

Abstract
Vaccinia DNA topoisomerase IB (TopIB) relaxes supercoils by forming and resealing a covalent DNA-(3′-phosphotyrosyl)-enzyme intermediate. Here we gained new insights to the TopIB mechanism through “chemical mutagenesis.” Meta-substituted analogs of Tyr274 were introduced by in vitro translation in the presence of a chemically misacylated tRNA. We report that a meta-OH reduced the rate of DNA cleavage 130-fold without affecting the rate of religation. By contrast, meta-OCH3 and NO2 groups elicited only a 6-fold decrement in cleavage rate. We propose that the meta-OH uniquely suppresses deprotonation of the para-OH nucleophile during the cleavage step. Assembly of the vaccinia TopIB active site is triggered by protein contacts with a specific DNA sequence 5′-C+5C+4C+3T+2T+1p↓N (where ↓ denotes the cleavage site). A signature α-helix of the poxvirus TopIB (132GKMKYLKENETVG144) engages the target site in the major groove and thereby recruits catalytic residue Arg130 to the active site. The effects of 11 missense mutations at Tyr136 highlight the importance of van der Waals interactions with the 3′-G+4pG+3p dinucleotide of the nonscissile strand for DNA cleavage and supercoil relaxation. Asn140 and Thr142 donate hydrogen bonds to the pro-(Sp)-oxygen of the G+3pA+2 phosphodiester of the nonscissile strand. Lys133 and Lys135 interact with purine nucleobases in the major groove. Whereas none of these side chains is essential per se, an N140A/T142A double mutation reduces the rate of supercoil relaxation and DNA cleavage by 120- and 30-fold, respectively, and a K133A/K135A double mutation slows relaxation and cleavage by 120- and 35-fold, respectively. These results underscore functional redundancy at the TopIB-DNA interface.
Type IB DNA topoisomerases (TopIB)2 relax DNA supercoils by repeatedly breaking and rejoining one strand of the DNA duplex through a covalent DNA-(3′-phosphotyrosyl)-enzyme intermediate. Vaccinia virus TopIB, which displays stringent specificity for cleavage at the sequence 5′(C/T)CCTT↓ in the scissile strand (1-5), has been an instructive model system for mechanistic studies of the TopIB family. Analyses of the effects of vaccinia TopIB mutations and DNA target site modifications revealed that the cleavage and religation transesterification reactions are driven by four conserved amino acid side chains (Arg130, Lys167, Arg223, and His265) that compose the active site and catalyze the attack of Tyr274 on the scissile phosphodiester (6-10). The two arginines and the histidine interact directly with the scissile phosphodiester (Fig. 1) and are proposed to stabilize the developing negative charge on a putative pentacoordinate phosphorane transition state (11-15). Lys167 serves together with Arg220 as a general acid to expel the 5′-OH leaving group during the cleavage reaction (16, 17). One of the outstanding problems in TopIB biochemistry concerns whether and how the enzyme might activate the tyrosine nucleophile via general base catalysis.
FIGURE 1.

Structural insights to TopIB transesterification chemistry. The active sites of the pentavalent vanadate complex of Leishmania TopIB (a transition state mimetic; PDB 2b9s; shown in green at left) and the covalent tyrosyl-DNA intermediate of poxvirus TopIB (PDB 2h7f; shown in beige at right) were superimposed and then offset laterally. The conserved active residues of the poxvirus TopIB are indicated. Atomic contacts are denoted by dashed lines. The catalytic contributions of the amino acid functional groups are discussed in the text.
The poxvirus TopIB active site is not preassembled prior to DNA binding. The apoenzyme crystal structure of vaccinia TopIB showed that three of the catalytic residues (Arg130, Lys167, and Tyr274) are either disordered or out of position to perform transesterification chemistry (18). Tyr274 in the apoenzyme is located far away from the scissile phosphate and the other catalytic amino acids (Fig. 2A). The act of DNA binding elicits a major rearrangement of Tyr274 and the surrounding α-helix that brings Tyr274 into the active site in proximity to Arg223. The conformational switch occurs by melting a long α-helix to form two smaller helices and a loop, and it is accompanied by a 16 Å movement of the tyrosine-OH (Fig. 2A).
FIGURE 2.

Structural insights to DNA recognition by poxvirus TopIB. A, apoenzyme structure of poxvirus TopIB (PDB 1a41 colored yellow) was superimposed on the covalent tyrosyl-DNA intermediate (PDB 2h7f; colored beige). Catalytic residues Arg223 and His265 overlap in both structures. The image highlights the large movement of Tyr274 and the rearrangement of the flanking secondary structure that is required to bring the nucleophile into the active site for attack on the scissile phosphodiester. B, fold of the C-terminal catalytic domain of poxvirus TopIB in the covalent intermediate is depicted as a ribbon diagram. The duplex DNA flanking the cleavage site is shown as a space-filling model. The image highlights the docking of the specificity helix (colored magenta) in the DNA major groove of the CCCTT recognition site. This segment is disordered in the poxvirus TopIB apoenzyme. C, stereo view of the specificity helix and its interactions with the nonscissile strand 3′-G+5G+4G+3A+2A+1. Ionic and hydrogen bonding interactions are indicated by black dashed lines. Van der Waals contacts of Tyr136 are depicted as magenta dashed lines. Close nonpolar contacts of Tyr136 to the +3 and +4 phosphates are rendered as green dashed lines.
The formation of a catalytically competent active site is triggered by recognition of specific nucleobases and backbone phosphates of the 5′-CCCTT/3′-GGGAA target sequence. DNA modification interference studies have generated an increasingly refined map of the functionally relevant (and irrelevant) DNA moieties flanking the scissile phosphodiester (4, 5, 19-25). In particular, the analysis of stereospecific methyl phosphonate (MeP) substitutions provided a single-atom resolution map of the functional interface of vaccinia TopIB with the flanking phosphates (26). Methyl groups at eight positions (four on the scissile strand and four on the nonscissile strand) inhibited the rate of cleavage by factors of 50-50,000. The cleavage-suppressing MePs had either no effect on the rate of religation or else had a much less pronounced slowing effect on religation compared with forward cleavage. A model for DNA-promoted active site assembly was proposed (26) as follows: (i) hydrogen bond contacts between the nonscissile strand +4 phosphate oxygens and the backbone amides of vaccinia TopIB residues Gly132 and Lys133 serve to recruit catalytic residue Arg130 (disordered in the apoenzyme) to the active site (Fig. 2C); (ii) contact of the backbone amide of Asp168 with the +3 pro-(Rp)-oxygen of the nonscissile strand anchors catalytic residue Lys167 in the active site; and (iii) a hydrogen bond from the backbone amide of Lys220 to the +5 pro-(Rp)-oxygen of the nonscissile strand tethers catalytic residue Arg223 in the active site. The crystal structure of the poxvirus TopIB-DNA complex (15) confirmed these contacts and also revealed many additional protein-DNA interactions that provide a blueprint for new rounds of mutagenesis experiments and their interpretation (27, 28).
In this study, we probe the TopIB mechanism via a “chemical mutagenesis” approach whereby unnatural amino acid analogs are introduced in lieu of a catalytic residue (Tyr274) by in vitro translation in the presence of a chemically misacylated tRNA (29-31). We thereby circumvent the limitations to the genetically programmable protein “tool kit” when it comes to modifying a functional group at the active site. We also continue to apply traditional mutagenesis, guided by the crystal structures of the TopIB-DNA complex, to gauge the roles of the many amino acids that contact the DNA bases and phosphates. Here we focus on a “specificity helix” that interacts with the DNA major groove (Fig. 2B).
EXPERIMENTAL PROCEDURES
Recombinant Vaccinia TopIB Produced in Bacteria—Mutations were introduced into the vaccinia topoisomerase gene by two-stage PCR overlap extension. NdeI-BglII restriction fragments containing the mutated TopIB genes were cloned into the T7-based expression vector pET16b (Novagen). The inserts of wild-type and mutant TopIB plasmids were sequenced completely to exclude the acquisition of unwanted changes during amplification and cloning. pET-TopIB plasmids were transformed into Escherichia coli BL21(DE3). Cultures (100 ml) derived from single transformants were grown at 37 °C in Luria-Bertani medium containing 0.1 mg/ml ampicillin until the A600 reached 0.6. The cultures were adjusted to 1 mm isopropyl β-d-thiogalactopyranoside and incubation was continued at 37 °C for 3 h with constant shaking. The cells were harvested by centrifugation, and the pellets were stored at -80 °C. All subsequent steps were performed at 4 °C. Thawed bacteria were resuspended in 10 ml of buffer A (50 mm Tris-HCl, pH 7.5, 0.25 m NaCl, 10% sucrose). Lysozyme and Triton X-100 were added to final concentrations of 1 mg/ml and 0.1%, respectively. The lysates were sonicated to reduce viscosity, and insoluble material was removed by centrifugation for 50 min at 15,000 rpm in a Sorvall SS34 rotor. The supernatants were applied to 2-ml columns of Ni2+-nitrilotriacetic acid-agarose (Qiagen) that had been equilibrated with buffer A. The columns were washed with 10 ml of the same buffer and then eluted stepwise with 2-ml aliquots of 50, 100, 200, and 500 mm imidazole in buffer B (50 mm Tris-HCl, pH 8.0, 50 mm NaCl, 10% glycerol). The polypeptide compositions of the column fractions were monitored by SDS-PAGE. Recombinant His10-TopIB was recovered predominantly in the 200 and 500 mm imidazole eluate fractions, which were combined and dialyzed against 50 mm NaCl in buffer C (50 mm Tris-HCl, pH 8.0, 2 mm EDTA, 5 mm dithiothreitol, 10% glycerol). The dialysates were applied to 1-ml columns of phosphocellulose that had been equilibrated with buffer C. The columns were washed with 10 ml of buffer C and then eluted stepwise with 4-ml aliquots of 0.2, 0.5, and 1 m NaCl in 50 mm Tris-HCl (pH 7.5), 10% glycerol. TopIB was recovered in the 1 m NaCl eluate fraction. The proteins were stored at -80 °C.
Synthesis of Chemically Modified Vaccinia TopIB by Translation in Vitro—The codon for Tyr274 of vaccinia TopIB was changed to a TAG stop codon by PCR-based mutagenesis. The mutated TopIB gene was inserted into plasmid pET21b under the control of a T7 promoter. The resulting plasmid encodes an in-frame fusion of the TopIB polypeptide with a C-terminal hexahistidine tag. Misacylated suppressor tRNAs were prepared as described previously (29). Syntheses of vaccinia TopIB and its analogs in vitro were performed using an S-30 extract from E. coli BL21(DE3) cells that had been induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside to produce T7 RNA polymerase. In a typical experiment, proteins were synthesized in a reaction mixture (100 μl) that contained 8 μg of plasmid DNA, 40 μl of premix solution (35 mm Tris acetate, pH 7.0, 190 mm potassium glutamate, 30 mm ammonium acetate, 2.0 mm dithiothreitol, 11 mm magnesium acetate, 20 mm phospho(enol)pyruvate, 0.8 mg/ml E. coli tRNA, 0.8 mm isopropyl β-d-thiogalactopyranoside, 20 mm ATP and GTP, 5 mm CTP and UTP, 4 mm cAMP), 100 μm of each of the 20 amino acids, 30 μCi of [35S]methionine, 10 μg/μl rifampicin, and 25 μl of S-30 extract. For chemical mutagenesis, the reaction mixtures contained 30 μg of deprotected misacylated tRNACUA. After incubation at 37 °C for 40 min, the in vitro translation reaction mixture was diluted with 300 μl of buffer D (50 mm Tris-HCl, pH 7.5, 0.3 m NaCl) and then mixed with 50 μl of a 50% slurry of Ni2+-nitrilotriacetic acid-agarose resin (Qiagen) for 2 h at 4 °C. The resin mixture was poured into a 0.5-ml column, which was then washed with 300 μl of buffer D containing 10% glycerol and 20 mm imidazole. Bound material was eluted with 150 mm imidazole in buffer D with 10% glycerol. Aliquots of the input material, the flow-through, and the serial 20 and 150 mm imidazole eluate fractions were analyzed by SDS-PAGE. Radiolabeled species were visualized by autoradiography. TopIB concentration was determined by SDS-PAGE analysis of an aliquot of the affinity-purified in vitro translated TopIB preparation in parallel with a range of known amounts of purified recombinant vaccinia TopIB.
RESULTS AND DISCUSSION
New Structural Insights to the TopIB Mechanism—Davies et al. (32) exploited vanadate as a mimetic to capture a crystal structure of the pentavalent transition state of TopIB. The active site in the Leishmania TopIB-vanadate-DNA complex is depicted in Fig. 1, with carbons colored green, alongside the superimposed active site of the poxvirus TopIB-DNA covalent intermediate (15), rendered with carbon atoms in beige. The transition state adopts a trigonal bipyramidal geometry expected for the proposed associative mechanism (11) with the attacking tyrosine and the O-5′ atom of the leaving strand at the apices. These structures confirmed most of the atomic contacts to the scissile phosphodiester that were surmised from functional studies of the vaccinia TopIB employing DNA substrates containing stereospecific MeP substitutions, i.e. that Arg223 and His265 coordinate the same nonbridging oxygen (corresponding to the (Sp)-MeP), whereas Arg130 contacts the other nonbridging oxygen (equivalent to the (Rp)-MeP) (12, 13). The O-5′ leaving atom in the transition state is coordinated simultaneously by the arginine (Arg130) and lysine (Lys167) side chains implicated as general acid catalysts (16, 17, 33). Although it had been speculated that a proton relay mechanism might apply (17), one could not surmise from the biochemical data the order of protein transfer (e.g. Arg130 → Lys167 → O-5′ versus Lys167 → Arg130 → O-5′) or exclude a concerted mechanism in which both Arg130 and Lys167 contact the O-5′ atom. It appears from the new structures that a concerted mechanism applies. Comparison of the transition state to the covalent intermediate reveals the mobility of the Lys167 side chain, which is positioned in the minor groove via its contact with the O-2 carbonyl atom of the +1T base and which moves toward the leaving O-5′ atom as the transition state is formed (Fig. 1), as was predicted (16).
Perhaps the most revealing aspect of the aligned active sites of the transition state and covalent intermediate is the contact between the Nε atom of Arg223 and the bridging oxygen atom of the tyrosyl phosphoester (Fig. 1), which suggests a role for Arg223 either in deprotonating the Tyr-OH or lowering its pKa value in the approach to the transition state. Whereas a low pKa value for Tyr274 in the vaccinia TopIB apoenzyme was not detected by NMR pH titration experiments performed with [ζ-13C]tyrosine-enriched topoisomerase (34), this result is mechanistically inconclusive given the subsequent finding that Tyr274 in the vaccinia apoenzyme structure is located far away from the active site and the Arg223 side chain (18). DNA binding triggers a major rearrangement of Tyr274 and the surrounding α-helix that brings Tyr274 into the active site in proximity to Arg223 (15) (Fig. 2A). Invoking a general base function for Arg223 on top of its role in transition state charge neutralization might account for the finding that the rate of transesterification by the conservative R223K mutant at a standard phosphodiester (although vastly better than R223A) is nonetheless 40-fold slower than wild-type TopIB (13).
Using Non-natural Amino Acid Analogs to Probe the TopIB Catalytic Mechanism—Whereas the effects of natural amino acid substitutions for the five catalytic amino acids of vaccinia TopIB (Arg130, Lys167, Arg223, His265, and Tyr274) have been quite informative, there are serious limitations to the genetically programmable protein tool kit when it comes to modifying the functional groups at the active site. Chemical mutagenesis methods allow unnatural amino acids to be introduced into proteins by in vitro translation in the presence of a chemically misacylated tRNA (29-31). In brief, the approach entails the following steps: (i) introduction of a stop codon at the position of interest in the gene encoding the protein; (ii) in vitro transcription of the stop codon-containing mRNA; (iii) in vitro transcription of a suppressor tRNA lacking the 3′-terminal CpA dinucleotide; (iv) chemical coupling of an unnatural amino acid to a pCpA dinucleotide; (v) enzymatic ligation of the unnatural aminoacylated dinucleotide to the suppressor tRNA; and (vi) in vitro translation of the stop codon-containing mRNA in the presence of the chemically misacylated tRNA, thereby resulting in the incorporation of an unnatural amino acid at the single site dictated by the stop codon (Fig. 3). Previously, this strategy was applied to introduce a variety of tyrosine analogs at the active sites of vaccinia and human TopIB (29, 30). Use of an N-terminal His-tagged version of vaccinia TopIB allowed for purification of the in vitro translation product away from the vast excess of components comprising the translation extract. This approach suffered from the problem that ∼70-90% of the in vitro translated TopIB polypeptide terminated at the Tyr274 stop codon (29). A potential caveat is that the truncated 273-amino acid vaccinia polypeptide, which is inert in DNA transesterification because it lacks the tyrosine nucleophile, might compete with the chemically modified full-length TopIB for binding to the DNA substrate. This prospect is of particular concern if one is interested in using short duplex DNA substrates to measure the rates of DNA cleavage by in vitro translated TopIB (see below). We have since redesigned the approach so that the His tag is incorporated at the C terminus of the translated TopIB polypeptide, which means that only the full-length TopIB is recovered during nickel-agarose affinity purification from the translation extract (Fig. 3). The yield of in vitro translated TopIB is much less than we obtain by conventional recombinant TopIB protein production in bacteria (35), but it sufficed for our biochemical experiments.
FIGURE 3.

Chemical mutagenesis of vaccinia TopIB. The mutagenesis strategy is outlined and described in detail under “Experimental Procedures.” The purification of the full-length [35S]methionine-labeled in vitro translated TopIB polypeptide by Ni2+-agarose chromatography is shown. Aliquots of the translation extract loaded on the column (L), the flow-though (F), and imidazole eluate fractions were resolved by SDS-PAGE; the polypeptides were visualized by autoradiography. The full-length His-tagged TopIB polypeptide is indicated by the arrow at right. The chemical structures of tyrosine and the tyrosine analogs introduced at position 274 are shown at bottom.
We analyzed DNA relaxation by in vitro translated vaccinia TopIB containing tyrosine or meta-substituted analogs with OCH3, NO2, F, or OH substituents (Fig. 3). Fig. 4A shows the rate of supercoil relaxation by equal amounts of in vitro translated TopIB containing the indicated side chains at position 274. The experiment shows that TopIB is surprisingly tolerant of the bulky OCH3 group and of fluorine (∼6-fold slowing), whereas the OH substitution is very deleterious (∼200-fold slower relaxation), even more so than the bulky NO2 (∼60-fold slowing). (Note the different reaction times, in seconds (″) or minutes (′), over the lanes for each enzyme.) The relative order of relaxation rates seen here for the purified C-terminal His-tagged TopIB polypeptides (wild-type > OCH3 = F > NO2 > OH) differed slightly from that reported previously (29) for comparatively impure preparations of N-terminal His-tagged in vitro translated TopIB (i.e. wild-type > OCH3 > F > OH > NO2). We suspect that differences in protein purity in the two studies could account for the reversal of order of the meta-OH versus NO2 substitution effects.
FIGURE 4.

Effects of Tyr274 analogs on DNA relaxation and single-turnover cleavage. A, relaxation reaction mixtures containing (per 20 μl) 50 mm Tris-HCl (pH 7.5), 100 mm NaCl, 2.5 mm EDTA, 100 ng of supercoiled pUC19 DNA, and 1 ng of in vitro translated TopIB (containing tyrosine or meta-substituted analogs at position 274) were incubated at 37 °C. The reactions were initiated by the addition of enzyme. Aliquots (20 μl) were withdrawn at the times specified and quenched immediately with SDS. The time 0 samples were taken prior to addition of enzyme. The reaction products were analyzed by 1% agarose horizontal gel electrophoresis. The gels were stained with ethidium bromide, and DNA was visualized under UV illumination. B, cleavage reaction mixtures (20 μl) containing 50 mm Tris-HCl (pH 7.5), 25 fmol of 5′ 32P-labeled 18-mer/30-mer DNA substrate (depicted at bottom with the cleavage site indicated by the arrow), and 8 ng of in vitro translated TopIB (containing tyrosine or meta-substituted analogs at position 274) were incubated at 37 °C for either 10 min (Tyr) or 1 h (analogs). The reactions were quenched with 1% SDS. Half of the sample was digested for 1 h at 37 °C with 10 μg of proteinase K, and the other half was not digested. The mixtures were adjusted to 47% formamide, heat-denatured, and then analyzed by electrophoresis through a 17% polyacrylamide gel containing 7 m urea in TBE (90 mm Tris borate, 2.5 mm EDTA). The reaction products were visualized by autoradiographic exposure of the gel. C, cleavage reaction mixtures containing (per 20 μl) 50 mm Tris-HCl (pH 7.5), 25 fmol of 5′ 32P-labeled 18-mer/30-mer DNA substrate, and 8 or 16 ng (0.2 or 0.4 pmol) of invitro translated TopIB (containing tyrosine or meta-substituted analogs at position 274) were incubated at 37 °C. The reactions were initiated by the addition of TopIB to prewarmed reaction mixtures. Aliquots (20 μl) were withdrawn at the times specified and quenched immediately with SDS (1% final concentration). The products were analyzed by electrophoresis though a 10% polyacrylamide gel containing 0.1% SDS. Free DNA migrated near the dye front. Covalent complex formation was revealed by transfer of radiolabeled DNA to the TopIB protein. The extent of covalent complex formation was quantified by scanning the dried gel with a Fujifilm BAS-2500 imager. A plot of the percentage of input DNA cleaved versus time is shown. The data were normalized to the end point values (redefined as 100%), and the cleavage rate constants (kcl) were calculated by fitting the normalized data to the equation 100 - %cleavage(norm) = 100e-kt.
Most importantly, we have adapted the single-turnover cleavage and religation assays to allow the use of lower DNA and TopIB concentrations so that we can determine the effects of unnatural amino acids on the rates of the component steps. A suicide substrate containing a single CCCTT cleavage site was used to examine the cleavage transesterification reaction under single-turnover conditions. The substrate (shown in Fig. 4B) consisted of a 5′ 32P-labeled 18-mer scissile strand 5′-pCGTGTCGCCCTTATTCCC annealed to an unlabeled 30-mer strand. The cleavage transesterification reaction of in vitro translated wild-type vaccinia TopIB resulted in covalent attachment of the 32P-labeled 12-mer 5′-pCGTGTCGCCCTTp to the enzyme. Digestion of the reaction product with proteinase K in the presence of SDS liberated a cluster of radiolabeled oligonucleotide-peptide adducts migrating faster than the input 32P-labeled 18-mer strand (Fig. 4B). Detection of the covalent oligonucleotide-peptide complex was dependent on prior digestion of the sample with proteinase K (Fig. 4B). This is because the labeled DNA does not enter the polyacrylamide gel when it is bound covalently to the full-sized TopIB polypeptide. The instructive finding was that the same cluster was produced by proteinase K digestion of the covalent complex formed by the tyrosine analog-containing TopIB proteins (Fig. 4B). Thus, the site of cleavage was unchanged by the Tyr274 chemical modifications. A shift in the cleavage site, and hence the size of the covalently bound oligonucleotide, would have been readily detected by an altered mobility of the array of labeled oligonucleotide-peptide complexes.
A key feature of the suicide cleavage reaction is that the unlabeled 6-mer 5′-OH leaving strand ATTCCC dissociates spontaneously from the protein-DNA complex. Loss of the leaving strand drives the reaction toward the covalent state, so that the reaction can be treated kinetically as a first-order unidirectional process. The reaction of excess in vitro translated wild-type vaccinia TopIB with the unmodified control substrate attained an end point in 5 min at which 90% of the DNA was covalently bound. The rate of single-turnover cleavage (Fig. 4C) fit well to a single exponential (not shown) with an apparent cleavage rate constant (kcl) of 0.04 s-1. This value is about 8-10-fold less than the rate of cleavage by purified recombinant vaccinia TopIB (lacking the C-terminal His-tag) assayed at 10-fold higher concentrations of protein and DNA (24). (The apparent kcl of recombinant tag-free bacterial TopIB assayed at the lower concentration of enzyme and DNA was 0.14 s-1; data not shown.) The tyrosine analogs had effects on cleavage rate that were generally consonant with their effects on supercoil relaxation, whereby kcl was reduced 6-fold by -OCH3 and 130-fold by -OH. It is striking that -OH is unique among the analog substituents tested insofar as it can act as a hydrogen bond donor.
The effects of the tyrosine modifications on the religation reaction were studied under single-turnover conditions by assaying the ability of preformed suicide intermediates to transfer the covalently held 5′ 32P-labeled 12-mer strand to a 5′ OH-terminated 18-mer strand to generate a 30-mer product (Fig. 5A). After forming the suicide intermediate on the 18-mer/30-mer DNA substrate, the religation reaction was initiated by adding a 60-fold molar excess of the 18-mer DNA acceptor strand. The sequence of the added 18-mer is fully complementary to the 5′ single-stranded tail of the suicide intermediate. The ionic strength was adjusted simultaneously to 0.5 m NaCl to promote dissociation of the topoisomerase after strand ligation and prevent recleavage of the 30-mer strand transfer product. Aliquots were withdrawn immediately prior to the addition of 18-mer and NaCl (defined as time 0) and at various times afterward. The religation reaction of wild-type in vitro translated TopIB revealed time-dependent depletion of the covalent adduct and concomitant accumulation of the 30-mer strand transfer product (Fig. 5A). The extent of religation at each time point was quantified as the fraction of the 32P-labeled DNA present as covalent adduct at time 0 that was converted to 30-mer strand transfer product (Fig. 5B). The reaction displayed pseudo first-order kinetics with an apparent rate constant (krel) of 0.11 s-1. Here too, the observed krel value for the His-tagged in vitro translation product was about 10-fold less than the krel value of purified recombinant TopIB measured at 10-fold higher concentrations of suicide intermediate and acceptor DNA strand. Note that the derived value for the cleavage equilibrium constant (Kcl) for in vitro translated wild-type vaccinia TopIB (calculated as kcl/krel) was nonetheless similar to the experimental value of Kcl measured for purified recombinant wild-type TopIB at the identical cleavage site (24).
FIGURE 5.
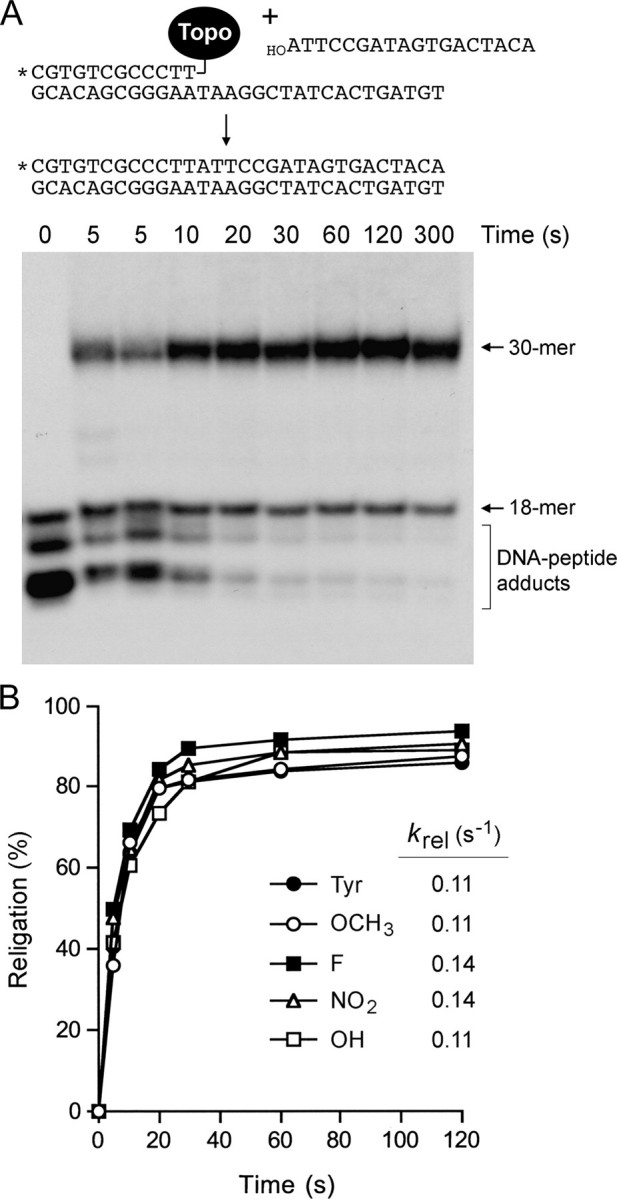
Effects of Tyr274 analogs on single-turnover religation. A, cleavage reaction mixtures containing (per 20 μl) 50 mm Tris-HCl (pH 7.5), 25 fmol of 5′ 32P-labeled 18-mer/30-mer DNA substrate, and 8 ng (0.2 pmol) of in vitro translated TopIB (containing tyrosine or meta-substituted analogs at position 274) were incubated at 37 °C for either 10 min (Tyr) or 1 h (analogs) to form the suicide intermediate. Religation was initiated by the simultaneous addition of NaCl to 0.5 m and a 5′-OH 18-mer acceptor strand d(ATTCCGATAGTGACTACA) to a concentration of 1.5 pmol/22 μl (a 60-fold molar excess over the input DNA substrate). Aliquots (22 μl) were withdrawn at 10, 20, 30, 60, 120, and 300 s and quenched immediately with 1% SDS. A time 0 sample was withdrawn before the addition of the acceptor strand. Two 5-s time points were taken from duplicate reactions performed in parallel. The samples were digested for 1 h at 37 °C with 10 μg of proteinase K and then mixed with an equal volume of 95% formamide, 20 mm EDTA, heat-denatured, and analyzed by electrophoresis through a 17% polyacrylamide gel containing 7 m urea in TBE. The products of the religation reaction of the Tyr274-containing TopIB are shown in the autoradiograph. Religation of the covalently bound 12-mer strand to the 18-mer acceptor DNA yielded a 5′ 32P-labeled 30-mer strand transfer product. B, extents of religation (expressed as the percent of the covalent in termediate converted into 30-mer) are plotted as a function of reaction time for each TopIB protein. The data were normalized to the end point values (redefined as 100%), and the religation rate constants (krel) were calculated by fitting the normalized data to the equation 100 - %religation(norm) = 100e-kt.
The salient finding was that the rates of single-turnover religation by the covalent intermediates of Tyr analog-containing TopIB proteins were virtually indistinguishable from that of the wild-type Tyr274 enzyme (Fig. 5B). Thus, we conclude that the OH moiety at the meta position specifically suppresses the forward cleavage reaction. A plausible explanation is that the extra hydroxyl vicinal to the para-OH nucleophile antagonizes the putative deprotonation of the OH nucleophile by a general base during the cleavage step, by donating a hydrogen bond to the para-oxygen. Of course, this would not impede the religation step, which presumably entails proton donation to the Tyr-O leaving group.
Mutational Analysis of Tyr136 in the TopIB Specificity Helix—An important insight from the poxvirus TopIB-DNA cocrystal (15) was the identification of a putative specificity helix (132GKMKYLKENETVG144) that binds the DNA target site in the major groove (Fig. 2B) and makes atomic contacts to nucleobases and phosphate oxygens (Fig. 2C). The specificity helix is conserved among viral TopIB enzymes and is a distinctive secondary structure feature of the viral/bacterial TopIB clade that is absent in human TopIB (36). The specificity helix is protease-sensitive and disordered in the apoenzyme (18, 37) but adopts a defined secondary structure and becomes protease-resistant when the poxvirus TopIB is in the DNA-bound state (15, 37). Whereas the specificity helix was one of the first segments of the vaccinia protein that was subjected to mutational analysis (38), those early studies were performed prior to the development of optimal cleavage substrates and a quantitative kinetic framework for the TopIB mechanism. Here we reevaluate the structure-function relationships of the specificity helix in light of the atomic contacts seen in the crystal structure.
We focused on Tyr136, a residue conserved in poxvirus topoisomerases (28), mimivirus TopIB (39), and the bacterial clade of poxvirus-like TopIB enzymes (40). Tyr136 projects into the DNA major groove, where it makes a bifurcated hydrogen bond to the guanine-N-7 atoms of the +3 and +4 bases of the 3′-GGGAA cleavage recognition site of the nonscissile DNA strand (Fig. 2C). The +3G and +4G bases are critical for DNA cleavage by poxvirus TopIB; single substitutions of the +3 or +4 guanosine by an abasic nucleoside suppressed the cleavage rate constant by 2 orders of magnitude (4). Replacing the +3 or +4 guanosine with a nonpolar indole analog provided little or no gain of function relative to abasic lesions (24), suggesting that TopIB makes essential contacts to one or more of the polar atoms of guanine: either N-1, N-2, N-3, O-6, or N-7. Inosine and 2-aminopurine substitutions for +3G or +4G had no deleterious effects on transesterification, indicating that the exocyclic amine in the minor groove and the O-6 in the major groove are not critical determinants of DNA cleavage (22, 24, 25). These findings jibe with the absence of protein contacts to those atoms in the TopIB-DNA cocrystal (15). Early studies had shown that guanine-N-7 methylation at the +3G and +4G bases interfered with TopIB binding to its cognate DNA; in turn, binding of TopIB to the target site resulted in protection of the +3G and +4G bases from N-7 methylation by dimethyl sulfate (3). Although the DNA modification data are consistent with the contacts between Tyr136 and guanine-N-7 in the crystal structure, the functional significance of the hydrogen bonding interactions is called into question by the findings of Nagarajan and Stivers (25) that single 7-deazaguanine substitutions at +3G and +4G had no effect on kcl compared with unmodified DNA. Thus, the simple loss of one N-7 atom in the major groove does not account for the cleavage-suppressing indole/abasic effects. It is possible that the +3 and +4 guanine-N-7 contacts of Tyr136 are functionally redundant during the forward cleavage reaction.
To probe the contributions of Tyr136, we replaced this residue by 11 different amino acids, produced the mutant proteins in bacteria as N-terminal His10 fusions, and purified them from soluble bacterial lysates in parallel with wild-type vaccinia TopIB (Fig. 6). We assayed single-turnover cleavage and religation by each protein; the apparent rate constants and reaction end points are reported in Fig. 6. The finding that phenylalanine supported transesterification at the same rate as the native tyrosine signified that hydrogen bonding to guanine N-7 is not the critical contribution of this amino acid. The benzyl moiety of the side chain packs closely against the +3 guanosine nucleoside and makes a plethora of van der Waals interactions as follows: (i) to the guanine C-8 atom from Cz (3.55 Å) and C-ε2 (3.76 Å); (ii) to the deoxyribose C-3 atom from C-ε1 (3.82 Å) and C-δ1 (3.88 Å); and (iii) to the deoxyribose C-5 atom from C-δ1 (3.76 Å), C-γ (3.65 Å), and C-β (3.94 Å) (Fig. 2C). We hypothesize that these van der Waals contacts promote binding of the specificity helix in the major groove. In particular, the presence of contacts between Tyr136 and the +3 guanine C-8 atom provides an explanation, via steric hindrance, for the 35-fold decrement in kcl elicited by replacing the +3G base with 8-oxoguanine (22).
FIGURE 6.

Effects of amino acid substitutions for Tyr136. Top panel, aliquots (4 μg) of the phosphocellulose preparations of vaccinia TopIB with the indicated amino acids at position 136 were analyzed by SDS-PAGE. The Coomassie Blue-stained gel is shown. The positions and sizes (kDa) of marker polypeptides are indicated on the left. Bottom panel, single-turnover transesterification. Cleavage reaction mixtures containing (per 20 μl) 50 mm Tris-HCl (pH 7.5), 0.3 pmol of 5′ 32P-labeled 18-mer/30-mer DNA substrate, and 75 ng (2 pmol) of vaccinia TopIB were incubated at 37 °C. The reactions were initiated by adding TopIB to prewarmed reaction mixtures. Aliquots (20 μl) were withdrawn at various times and quenched immediately with SDS (1% final concentration). The samples were digested with proteinase K and then analyzed by urea-PAGE as described in Fig. 4. The apparent cleavage rate constants (kcl) and the cleavage reaction end points (percent of input 32P-DNA covalently attached to TopIB) are shown. Single-turnover religation was assayed as follows. Reaction mixtures containing (per 20 μl) 50 mm Tris-HCl (pH 7.5), 0.3 pmol of 5′ 32P-labeled 18-mer/30-mer DNA substrate, and 75 ng of wild-type or mutant TopIB were incubated at 37 °C for either 5 min (Tyr, His, Phe), 15 min (Asn, Gln), 60 min (Ala, Ser, Val), or 240 min (Thr, Arg, Asp, Glu) to form the suicide intermediate. Religation was initiated by the simultaneous addition of NaCl to 0.5 m and a 5′-OH 18-mer acceptor strand d(ATTCCGATAGTGACTACA) to a concentration of 15 pmol/22 μl (a 50-fold molar excess over the input DNA substrate). Aliquots (22 μl) were withdrawn at the various times thereafter and quenched immediately with SDS. A time 0 sample was withdrawn before the addition of the acceptor strand. The samples were digested with proteinase K and then analyzed by urea-PAGE. The apparent religation rate constants (krel) and the religation reaction end points (percent of the covalent intermediate converted into 30-mer) are shown. aa, amino acids.
Introducing histidine in lieu of Tyr136 had little effect on transesterification rates (Fig. 6). Histidine, although capable of forming two hydrogen bonds via N-δ and N-ε, is unlikely to penetrate deep enough into the major groove to sustain either of the two guanine-N-7 hydrogen bonds observed for the native Tyr136 hydroxyl. However, the histidine ring could pack against the guanosine in a manner similar to tyrosine and would allow for many of the van der Waals contacts made by the tyrosine benzyl group. It is worth noting that the native Tyr136 is also packed closely against the +3 and +4 phosphates of the nonscissile strand 3′-GgpGpAA as follows: (i) the pro-(Sp)-oxygen of the +3 phosphate is proximal to C-β (3.4 Å) and C-δ2 (3.72 Å), and (ii) the pro-(Sp)-oxygen of the +4 phosphate is proximal to C-δ1 (3.22 Å) (Fig. 2C). Modeling a histidine into the structure suggests that a new hydrogen bond could arise from one of the phosphate oxygens (likely the +4 phosphate) to the histidine N-δ. This notion is supported by the benign effects of the asparagine change, which caused only a 2-fold decrement in kcl (Fig. 6). Although asparagine modeled into the structure would not make the van der Waals contacts to guanine C-8, it could sustain the proposed new hydrogen bond invoked for histidine N-δ, of which asparagine N-δ is an isosteric mimetic. The model is sustained further by considering the severely deleterious effects of an aspartate at position 136, which reduced kcl to 0.0001 s-1 (Fig. 6). The 1600-fold difference in kcl between asparagine and aspartate signifies that the carboxylate is responsible for inhibiting activity, most likely because aspartate (which is superimposable on the γ-branched tyrosine so that the carboxylate oxygens overlap the tyrosine C-δ atoms) electrostatically repels the +3 and +4 phosphate anions and thwarts the close contacts of the specificity helix with the DNA major groove that are essential to recruit Arg130 to the active site. The aspartate change also reduced the rate of religation to 0.01 s-1 (Fig. 6). We surmise that the charge repulsion and ensuing effects on the specificity helix and Arg130 can also destabilize the catalytically competent active site configuration of the covalent TopIB(Y136D)-DNA intermediate. The effects of Y136D on cleavage and religation agree with an early finding that the same mutation suppressed the supercoil relaxation activity of vaccinia TopIB (38).
Glutamine at position 136 slowed kcl to 0.009 s-1 (a 33-fold decrement compared with tyrosine) but had relatively little effect on krel (Fig. 6). Pairwise comparison of glutamine and asparagine highlighted a 17-fold lower kcl when the distance from the main chain to the amide was extended by a single methylene group. This could reflect any of the following factors: (i) steric hindrance by the longer glutamine side chain; (ii) differences between γ-branched and δ-branched side chains; or (iii) displacement of the putative hydrogen-bonding interaction of the amide nitrogen to the DNA phosphate. It is noteworthy that glutamate (kcl to 0.0009 s-1) was an order of magnitude slower than glutamine in DNA cleavage, but nearly an order of magnitude faster than the aspartate (Fig. 6). Thus, extending the distance from the main chain to the carboxylate likely ameliorated some of the electrostatic repulsion effects at the DNA phosphates.
Replacing Tyr136 with alanine eliminates the side chain beyond the β-carbon; this maneuver reduced kcl to 0.008 s-1 (a 38-fold decrement compared with tyrosine) but had little impact on krel (Fig. 6). A previous study of Y136A from this laboratory reported a lower kcl value of 4.9 × 10-4 s-1 (9). In light of this discrepancy, we tested several independent preparations of recombinant Y136A and found that the present higher value is correct. Serine (kcl = 0.009 s-1) phenocopied alanine, signifying that potential for hydrogen bonding by the hydroxyl group could not restore wild-type cleavage activity. The β-branched amino acids threonine and valine (kcl = 0.003 s-1) were both 100-fold less effective than tyrosine in the forward cleavage reaction. Arginine (kcl = 0.005 s-1) had no salutary effect compared with alanine (Fig. 6). In sum, these results underscore the importance of the van der Waals interactions of the Tyr136 side chain with the +3 nucleoside seen in the crystal structure of the TopIB-DNA complex as a key factor in promoting active site assembly prior to DNA cleavage.
Selected Tyr136 mutants were also assayed for their rates of supercoil relaxation (Fig. 7A). The hierarchy of mutational effects on relaxation was generally concordant with mutational effects on DNA cleavage. For example, the Phe and His mutants, which were the most active in transesterification, relaxed the supercoiled plasmid at about one-half and one-fourth of the wild-type rate, whereas the Asn mutant, which was modestly slowed in transesterification, relaxed supercoiled DNA at about one-tenth the wild-type rate (as gauged by the time required to relax all of the input plasmid). Gln, Ser, and Ala, which exerted more significant effects on single-turnover cleavage, were slowed in relaxation by ≥60- to 120-fold compared with wild-type TopIB (Fig. 7A). The apparently greater relative impact of the mutations on relaxation versus cleavage likely reflects several factors. (i) Relaxation is a multiple turnover reaction and was assayed at a 2:1 molar ratio of plasmid DNA to TopIB (which represents a large excess of possible cleavage sites over input enzyme). (ii) The DNA cleavage assay scores a single catalytic cycle and was performed under conditions of TopIB excess. (iii) The relaxation assay is performed in 100 mm NaCl, whereas the cleavage assay mixture has no added salt. Thus, although the cleavage assay is explicitly designed to minimize mutational effects on initial noncovalent DNA binding, the relaxation assay is sensitive to DNA binding effects (42, 43).
FIGURE 7.

DNA relaxation by specificity helix mutants. Reaction mixtures containing (per 20 μl) 50 mm Tris-HCl (pH 7.5), 100 mm NaCl, 2.5 mm EDTA, 300 ng of supercoiled pUC19 DNA, and 4 ng of recombinant TopIB with the indicated amino acid at position 136 (A) or double mutations K133A/K135A or N140A/T142A (B) were incubated at 37 °C. Aliquots (20 μl) were withdrawn at the times specified and quenched immediately with SDS. The reaction products were analyzed by 1% agarose horizontal gel electrophoresis. The gels were stained with ethidium bromide, and DNA was visualized under UV illumination. WT, wild type.
Contributions of Other Contacts of the Specificity Helix to DNA Bases—Two other amino acid side chains in the specificity helix make contacts to the nucleobases; Lys133 makes a bifurcated hydrogen bond to the O-6 and N-7 atoms of the +5G of the nonscissile strand (Fig. 2C), and Lys135 donates a hydrogen bond to the N-7 atom of the +6 purine base of the scissile strand (15). Lys133 is conserved in all poxvirus topoisomerases and mimivirus TopIB (which can cleave at CCCTT) but not in bacterial TopIB enzymes such as Deinococcus TopIB (which does not cleave CCCTT) (39, 40). Lys135 is conserved only in the poxvirus enzymes. The involvement of these lysines in DNA contacts had been suggested by several lines of biochemical studies, including the following: (i) protection of Lys135 from proteolysis by trypsin when vaccinia TopIB is bound to DNA (37), and (ii) protection of the Lys133/Lys135/Lys138 cluster from chemical modification when vaccinia TopIB is bound to DNA (41). Neither the Lys133 nor the Lys135 DNA contact is critical per se, insofar as an abasic lesion in place of +5G has virtually no effect on cleavage rate (4), and changing Lys135 to alanine has little or no effect on supercoil relaxation or cleavage (27, 38). Our speculation is that these two lysines and their base contacts might be functionally redundant. Thus we studied the consequences of a K133A/K135A double mutation.
The forward cleavage reaction of the K133A/K135A protein (kcl = 0.011 s-1) was slowed by a factor for 35 compared with wild-type enzyme assayed in parallel (kcl =0.38 s-1) (Fig. 8A), but religation was apparently unaffected within the limits of detection of the manual assay (Fig. 8B). Sensitivity of the single-turnover cleavage reaction to inhibition by salt and magnesium is a reliable indicator of mutational effect on pre-cleavage binding (28, 42, 43). We measured the amounts of covalent adduct formed in the presence of 50, 100, 150, 200, and 250 mm NaCl or 1, 2, 4, 6, 8, and 10 mm MgCl2 and normalized to the extent of cleavage in unsupplemented control reactions (Fig. 8, C and D). The reaction times differed for the wild-type and mutant enzymes; wild-type cleavage reactions were quenched after 15 s. K133A/K135A mutant reactions were terminated after 5 min. The reaction times were chosen to attain comparable sensitivity for the effects of solution parameters on the cleavage reaction. We observed that the wild-type TopIB was unaffected by up to 100 mm NaCl but was inhibited progressively by 150, 200, and 250 mm NaCl (Fig. 8C). In contrast, covalent adduct formation by K133A/K135A was salt-sensitive. K133A/K135A was inhibited by 70% at 100 mm NaCl and by 95% at 200 mm NaCl, effectively shifting the salt inhibition curve to the left by 100 mm (Fig. 8C). Wild-type TopIB was insensitive to magnesium up 10 mm, whereas K133A/K135A was inhibited progressively at 1-10 mm MgCl2 (Fig. 8D). In the experiment shown in Fig. 8E, the cleavage reactions containing 100 mm NaCl were supplemented with increasing concentrations of magnesium. The inclusion of salt rendered the wild-type TopIB susceptible to inhibition by magnesium. Again, the K133A/K135A mutant was hypersensitized. These findings suggested that K133A/K135A bound less avidly to the CCCTT-containing DNA substrate. The rate of supercoil relaxation by the K133A/K135A mutant (in 100 mm NaCl) was slower than wild-type by ≥120-fold (Fig. 7B). We surmise that Lys133 and Lys135 together are important for vaccinia TopIB activity.
FIGURE 8.

Characterization of mutants K133A/K135A and N140A/T142A. A, single-turnover DNA cleavage. Reaction mixtures contained (per 20 μl) 50 mm Tris-HCl (pH 7.5), 0.3 pmol of 18-mer/30-mer DNA, and 75 ng of wild-type (WT) or mutant TopIB. The extent of covalent complex formation is plotted as a function of time. B, single-turnover religation. Reaction mixtures containing (per 20 μl) 50 mm Tris-HCl (pH 7.5), 0.3 pmol of 5′ 32P-labeled 18-mer/30-mer DNA substrate, and 75 ng wild-type or mutant TopIB were incubated at 37 °C for either 5 min (wild-type TopIB) or 15 min (K133A/K135A and N140A/T142A) to form the suicide intermediate. Religation was initiated by the simultaneous addition of NaCl to 0.5 m and a 5′-OH 18-mer acceptor strand d(ATTCCGATAGTGACTACA) to a concentration of 15 pmol/22 μl (a 50-fold molar excess over the input DNA substrate). Aliquots (22 μl) were withdrawn at the times specified and quenched immediately with SDS. A time 0 sample was withdrawn before the addition of the acceptor strand. The samples were digested with proteinase K and then analyzed by urea-PAGE. The extents of religation (expressed as the percent of the covalent intermediate converted into 30-mer) are plotted as a function of reaction time for each TopIB protein. C-E, cleavage reaction mixtures (20 μl) containing 50 mm Tris-HCl (pH 7.5), 0.3 pmol of 18-mer/30-mer DNA, 75 ng of TopIB, and either 0, 50, 100, 150, 200, or 250 mm NaCl (C), or 0, 1, 2, 4, 6, 8, or 10 mm MgCl2 (D), or 100 mm NaCl plus 0, 1, 2, 4, 6, 8, or 10 mm MgCl2 (E) were incubated at 37 °C for either 10 s (WT) or 5 min (K133A/K135A or N140A/T142A) and then quenched with SDS. The extents of covalent complex formation were normalized to that of a control reaction without added salt or magnesium (defined as 100%) and plotted as a function of NaCl (C) or MgCl2 (D and E) concentration.
Contributions of Contacts of the Specificity Helix to the Nonscissile Strand +3 Phosphate—The poxvirus TopIB-DNA structure reveals a rich network of interactions with the phosphodiester backbone of both strands. It is satisfying to note that the phosphate oxygens at which we observed significant MeP interference with cleavage (26) are those that make direct contacts with the topoisomerase. The most severe MeP effects are noted at atoms that hydrogen bond to main chain amides, including those of the specificity helix where Gly132 NH and Lys133 NH contact the pro-(Rp) and pro-(Sp)-oxygens of the +4 phosphate on the nonscissile strand, respectively (Fig. 2C). The structure also revealed how the nonscissile strand +3 pro-(Sp)-oxygen receives amino acid side chain hydrogen bonds from both Asn140 and Thr142 of the specificity helix. Yet, because an (Sp)-MeP at this position had no virtually effect on single turnover cleavage (26), and N140Y and T142A mutations did not significantly affect supercoil relaxation (38), we suspected that these particular phosphate contacts are either not critical for active site assembly, or they might play functionally redundant roles in DNA recognition. Thus, we characterized a N140A/T142A double mutant.
DNA cleavage by N140A/T142A protein (kcl = 0.012 s-1) was ∼30-fold slower than wild-type TopIB (Fig. 8A), but religation was apparently unaffected (Fig. 8B). N140A/T142A cleavage activity was sensitized to inhibition by salt (∼50 mm shift to the left compared by wild-type TopIB; Fig. 8C) and magnesium (Fig. 8D) and to the combination of 100 mm salt plus magnesium (Fig. 8E), indicative of diminished binding to the CCCTT-containing DNA. The rate of supercoil relaxation by N140A/T142A was slower than wild-type by ∼120-fold (Fig. 7B). We surmise that Asn140 and Thr142 are functionally redundant but, as a pair, are important for vaccinia TopIB activity. Given the a +3(Sp)-MeP modification had little effect on transesterification, we infer that the redundant hydrogen bonding network of Asn140 and Thr142 with the nonbridging phosphate oxygen is not the critical contribution. Rather, we speculate that van der Waals contacts between the Asn140 and Thr142 side chains and the nonbridging substituent (either the polar O or the apolar CH3) are the key factors. Asn140 and Thr142 line a deep groove on the surface of the enzyme into which the DNA phosphate docks; thus, we surmise that the contour of +3 pro-(Sp) binding pocket is a more important feature than its native polarity. The Asn140-Thr142 pair is present in nearly all poxvirus and bacterial TopIB proteins, with occasional conservative substitution of one of the residues (e.g. Ser for Thr142).
Conclusions—This study ushers in a new phase of functional analysis of the TopIB mechanism, guided by crystal structures of DNA-bound reaction intermediates and driven by both chemical and classical mutagenesis. Initial studies of the effects of tyrosine analogs on supercoil relaxation (29, 30) have been extended here to include transient state kinetic studies of the forward and reverse transesterification steps. The selective inhibitory effects of a meta-OH in the Tyr274 nucleophile on the forward DNA cleavage reaction are consistent with a mechanism of general base catalysis. We suggest Arg223 as a candidate for this role, specifically via its N-ε atom. A critical test of this hypothesis is not feasible via conventional mutagenesis but is approachable, in principle, by the chemical route using unnatural amino acids. Future studies in that direction will require the synthesis of suppressor tRNAs aminoacylated with arginine analogs containing single-atom substitutions at each guanidinium nitrogen. Conventional mutagenesis was applied herein to illuminate the role of the specificity helix, a defining feature of the poxvirus/mimivirus/bacterial TopIB clade. Mutational effects at Tyr136 can be usefully interpreted in light of the TopIB-DNA crystal structure and available DNA modification interference data. Conventional mutagenesis of vaccinia TopIB by the “one residue at a time” strategy, although greatly informative, has reached the point of diminishing returns. The crystal structure empowers future studies by highlighting redundant atomic contacts, which can be probed functionally to good effect, as exemplified by the K133A/K135A and N140A/T142A mutants.
This work was supported, in whole or in part, by National Institutes of Health Grants GM46330 (to S. S.) and CA077359 (to S. M. H.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: TopIB, topoisomerase IB; PDB, Protein Data Bank; MeP, methyl phosphonate.
References
- 1.Shuman, S., and Prescott, J. (1990) J. Biol. Chem. 265 17826-17836 [PubMed] [Google Scholar]
- 2.Shuman, S. (1991) J. Biol. Chem. 266 11372-11379 [PubMed] [Google Scholar]
- 3.Shuman, S., and Turner, J. (1993) J. Biol. Chem. 268 18943-18950 [PubMed] [Google Scholar]
- 4.Tian, L., Sayer, J. M., Jerina, D. M., and Shuman, S. (2004) J. Biol. Chem. 279 39718-39726 [DOI] [PubMed] [Google Scholar]
- 5.Minkah, N., Hwang, Y., Perry, K., Van Duyne, G. D., Hendrickson, R., Lefkowitz, E. J., Hannenhalli, S., and Bushman, F. D. (2007) Virology 365 60-69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shuman, S., Kane, E. M., and Morham, S. G. (1989) Proc. Natl. Acad. Sci. U. S. A. 86 9793-9797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morham, S. G., and Shuman, S. (1990) Genes Dev. 4 515-524 [DOI] [PubMed] [Google Scholar]
- 8.Cheng, C., Wang, L. K., Sekiguchi, J., and Shuman, S. (1997) J. Biol. Chem. 272 8263-8269 [DOI] [PubMed] [Google Scholar]
- 9.Wittschieben, J., and Shuman, S. (1997) Nucleic Acids Res. 25 3001-3008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petersen, B. Ø., and Shuman, S. (1997) J. Biol. Chem. 272 3891-3896 [DOI] [PubMed] [Google Scholar]
- 11.Stivers, J. T., Jagadeesh, G. J., Nawrot, B., Stec, W. J., and Shuman, S. (2000) Biochemistry 39 5561-5572 [DOI] [PubMed] [Google Scholar]
- 12.Tian, L., Claeboe, C. D., Hecht, S. M., and Shuman, S. (2003) Mol. Cell 12 199-208 [DOI] [PubMed] [Google Scholar]
- 13.Tian, L., Claeboe, C. D., Hecht, S. M., and Shuman, S. (2005) Structure (Lond.) 13 513-520 [DOI] [PubMed] [Google Scholar]
- 14.Nagarajan, R., Kwon, K., Nawrot, B., Stec, W. J., and Stivers, J. T. (2005) Biochemistry 44 11476-11485 [DOI] [PubMed] [Google Scholar]
- 15.Perry, K., Hwang, Y., Bushman, F. D., and Van Duyne, G. D. (2006) Mol. Cell 23 343-354 [DOI] [PubMed] [Google Scholar]
- 16.Krogh, B. O., and Shuman, S. (2000) Mol. Cell 5 1035-1041 [DOI] [PubMed] [Google Scholar]
- 17.Krogh, B. O., and Shuman, S. (2002) J. Biol. Chem. 277 5711-5714 [DOI] [PubMed] [Google Scholar]
- 18.Cheng, C., Kussie, P., Pavletich, N., and Shuman, S. (1998) Cell 92 841-850 [DOI] [PubMed] [Google Scholar]
- 19.Cheng, C., and Shuman, S. (1999) Biochemistry 38 16599-16612 [DOI] [PubMed] [Google Scholar]
- 20.Krogh, B. O., Claeboe, C. D., Hecht, S. M., and Shuman, S. (2001) J. Biol. Chem. 276 20907-20912 [DOI] [PubMed] [Google Scholar]
- 21.Tian, L., Sayer, J. M., Kroth, H., Kalena, G., Jerina, D. M., and Shuman, S. (2003) J. Biol. Chem. 278 9905-9911 [DOI] [PubMed] [Google Scholar]
- 22.Yakovleva, L., Tian, L., Sayer, J. M., Kalena, G. P., Kroth, H., Jerina, D. M., and Shuman, S. (2003) J. Biol. Chem. 278 42170-42177 [DOI] [PubMed] [Google Scholar]
- 23.Yakovleva, L., Handy, C. J., Yagi, H., Sayer, J. M., Jerina, D. M., and Shuman, S. (2006) Biochemistry 45 7644-7653 [DOI] [PubMed] [Google Scholar]
- 24.Yakovleva, L., Lai, J., Kool, E. T., and Shuman, S. (2006) J. Biol. Chem. 281 35914-35921 [DOI] [PubMed] [Google Scholar]
- 25.Nagarajan, R., and Stivers, J. T. (2006) Biochemistry 45 5775-5782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tian, L., Claeboe, C. D., Hecht, S. M., and Shuman, S. (2004) Structure (Lond.) 12 31-40 [DOI] [PubMed] [Google Scholar]
- 27.Hwang, Y., Minkah, N., Perry, K., Van Duyne, G. D., and Bushman, F. D. (2006) J. Biol. Chem. 281 38052-38060 [DOI] [PubMed] [Google Scholar]
- 28.Tian, L., and Shuman, S. (2006) Virology 359 466-476 [DOI] [PubMed] [Google Scholar]
- 29.Gao, R., Zhang, Y., Choudhury, A. K., Dedkova, L. M., and Hecht, S. M. (2005) J. Am. Chem. Soc. 127 3321-3331 [DOI] [PubMed] [Google Scholar]
- 30.Gao, R., Zhang, Y., Dedkova, L. M., Choudhury, A. K., Rahier, N. J., and Hecht, S. M. (2005) Biochemistry 45 8402-8410 [DOI] [PubMed] [Google Scholar]
- 31.Choudhury, A. K., Golovine, S. Y., Dedkova, L. M., Laikhter, A. L., and Hecht, S. M. (2007) Biochemistry 46 4066-4076 [DOI] [PubMed] [Google Scholar]
- 32.Davies, D. R., Mushtaq, A., Interthal, H., Champoux, J. J., and Hol, W. G. J. (2006) J. Mol. Biol. 357 1202-1210 [DOI] [PubMed] [Google Scholar]
- 33.Interthal, H., Quigley, P. M., Hol, W. G., and Champoux, J. J. (2004) J. Biol. Chem. 279 2984-2992 [DOI] [PubMed] [Google Scholar]
- 34.Stivers, J. T., Shuman, S., and Mildvan, A. S. (1994) Biochemistry 33 15449-15458 [DOI] [PubMed] [Google Scholar]
- 35.Shuman, S., Golder, M., and Moss, B. (1988) J. Biol. Chem. 263 16401-16407 [PubMed] [Google Scholar]
- 36.Redinbo, M. R., Stewart, L., Kuhn, P., Champoux, J. J., and Hol, W. G. J. (1998) Science 279 1504-1513 [DOI] [PubMed] [Google Scholar]
- 37.Sekiguchi, J., and Shuman, S. (1995) J. Biol. Chem. 270 11636-11645 [DOI] [PubMed] [Google Scholar]
- 38.Wittschieben, J., and Shuman, S. (1994) J. Biol. Chem. 269 29978-29983 [PubMed] [Google Scholar]
- 39.Benarroch, D., Claverie, J. M., Raoult, D., and Shuman, S. (2005) J. Virol. 80 314-321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krogh, B. O., and Shuman, S. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 1853-1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hanai, R., and Wang, J. C. (1994) Proc. Natl. Acad. Sci. U. S. A. 91 11904-11908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sekiguchi, J., and Shuman, S. (1997) Nucleic Acids Res. 25 3649-3656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krogh, B. O., and Shuman, S. (2001) J. Biol. Chem. 276 36091-36099 [DOI] [PubMed] [Google Scholar]