the second-generation conditions not feasible. The hydroxy group at C4 would appear to assist in the ionization of the bis-spiroketals, presumably through increasing the acidity of the local environment and/or a hydrogen-bonding interaction. Finally, debenzylation and oxidation/reduction at C19 provided the tetracycle 5 (Scheme 4).
Scheme 4.

Equilibration of C4-hydroxy bis-spiroketals. Reagents and conditions: a) TBAF, THF; b) CSA, tBuOH/PhMe (1:1), 18 h; c) TBSOTf, 2,6-lutidine, CH2Cl2, 78°C, 99%; d) LiDBB, THF, −78°C, 95%; e) TPAP, NMO, CH2Cl2, M.S.; 67%; f) DIBAL-H, CH2Cl2, 90%. TBAF = tetra-n-butylammonium fluoride; DBB = lithium 4,4–di-tert-butylbiphenylide.
Completion of the northern portion of azaspiracid-1 (2) is shown in Scheme 5. Wadsworth–Emmons coupling of 5 and 6 using KHMDS with in situ, intramolecular hetero-Michael addition yielded 27 with single stereochemistry at C19 of the furan D ring. Deprotonation using NaHMDS followed by a large excess of the Davis oxaziridine provided the hydroxyketone 29 as a single stereoisomer. Mosher ester analysis[10] confirmed that 30 displayed the undesired α stereochemistry. The stereochemical outcome in the hydroxylation can be explained through chelation of the sodium Z-enolate 28 to the oxygen atom of the furan D ring. Initial attempts to invert the stereochemistry at C20 using Mitsunobu-type conditions proved unsuccessful. Fortunately, triflation followed by displacement using the potassium salt of p-nitrobenzoic acid in DMF cleanly provided the desired β stereochemistry. The stereochemistry of 31 was once again confirmed by means of Mosher ester analysis. Desilylation using CSA followed selenation, and oxidation/elimination using TPAP yielded the reactive bisallylic pyran 34. To the best of our knowledge, the TPAP-mediated oxidation of selenium has not been previously reported.[11] Attempted oxidation of the selenide using traditional conditions (e.g. H2O2, THF)[12] led to multiple products. Finally, selective olefin metathesis with 35 using the second-generation Grubbs catalyst[13] gave the target 3.
Scheme 5.

Completion of the northern portion of azaspiracid-1 (2). Reagents and conditions: a) 6, KHMDS, THF, −78°C→RT, 84%; b) NaHMDS, THF, then Davis oxaziridine, 70%, > 20:1 d.r.; c) Tf2O, 2,6-lutidine, CH2Cl2, −78°C, 88%; d) KO2C-p-NO2-C6H4 (20 equiv), DMF, 90%; e) CSA, MeOH/CH2Cl2, 93%; f) o-NO2C6H4SeCN, Bu3P, THF 93%; g) TPAP, NMO, Et3N, CH2Cl2, 66%; h) second-generation Grubbs catalyst, 35, CH2Cl2, 95% (BORSM), > 10:1 E/Z; i)(R)/(S)-Mosher acid chloride, DMAP, CH2Cl2. MTPA = α-methoxy-α-trifluoromethylphenylacetic acid (Mosher); DMAP = 4-(dimethylamino)pyridine. The stereochemistry at C20 was confirmed by Mosher ester analysis. Representative data points for the difference in the chemical shift values [(S)-Mosher ester (R)-Mosher ester (in ppm); CDCl3, 300 or 400 MHz] are shown for structures 30 and 33.
In summary, an efficient approach to the entire C1–C26 northern portion of azaspiracid-1 has been described (27 steps from known ester 12). We have unearthed novel controlling factors for formation of the bis-spiroketal under kinetic versus thermodynamic control. The proper choice of substitution patterns and acidic media allows for the tuning of the equilibration of the bis-spiroketal. In addition, key steps in this synthetic sequence include the oxidation of a sulfone at C10 to form the β,γ-unsaturated ketone, tandem Wadsworth–Emmons/intramolecular hetero-Michael addition to construct ring D, Davis oxidation to incorporate the hydroxy moiety at C20, and selective olefin metathesis to incorporate the sidearm at C5. Application of this strategy to the total synthesis of azaspiracid-1 will be reported in due course.
Footnotes
Dedicated to Professor James D. White on the occasion of his 70th birthday
Financial support was provided by the National Institutes of Health (GM63723). This publication was made possible in part by grant number P30 ES00210 from the National Institute of Environmental Health Sciences, NIH. The authors would also like to thank Professor Max Deinzer (OSU) and Dr. Jeff MorrC (OSU) for mass spectral data, Rodger Kohnert (OSU) and Dr. Clemens Anklin (BrEker Biospin) for NMR assistance, Damien Kuiper (OSU) for synthetic assistance with sulfone 14 and aldehyde 9, and Dr. Roger Hanselmann (Rib-X Pharmaceuticals) for helpful discussions.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.a Carter RG, Weldon DJ. Org. Lett. 2000;2:3913–3916. doi: 10.1021/ol006674w. [DOI] [PubMed] [Google Scholar]; b Carter RG, Weldon DJ, Bourland TC. 221st National ACS Meeting; San Diego. 2001.pp. ORG–479. [Google Scholar]; c Carter RG, Graves DE. Tetrahedron Lett. 2001;42:6035–6039. [Google Scholar]; d Carter RG, Bourland TC, Graves DE. Org. Lett. 2002;4:2177–2179. doi: 10.1021/ol026033w. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Carter RG, Graves DE, Gronemeyer MA, Tschumper GS. Org. Lett. 2002;4:2181–2184. doi: 10.1021/ol026034o. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Carter RG, Bourland TC, Zhou X-T, Gronemeyer MA. Tetrahedron. 2003;59:8963–8974. [Google Scholar]
- 2.a Dounay AB, Forsyth CJ. Org. Lett. 2001;3:975–978. [PubMed] [Google Scholar]; b Aiguade J, Hao J, Forsyth CJ. Org. Lett. 2001;3:979–982. [PubMed] [Google Scholar]; c Aiguade J, Hao J, Forsyth CJ. Tetrahedron Lett. 2001;42:817–820. [Google Scholar]; d Hao J, Aiguade J, Forsyth CJ. Tetrahedron Lett. 2001;42:821–824. [Google Scholar]; e Nicolaou KC, Pihko PM, Diedrichs N, Zou N, Bernal F. Angew. Chem. 2001;113:1302–1305. [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2001;40:1262–1265. [PubMed] [Google Scholar]; f Buszek KR. 221st National ACS Meeting; San Diego. 2001.pp. ORGN–570. [Google Scholar]; g Forsyth CJ, Hao J, Aiguade J. Angew. Chem. 2001;113:3775–3779. doi: 10.1002/1521-3773(20011001)40:19<3663::aid-anie3663>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2001;40:3663–3667. doi: 10.1002/1521-3773(20011001)40:19<3663::aid-anie3663>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]; h Nicolaou KC, Qian W, Bernal F, Uesaka N, Pihko PM, Hinrichs J. Angew. Chem. 2001;113:4192–4195. doi: 10.1002/1521-3773(20011105)40:21<4068::AID-ANIE4068>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2001;40:4068–4071. [Google Scholar]; i Sasaki M, Iwamuro Y, Nemoto J, Oikawa M. Tetrahedron Lett. 2003;44:6199–6201. [Google Scholar]; j Buszek KR, Gibson TS, Reinhardt BC, Sunde JR. 226th ACS National Meeting; New York. 2003.pp. ORGN–179. [Google Scholar]; k Ishikawa Y, Nishiyama S. Tetrahedron Lett. 2004;45:351–354. [Google Scholar]; l Ishikawa Y, Nishiyama S. Heterocycles. 2004;63:539–565. [Google Scholar]; m Ishikawa Y, Nishiyama S. Heterocycles. 2004;63:885–893. [Google Scholar]; n Geisler LK, Nguyen S, Forsyth CJ. Org. Lett. 2004;6:4159–4162. doi: 10.1021/ol048581a. [DOI] [PubMed] [Google Scholar]
- 3.a Nicolaou KC, Li Y, Uesaka N, Koftis TV, Vyskocil S, Ling T, Govindasamy M, Qian W, Bernal F, Chen DY-K. Angew. Chem. 2003;115:3771–3776. doi: 10.1002/anie.200351825. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2003;42:3643–3648. doi: 10.1002/anie.200351825. [DOI] [PubMed] [Google Scholar]; b Nicolaou KC, Chen DY-K, Li Y, Qian W, Ling T, Vyskocil S, Koftis TV, Govindasamy M, Uesaka N. Angew. Chem. 2003;115:3777–3781. doi: 10.1002/anie.200351826. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2003;42:3649–3653. doi: 10.1002/anie.200351826. [DOI] [PubMed] [Google Scholar]
- 4.a Nicolaou KC, Vyskocil S, Koftis TV, Yamada YMA, Ling T, Chen DY-K, Tang W, Petrovic G, Frederick MO, Satake YM. Angew. Chem. 2004;116:4412–4418. doi: 10.1002/anie.200460695. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2004;43:4312–4318. doi: 10.1002/anie.200460695. [DOI] [PubMed] [Google Scholar]; b Nicolaou KC, Koftis TV, Vyskocil S, Petrovic G, Ling T, Yamada YMA, Tang W, Frederick MO. Angew. Chem. 2004;116:4418–4424. doi: 10.1002/anie.200460696. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2004;43:4318–4324. doi: 10.1002/anie.200460696. [DOI] [PubMed] [Google Scholar]
- 5.Zhou X-T, Carter RG. Chem. Commun. 2004:2138–2140. doi: 10.1039/b410092a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a Flögel O, Amombo MGO, Reißig H-U, Zahn G, Brödgam I, Hartl H. Chem. Eur. J. 2003;9:1405–1415. doi: 10.1002/chem.200390160. [DOI] [PubMed] [Google Scholar]; b Herradon B. Tetrahedron: Asymmetry. 1991;2:191–194. [Google Scholar]
- 7.Hwu JR. J. Org. Chem. 1983;48:4432–4433. [Google Scholar]
- 8.Guertin KR, Kende AS. Tetrahedron Lett. 1993;34:5369–5372. [Google Scholar]
- 9.Ley SV, Norman J, Griffith WP, Marsden SP. Synthesis. 1994:639–666. [Google Scholar]
- 10.a Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J. Am. Chem. Soc. 1991;113:4092–4096. [Google Scholar]; b Dale JA, Mosher HS. J. Am. Chem. Soc. 1973;95:512–519. [Google Scholar]; c Sullivan GR, Dale JA, Mosher HS. J. Org. Chem. 1973;38:2143–2147. [Google Scholar]
- 11. For the first example of a metal-catalyzed oxidation of aryl allylic selenides, see:; a Carter RG, Bourland TC. Chem. Commun. 2000:2031–2032. [Google Scholar]; b Bourland TC, Carter RG, Yokochi AFT. Org. Biomol. Chem. 2004;2:1315–1329. doi: 10.1039/b316502g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grieco PA, Gilman S, Nishizawa M. J. Org. Chem. 1976;41:1485–1486. [Google Scholar]
- 13.Chatterjee AK, Choi T-L, Sanders DP, Grubbs RH. J. Am. Chem. Soc. 2003;125:11 360–11 370. doi: 10.1021/ja0214882. [DOI] [PubMed] [Google Scholar]