

Abstract
Type I procollagen is a heterotrimer composed of two proα1(I) chains and one proα2(I) chain, encoded by the COL1A1 and COL1A2 genes, respectively. Mutations in these genes usually lead to dominantly inherited forms of osteogenesis imperfecta (OI) by altering the triple helical domains, but a few affect sequences in the proα1(I) C-terminal propeptide (C-propeptide), and one, which has a phenotype only in homozygotes, alters the proα2(I) C-propeptide. Here we describe four dominant mutations in the COL1A2 gene that alter sequences of the proα2(I) C-propeptide in individuals with clinical features of a milder form of the disease, OI type IV. Three of the four appear to interfere with disulfide bonds that stabilize the C-propeptide conformation and its interaction with other chains in the trimer. Cultured cells synthesized proα2(I) chains that were slow to assemble with proα1(I) chains to form heterotrimers and that were retained intracellularly. Some alterations led to the uncharacteristic formation of proα1(I) homotrimers. These findings show that the C-propeptide of proα2(I), like that of the proα1(I) C-propeptide, is essential for efficient assembly of type I procollagen heterotrimers. The milder OI phenotypes likely reflect a diminished amount of normal type I procollagen, small populations of overmodified heterotrimers, and proα1(I) homotrimers that are compatible with normal skeletal growth.
Osteogenesis imperfecta (OI)4 (1, 2), commonly known as brittle bone disease, is usually caused by mutations in the COL1A1 and COL1A2 genes that encode the proα1(I) and proα2(I) chains, respectively, of type I procollagen. This heterotrimeric collagen is composed of two proα1(I) chains and one proα2(I) chain. Molecular assembly of the trimer is a multistep process that occurs following synthesis of the full-length chains and release from the ribosome. The C-terminal propeptide (C-propeptide) of each chain folds into a structure that is stabilized by intra-chain disulfide bonds and exposes a chain selectivity domain (3) that directs the interaction of the correct three chains into trimers. Following stabilization of the trimer by inter-chain disulfide bonds, the collagen triple helical domains are nucleated by sequences at their C-terminal end and the helix then propagates in an N-terminal direction.
The vast majority of OI-causing mutations (4, 5)5 affect the triple helical domains and perturb either the nucleation or the propagation of the triple helix N-terminal to the altered sequence but do not affect initial assembly of the proα chains (6). The delay in helix propagation permits prolonged access of modifying enzymes to the chains N-terminal of the alteration (7), whereas the sequence alterations themselves alter the helical structure (8, 9) and reduce molecular thermal stability (10, 11). The phenotypic outcome of these mutations is, in part, due to the synthesis of structurally altered triple helices, and reflects both the location and nature of the change. These mutations result in the full spectrum of OI severity, ranging from lethality in the perinatal period to only an increase in propensity for fractures.
A small number of mutations that disrupt the initial phase of chain folding or association have been identified in the C-propeptide coding domains. All but one were found in COL1A1 and include point mutations (12-15), a small in-frame deletion (12), and deletions and insertions that lead to frameshifts (16, 17). The only COL1A2 mutation in this domain was a homozygous 4-bp deletion that resulted in the synthesis of proα2(I) chains of near normal length in which the terminal 33 residues were changed (18). In this case, the altered chains were excluded from trimer formation, and cells appeared to synthesize only proα1(I) homotrimers. Unlike helical defects, proα(I) C-propeptide alterations impede the initial oligomerization event, and allow modifying enzymes access of the entire helical domain for a prolonged period, resulting in uniform overmodification. Once chain association does occur, helix formation is thought to proceed with-out interruption, and thermal stability of the trimer is usually normal (14, 15). Therefore, the phenotypic consequences of these defects appear to be due to decreased amounts of normal procollagen molecules, and the presence of a small population of structurally normal but overmodified trimers. Most of these mutations lead to death in the perinatal period.
Herein we report dominant mutations in COL1A2 that alter the sequences of the proα2(I) C-propeptide domain. These observations suggest functions for the proα2(I) C-propeptide domain and provide insight into the molecular basis of OI and the mechanisms governing procollagen assembly.
EXPERIMENTAL PROCEDURES
Clinical Features of the Probands and Family Members—Patient 1 (P1) was identified at 39 years of age because of multiple fractures (>40), all of which occurred in adulthood. He was 105 cm tall (≪5th centile), had normal scleral hue, and no evidence of dentinogenesis imperfecta. He was part of a family in which clinical characteristics of OI, including bone fractures, blue sclerae, and dentinogenesis imperfecta, had been noted in members of three generations among whom there was clinical heterogeneity (Fig. 1a and Table 1).
FIGURE 1.

Pedigrees of patients 1 and 2. P1 (a) and P2 (b) (arrows) are part of families in which there is history of disease. Several family members were diagnosed by molecular tests (*). In the family of P1 the condition was detected in three generations. In each family the mode of inheritance is dominant and there is significant clinical heterogeneity between family members (see Tables 1 and 2).
TABLE 1.
Clinical features of the family members of P1
| Individuala | Ageb | Affected | Height (cm)/[percentile] | Number of fractures | Change in scleral hue | Dentinogenesis imperfecta |
|---|---|---|---|---|---|---|
| years | ||||||
| I-1 | Deceased | NAc | 160 [<5th] | 0 | NA | NA |
| I-2 | Deceased | NA | 153 [5th] | 0 | NA | NA |
| II-2 | NA | +d | NA | 0 | − | − |
| II-7 | NA | + | 160 [<5th] | 4-5 | − | + |
| III-4e | 39 | + | 105 [«5th] | >40 | − | − |
| III-6 | 22 | − | 173 [25th] | 0 | − | − |
| III-7 | 19 | − | 173 [25th] | 1 | − | − |
| III-8 | 12 | − | NA | 0 | − | − |
| III-9 | 9 | + | 134 [50th] | 1 | + | + |
| III-10 | 4 | + | 103 [75th] | 3 | + | + |
| IV-1 | 8 | + | NA | 2 | + | + |
| IV-2 | 5 | + | NA | 0 | − | − |
Individual designations are from pedigree (Fig. 3a).
Age at clinical evaluation.
NA, not available.
Plus sign (+) denotes that the trait was present; minus sign (−) denotes the trait was absent.
Proband.
Patient 2 (P2) was identified in early childhood because of five fractures after minimal trauma. Further examination showed the presence of wormian bones in the skull and blue sclerae. At age 6 he was 117 cm tall (50th centile). His father had only one fracture by 33 years of age and had leg asymmetry and back pain. The proband's two younger siblings both had blue sclerae. One also had back pain, whereas the other was born with club feet (Fig. 1b and Table 2).
TABLE 2.
Clinical features of the family members of P2
| Individuala | Ageb | Affected | Height (cm)/[percentile] | Number of fractures | Change in scleral hue | Other |
|---|---|---|---|---|---|---|
| years | ||||||
| I-1 | 33 | +c | 172 [25th] | 1 | − | Leg asymmetry, back pain |
| I-2 | NAd | − | 160 [25th] | 0 | − | − |
| II-1e | 6 | + | 117 [50th] | 5 | + | Wormian bones |
| II-2 | 8 | + | 0 | + | Back pain | |
| II-3 | 1 | + | 0 | + | Club feet |
Individual designations are from pedigree (Fig. 3b).
Age at clinical evaluation.
Plus sign (+) denotes that the trait was present; minus sign (−) denotes the trait was absent.
NA, not available.
Proband.
Patient 3 (P3) was a 33-year-old woman who presented with severe osteoporosis. She had had at least 17 fractures, the first of which occurred at 8 years of age, mostly of the fingers, wrists, and toes. She was 162 cm tall (75th centile) with no alterations in scleral color, teeth, or hearing. Her father and brother each had sustained five or six fractures of ribs, fingers, and wrists.
Patient 4 (P4) was evaluated at 33 years of age because of multiple fractures. X-rays showed thin diaphyses and funneled metaphyses and overall decreased bone density. His teeth appeared somewhat translucent and his sclerae were white. He was 164 cm tall (5th centile). There was no family history of OI.
Cell Culture and Metabolic Labeling of Proteins—Dermal fibroblast cultures were established from explanted skin biopsies of four OI patients (P1, P2, P3, and P4) and one normal individual (control) with appropriate consent. For each experiment, 250,000 cells were seeded into 35-mm dishes, incubated with ascorbic acid (50 μg/ml), and were proline-deprived prior to radiolabeling as described (19). For analysis of collagenous proteins, cells were incubated with 20 μCi of [3H]proline (Amersham Biosciences) for 18 h. To characterize hetero- and homotrimer populations, cells were incubated with 5 μCi of [14C]proline for 18 h. For pulse-chase studies, cells were incubated for 80 min with either 100 μCi of [3H]proline or 5 μCi of [14C]proline and then chased for indicated times with excess non-labeled proline as described (15).
Isolation and Analysis of Collagenous Proteins—Cell layer and medium fractions were collected separately, and proteins were ethanol-precipitated in the presence of protease inhibitors as detailed elsewhere (20). For pulse-chase analysis, 15 mm iodoacetamide (Sigma) was added to the cell fractions at the time of harvesting to prevent additional disulfide bond formation. To generate collagen-sized molecules, procollagen was partially digested with pepsin (19) to remove the propeptides. Samples were dissolved in SDS buffer, in some cases reduced with addition of dithiothreitol, and boiled for 3 min prior to separation by SDS-PAGE (3% stacking gel, pH 6.8, and 5% separating gel, pH 8.8). To separate unreduced trimers of type I procollagen, a lower percentage SDS-PAGE gel was used (2.5% stacking gel, pH 6.8, and 3% separating gel, pH 8.8) with-out reduction. For fluorographic detection of radiolabeled protein, gels were equilibrated with Enhance® (New England Nuclear), vacuum-dried, and exposed to film (X-Omat AR Kodak) at -80 °C.
To measure the proportion of proα1(I) homotrimers secreted by the cells, ammonium sulfate precipitates of medium procollagens made by control cells, cells from an individual whose cells synthesized only homotrimers (resulting from homozygosity for a nonsense codon in COL1A2 (21)), and those made by cells from P2 were suspended in 0.5 m acetic acid and digested by pepsin (∼0.1 mg/ml) overnight at 4 °C. After removal of insoluble material by centrifugation, pepsin-soluble collagen was precipitated by addition of NaCl to 0.9 m final concentration. The precipitate was collected by centrifugation and redissolved in 2 mm HCl, 0.03% Brij. Concentrations of normal control and homotrimer control collagens were evaluated by CD measurements on a J-810 spectropolarimeter (Jasco). Calibration mixtures of normal heterotrimers and homotrimers were prepared and compared with proband collagen by gel electrophoresis. Briefly, 10 μl of collagen solution (0.5-60 μg/ml) in 2 mm HCl was mixed with 2.5 μl of 0.5 m sodium carbonate/2.5 m NaCl, pH 9.3. Each sample was fluorescently labeled by addition of 0.5 μl of freshly prepared 10 mg/ml solution of Cy5 NHS ester (Amersham Biosciences) in anhydrous dimethylformamide followed by rapid mixing and incubation for 30 min at room temperature with gentle shaking. After labeling, each sample was mixed with 4× lithium deodecyl sulfate sample loading buffer (Invitrogen), denatured by heating to 65 °C for several minutes and loaded into two lanes on a pre-cast 3-8% gradient Tris acetate gel (Invitrogen). After electrophoretic separation, each gel was rinsed for 30 min in 20% ethanol and scanned on an FLA5000 fluorescence scanner (Fuji Medical Systems) at 50-μm resolution. The gels were analyzed with the ScienceLab software supplied with the scanner. To ensure identical labeling conditions and optimal labeling intensity, all samples intended for comparison were adjusted to the same collagen concentration and labeled simultaneously using the same stock solution of Cy5. The concentration of the proband collagen solution was evaluated from the fluorescence intensity of the corresponding gel bands in a preceding calibration experiment.
Preparation of DNA and Mutation Characterization—Genomic DNA was prepared from cultured fibroblasts or blood using the QIAamp DNA Mini Kit (Qiagen). Total cellular RNA was isolated from cultured dermal fibroblasts using the RNeasy Mini Kit (Qiagen), and cDNA was made using the Superscript II First-Strand Synthesis System (Invitrogen). PCR products that span the COL1A1 and COL1A2 genes were screened for base mismatches by conformation sensitive gel electrophoresis (22), and the results were used to direct sequencing efforts. Mutant sequences were determined using the ABI Prism™ BigDye Terminator Cycle Sequencing Reaction Kit and the ABI Prism™ 310 Genetic Analyzer according to manufacturer's recommendations (PerkinElmer Life Sciences), and the mutation identities were confirmed by sequencing opposite strands or by restriction endonuclease digestion. The primer sequences are available along with PCR conditions on request from the authors. Family members of P1 and P2 were screened for mutations by isolating genomic DNA from peripheral blood samples using the PureGene kit as recommended by the manufacturer (Gentra Systems), PCR, and sequencing as described above.
Immunocytochemistry—Procollagen accumulation in cultured dermal fibroblasts from P3 was evaluated by immunocytochemistry as described (23), with some minor changes. Briefly, cultured cells were washed with phosphate-buffered saline and treated with acetone/methanol (1:1) for 5 min at -20 °C. After three 5-min washes with Tris-buffered saline, cells were blocked with 2% goat and 0.5% horse sera (Jackson ImmunoResearch Laboratories) for 1 h. Fibroblasts were then incubated for 2 h with LF-116, a polyclonal antibody directed against the N-propeptide of proα2(I) (24), at a 1:40 dilution. The remaining steps were done as described (23).
RESULTS
Abnormal Type I Procollagen Was Made by Cultured Cells—Alterations in the amount of type I procollagen and the mobilities of the proα2(I) chains were subtle and differed among the four cell strains (Fig. 2). P1 cells made a population of proα2(I) chains, visible in the cell layer that migrated just above the normal chains. P2 cells made proα2(I) chains that migrated slightly faster than the normal chains. Following partial proteolysis with pepsin, the α2(I) chains in the medium and the cell layer migrated slightly faster than control and the α1(I) to α2(I) ratio was slightly altered (see below). P3 cells synthesized a population of proα2(I) chains with slowed mobility and following partial proteolysis with pepsin a distinct band that migrated more slowly than α2(I) was obvious in both the medium and the cell layer. The P4 cells also synthesized proα2(I) chains that had slow mobilities and were located primarily in the cell layer. In contrast to the other cell strains, these cells appeared to secrete less type I procollagen than control cells with-out distinct retention of abnormal molecules.
FIGURE 2.

Cells synthesized defective proα2(I) chains. Skin fibroblasts from a normal control individual (C) and P1-P4 were cultured, and proteins were metabolically labeled. Separation of collagenous proteins from cell and medium fractions by SDS-PAGE showed subtle alterations (indicated by arrows) in the amount of type I procollagen made and in the mobilities of the proα2(I) and α2(I) chains. P1 cells made chains, visible in the cell layer that migrated slowly. P2 cells made chains with faster mobilities. P3 cells synthesized chains with slower mobilities that were visible in all preparations. P4 cells made chains that had slow mobilities that were restricted to the cell layer.
Mutations Were Located in the C-propeptide Coding Region of COL2A1—We identified mutations in one COL1A2 allele in all four individuals (Fig. 3). In P1, a c.3944A → T transversion in exon 51 predicted the substitution of aspartic acid by valine at position 1315 (D1315V), measured from the initiator methionine. All affected family members (Fig. 1) had the same mutation. In P2, insertion of a single nucleotide (c.3952_3953insT) in exon 51 resulted in a frameshift that substituted phenylalanine for the serine at position 1318 (S1318F) and created a premature termination codon (TAA) as the next downstream codon. This resulted in the deletion of 48 amino acids from the chain, including a cysteine involved in stabilizing the folded conformation of the chain. The mRNA from this allele was stable (data not shown). In P3, a c.3527G → T transversion in exon 50 predicted the substitution of glycine at position 1176 to valine (G1176V). P4 had a c.3487T → C transition that led to substitution by arginine for the first of the cysteines involved in interchain disulfide bonds (C1163R). The three missense mutations changed evolutionarily conserved amino acids and the frameshift mutation deleted several (Fig. 4).
FIGURE 3.
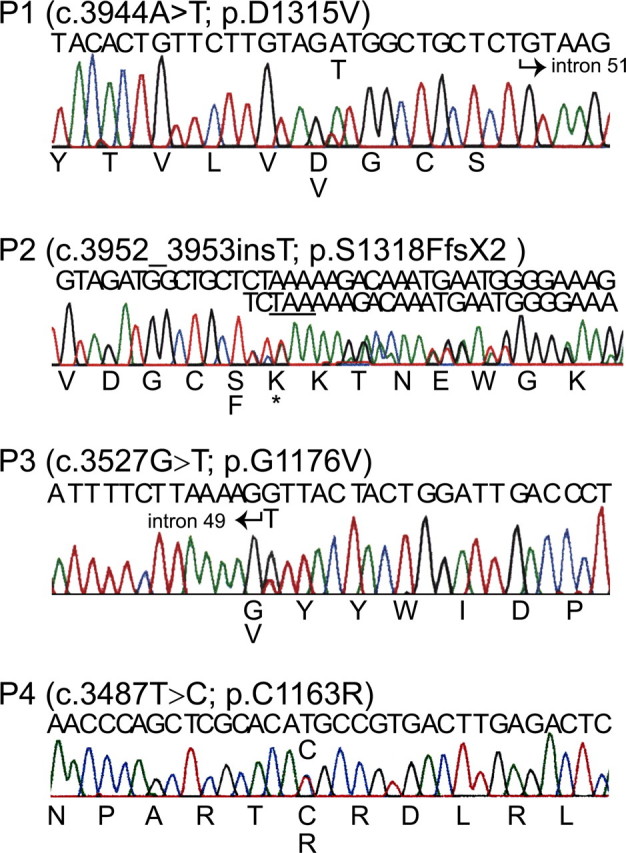
Mutations detected in the COL1A2 C-propeptide coding region. Genomic DNA was sequenced for P1, P3, and P4 and cDNA was sequenced for P2. The nucleotide sequence is above each chromatogram, and amino acid sequence is below. All but P2 had a single base change that altered an amino acid residue. P2 had an insertion of a single thymidine that changed the reading frame and resulted in a premature stop codon (*), predicting synthesis of a proα2(I) chain truncated by 48 residues.
FIGURE 4.

Mutations changed conserved amino acid residues. The C-propeptide amino acid sequences of the 11 known proα chains were aligned. The chain selectivity domain (gray), cysteines, and intramolecular disulfide bonds are indicated. Residues that correspond to the single amino acid substitutions (P1, P3, and P4) are in bold font. The mutation of P2 deletes the terminal amino acids of the C-propeptide (indicated with dots). Each alteration involves residues that are completely conserved among the fibrillar collagen genes and, therefore, likely biologically important.
Type I Procollagen Assembly Was Hindered, and Proα2(I) Chains Accumulated Intracellularly—The C-propeptides direct the initial assembly of proα chains into dimers and then into trimers. To see if molecular assembly was perturbed, we analyzed the kinetics of folding. Monomeric proα1(I) and proα2(I) chains were present in both control and patient cells (time 0, Fig. 5), and proα1(I) chains assembled into dimers and trimers by 20-40 min. In control cells excess proα2(I) chains present at time 0 left the monomeric pool by 60-80 min. In contrast, free proα2(I) chains were apparent in each patient cell strain for the duration of the chase period of 80 min.
FIGURE 5.

Defects hindered assembly of proα2(I) chains that accumulated intracellularly. Control (C) and patient (P1-P4) cells were pulse-labeled and then chased for up to 80 min. Control cells made proα2(I) monomers that gradually disappeared through time and were largely gone by 60-80 min, moving to the trimeric pool of molecules. The OI cells made proα2(I) chains that remained in the monomeric pool for the entire 80-min chase period. Immunofluorescence microscopy with an antibody against proα2(I) showed markedly increased intracellular levels of proα2(I) chains in the representative cell line, P3, relative to control cells.
Intracellular retention of proα2(I) chains was confirmed by immunolabeling cultured cells from a normal control and P3. Cells were stained with an antibody directed against the N-propeptide of proα2(I). Both normal and patient cells were stained in a pattern consistent with the presence of protein in the secretory pathway, but staining of P3 cells was markedly increased (Fig. 5).
Proα1(I) Homotrimers Were Made—Type I procollagen exists as two species: the major one has two proα1(I) chains and a single proα2(I) chain, but a minor population made in some tissues has three proα1(I) chains. Because chain-chain association is determined by sequences in the C-propeptides of proα chains, we examined the secreted procollagens to determine if any of the mutations resulted in failure of the abnormal proα2(I) chains to associate. The control cells made only heterotrimeric type I procollagen. Cells from two of the four individuals (P2 and P4) made significant amounts of proα1(I) homotrimer (Fig. 6a).
FIGURE 6.

Cells made proα1(I) homotrimers. a, proteins from the four cell lines (P1-P4) were separated on a low percentage gel to resolve hetero- and homotrimeric type I procollagen. Protein from normal cells that make only heterotrimers and from another cell line that makes only homotrimers were separated on the same gel as standards. All OI cells made heterotrimeric procollagen, but in addition, P2 and P4 synthesized and secreted proα1(I) homotrimers (arrows). b, to determine relative levels of homotrimers made by P2 cells, control mixtures of fluorescently labeled, pepsin-treated hetero- and homotrimers with known composition (0 and 50% homotrimer) were compared with a sample from P2 (c and d). These and other calibration mixtures (symbols) were used to generate a calibration curve (fitted solid line) by measuring the ratio of fluorescence intensities of either monomer or dimer bands. An average of measurements indicated that 25 ± 5% of the procollagen trimers made by P2 cells were homotrimeric. Each calibration mixture and the OI sample was separated into two samples and loaded into two gel lanes. An apparent single calibration point at a given concentration and a single dashed line for the monomer bands indicate identical measured fluorescent intensity ratios for the two samples. The homotrimer content was determined by comparison of the measured ratio with the corresponding fitted calibration curve. The labeling and measurements were repeated twice producing the same result for the homotrimer content in the proband.
To determine the proportion of type I collagen that was made up of α1(I) trimers, we prepared mixtures of fluorescently labeled, hetero- and homotrimers and compared them by gel electrophoresis to similarly prepared P2 collagen (Fig. 6, b-d). Based on the calibration curves (solid lines) built for the control mixtures of hetero- and homotrimers, homotrimers accounted for 25 ± 5% of the type I collagen secreted by the P2 cells, whether measured by comparing α1(I)/α2(I) ratios (Fig. 6c) or the proportion of cross-linked dimer species α1(I)-α1(I)/α1(I)-α2(I) (Fig. 6d).
DISCUSSION
The closely related proα chains of the fibrillar collagens are encoded by 11 homologous genes (COL1A1, COL1A2, COL2A1, COL3A1, COL5A1, COL5A2, COL5A3, COL11A1, COL11A2, COL24A1, and COL27A1) and assemble into trimeric molecules in a collagen type-specific manner. In some cells as many as six of these genes are co-expressed, and as a consequence, cells must organize the proα chains to the correct procollagen trimer in proper ratios. For type I procollagen, the predominant end-product of this assembly process comprises two proα1(I) chains and one proα2(I) chain, although small levels of proα1(I) homotrimers are also made in some tissues (25, 26). Other combinations of these chains have not been detected in tissue or in proteins from cultured cells but have been identified in cell-free systems that expressed only the recombinant C-propeptides of both type I procollagen proα chains (27).
The sequences that govern directed assembly reside in the C-propeptide domains of the proα chains. The propeptides can be looked at as having several domains: a proteolytic conversion site at which the BMP1 protein cleaves, an α-helical domain that precedes the first cysteine and may assist in trimerization and nucleation for triple helix formation (28), a set of four or three cysteine residues that are involved in interchain disulfide cross-link formation, a second set of four cysteine residues that stabilize the folded domain of each propeptide, and a region which, in contrast to each of the above, is not conserved among the chains and appears to function in specification of partners for each chain (Fig. 7). It has been suggested that molecular chaperones also play a role in correct assembly (27).
FIGURE 7.
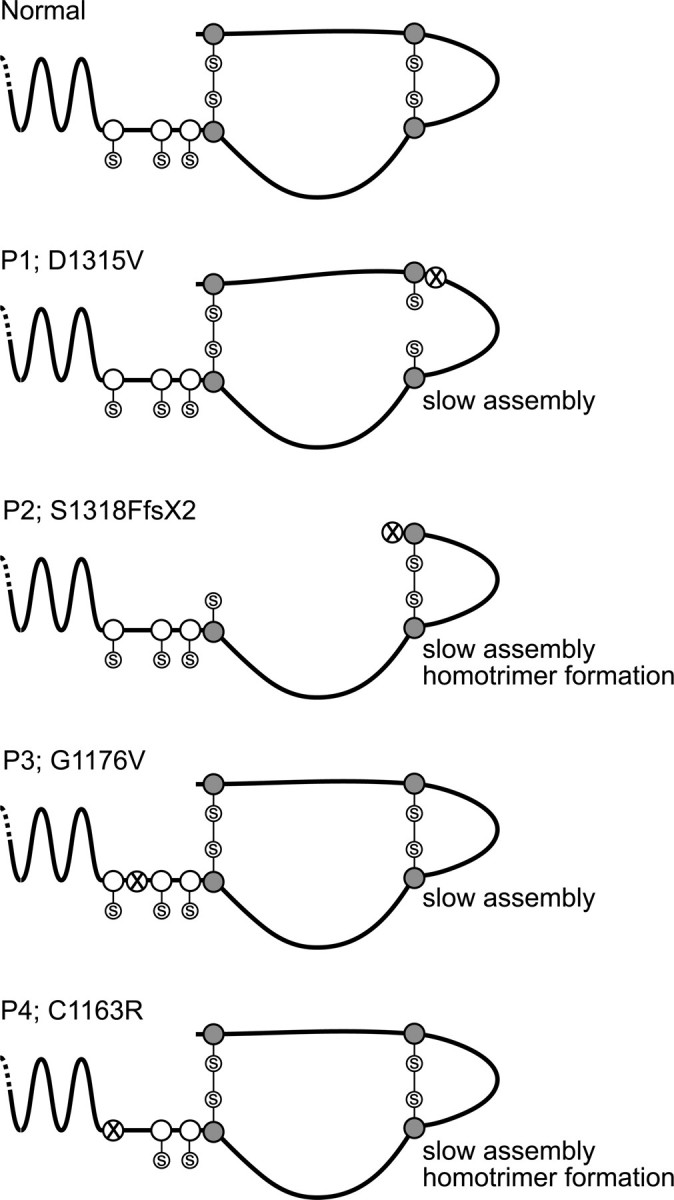
Effects of defects on folding, assembly, and propeptide cleavage. The proα2(I) C-propeptide is stabilized by cysteines (shaded circles) that form intra-chain disulfide bonds, and interaction between neighboring chains in a trimer is stabilized by cysteines (open circles) that form inter-chain disulfide bonds. The alterations of P2 and P4 perturb formation of these bonds and lead to slow assembly, abnormal formation of proα1(I) homotrimers, and slow cleavage of propeptides to form collagens. The D1315V change of P1 may hinder formation of an intra-chain bond by creating a hydrophobic pocket; slow assembly and conversion result from it. The effect of the G1176V alteration in P3 is unknown, but it leads to hindered assembly of the proα2(I) chain.
Naturally occurring mutations have been identified in the gene that encodes the proα2(I) chains in humans (18), oim/oim mice (29), and dogs (30). All are symptomatic only in the homozygote, and the heterozygotes are reported to be clinically normal. In each case these are frameshift mutations that alter the last 48 amino acids (mouse and human) or the last 30 residues (dog) and eliminate the ability of the chains to associate with proα1(I) chains. As a consequence, only proα1(I) trimers are formed, and all have a moderately severe form of osteogenesis imperfecta. In each instance, the mRNA is stable so that the proteins are probably made but very rapidly degraded, although this last point has not been conclusively demonstrated. The phenotype has been generally attributed to the production of only proα1(I) trimers, but the identification of individuals with a form of Ehlers-Danlos syndrome whose cells make only proα1(I) trimers as a consequence of unstable COL1A2 mRNA (21) raises doubt about the proposed mechanism of pathogenesis. These findings suggest that, in part, cellular alterations from a high rate of proα2(I) destruction, presumably by the proteasome (31), may contribute to the phenotype.
The mutations we describe here are diverse and lead to varied effects on procollagen assembly (Fig. 7), but all result in a similar mild OI phenotype in the heterozygous individual. In two instances, for P1 and P2, the clinical picture, with some heterogeneity, is shared among individuals in several generations. P2 is most like the mutations previously reported in that it appears to dramatically decrease the inclusion of the altered chain into trimers. In those cells a frameshift results in synthesis of a truncated protein that appears to be very unstable and poorly, if at all, incorporated into trimers. The truncation occurs just distal to the third of the four cysteine residues involved in stabilizing the intra-chain fold. Experimentally, deletion of the last 10 residues of the propeptide, which removes the last cysteine of the chain, stops chain association (32, 33). In cells from P2 the expectation would be that none of the abnormal proα2(I) chains are incorporated into trimers. If this were correct, then the relative ratio of heterotrimer to homotrimer molecules should be ∼3:2, and the α1(I) to α2(I) ratio in intact molecules should be ∼4:1. Instead, we estimated the latter ratio to be closer to 3:1, which suggests that there is some incorporation of the abnormal chain or, alternatively, that there is excess proα2(I) chains available for molecule heterotrimer generation. From the studies of the control cells in this report and studies by Chessler et al. (34) there appears to be an excess of proα2(I) in cells that could compensate for failure of abnormal chains to incorporate into trimers.
Two other mutations we identified, D1315V in P1 and C1163R in P4, are also likely to alter disulfide bonding. C1163R results in substitution of arginine for the first cysteine in the propeptide. Experimental substitution of this residue by alanine in either the proα2(I) chain or the proα1(III) chain resulted in failure of trimer formation and of interchain disulfide bond formation (35). In cells from P4 it is clear that proα2(I) chains are slow to incorporate into trimers and that proα1(I) trimers form, reflecting the functional excess of those chains. The relative amount of the proα1(I) trimers is less than in P2 suggesting that another factor may be at work. The relative amount of type I procollagen produced by those cells appears to be reduced compared with the control and the other OI cell strains. This suggests that the abnormal proα2(I) chains could “capture” proα1(I) chains during ineffective trimer formation and that those would then be degraded. In this individual, the phenotype could reflect the diminished amount of type I procollagen produced. The defect of P1 (D1315V) changes the charged aspartic acid residue to the branched hydrophobic residue valine, two amino acids N-terminal to one of the cysteine residues involved in intra-chain disulfide bond formation. This change could create a local hydrophobic pocket in which the cysteine could be hidden from access. Analysis of procollagens from the cells support this idea; they show an extra proα2(I) band with decreased mobility, which could be caused by an extended, less compact formation, owing to failure of disulfide bond formation. It appears that the G1176V substitution has an effect on helix formation similar to those seen by mutations within the helical domain itself, because overmodified α2(I) chains can be seen in both the medium and cell layer. This suggests that many of these chains are effectively incorporated into trimers, despite what appears to be a significant sequence change and, in contrast to some of the other mutations, allows helix to form, but apparently more slowly than normal so that additional modification occurs.
Unlike missense sequence alteration in the proα1(I) C-propeptide, which often leads to lethal forms of OI, the proα2(I) defects resulted in a relatively mild phenotype. The moderated severity probably reflects two factors. First, proα1(I) chains can substitute for proα2(I) chains in type I procollagen molecules (that is, proα1(I) trimers) and thus could compensate for the loss of the chain. Further, proα1(I) homotrimers are compatible with normal skeletal development and function (although when made, they can lead to skin and vessel abnormalities seen in forms of Ehlers-Danlos syndrome). Proα2(I) cannot substitute for proα1(I) chains, and so alterations in the amount or structure of those chains should be far more deleterious. Second, the proα2(I) chain represents just one of three subunits in the fully assembled trimer, whereas the proα1(I) chain occupies two of three positions. This means that, in the case of a proα1(I) defect, three-quarters of possible trimers will have one or two defective chains: a proα2(I) defect will affect only half of the possible procollagen molecules.
Acknowledgments
We thank Russell J. Fernandes for valuable discussions concerning the work; Drs. Diane Broome, Virginia V. Michels, and Joan Stoler for referring the patients to us; and the patients for their kind cooperation.
This work was supported, in whole or in part, by National Institutes of Health Grant 5R01AR041223-13. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: OI, osteogenesis imperfecta; C-propeptide; C-terminal propeptide; P, patient.
Database of Human Type I and Type III Collagen Mutations, The University of Leicester (available on the web).
References
- 1.Byers, P. H., and Cole, W. G. (2002) Osteogenesis Imperfecta. In: Royce, P., and Steinmann, B. (eds). Connective Tissue and Its Heritable Disorders: Molecular, Genetic and Medical Aspects, 2nd Ed., Wylie
- 2.Myllyharju, J., and Kivirikko, K. I. (2001) Ann. Med. 33 7-21 [DOI] [PubMed] [Google Scholar]
- 3.Lees, J. F., Tasab, M., and Bulleid, N. J. (1997) EMBO J. 16 908-916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dalgleish, R. (1997) Nucleic Acids Res. 25 181-187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dalgleish, R. (1998) Nucleic Acids Res. 26 253-255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raghunath, M., Bruckner, P., and Steinmann, B. (1994) J. Mol. Biol. 236 940-949 [DOI] [PubMed] [Google Scholar]
- 7.Kivirikko, K. I., and Pihlajaniemi, T. (1998) Adv. Enzymol. Relat. Areas Mol. Biol. 72 325-398 [DOI] [PubMed] [Google Scholar]
- 8.Vogel, B. E., Doelz, R., Kadler, K. E., Hojima, Y., Engel, J., and Prockop, D. J. (1988) J. Biol. Chem. 263 19249-19255 [PubMed] [Google Scholar]
- 9.Forlino, A., Keene, D. R., Schmidt, K., and Marini, J. C. (1998) Matrix Biol. 17 575-584 [DOI] [PubMed] [Google Scholar]
- 10.Vogel, B. E., Minor, R. R., Freund, M., and Prockop, D. J. (1987) J. Biol. Chem. 262 14737-14744 [PubMed] [Google Scholar]
- 11.Yang, W., Battineni, M. L., and Brodsky, B. (1997) Biochemistry 36 6930-6935 [DOI] [PubMed] [Google Scholar]
- 12.Chessler, S. D., and Byers, P. H. (1993) J. Biol. Chem. 268 18226-18233 [PubMed] [Google Scholar]
- 13.Oliver, J. E., Thompson, E. M., Pope, F. M., and Nicholls, A. C. (1996) Hum. Mutat. 7 318-326 [DOI] [PubMed] [Google Scholar]
- 14.Pace, J. M., Chitayat, D., Atkinson, M., Wilcox, W. R., Schwarze, U., and Byers, P. H. (2002) J. Med. Genet. 39 23-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pace, J. M., Kuslich, C. D., Willing, M. C., and Byers, P. H. (2001) J. Med. Genet. 38 443-449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bateman, J. F., Lamande, S. R., Dahl, H. H., Chan, D., Mascara, T., and Cole, W. G. (1989) J. Biol. Chem. 264 10960-10964 [PubMed] [Google Scholar]
- 17.Willing, M. C., Cohn, D. H., and Byers, P. H. (1990) J. Clin. Invest. 85 282-290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pihlajaniemi, T., Dickson, L. A., Pope, F. M., Korhonen, V. R., Nicholls, A., Prockop, D. J., and Myers, J. C. (1984) J. Biol. Chem. 259 12941-12944 [PubMed] [Google Scholar]
- 19.Bonadio, J., and Byers, P. H. (1985) Nature 316 363-366 [DOI] [PubMed] [Google Scholar]
- 20.Bonadio, J., Holbrook, K. A., Gelinas, R. E., Jacob, J., and Byers, P. H. (1985) J. Biol. Chem. 260 1734-1742 [PubMed] [Google Scholar]
- 21.Schwarze, U., Hata, R., McKusick, V. A., Shinkai, H., Hoyme, H. E., Pyeritz, R. E., and Byers, P. H. (2004) Am. J. Hum. Genet. 74 917-930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Korkko, J., Annunen, S., Pihlajamaa, T., Prockop, D. J., and Ala-Kokko, L. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 1681-1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chamberlain, J. R., Schwarze, U., Wang, P. R., Hirata, R. K., Hankenson, K. D., Pace, J. M., Underwood, R. A., Song, K. M., Sussman, M., Byers, P. H., and Russell, D. W. (2004) Science 303 1198-1201 [DOI] [PubMed] [Google Scholar]
- 24.Fisher, L. W., Stubbs, J. T., 3rd, and Young, M. F. (1995) Acta Orthop. Scand. Suppl. 266 61-65 [PubMed] [Google Scholar]
- 25.Jimenez, S. A., Bashey, R. I., Benditt, M., and Yankowski, R. (1977) Biochem. Biophys. Res. Commun. 78 1354-1361 [DOI] [PubMed] [Google Scholar]
- 26.Uitto, J. (1979) Arch. Biochem. Biophys. 192 371-379 [DOI] [PubMed] [Google Scholar]
- 27.Alvares, K., Siddiqui, F., Malone, J., and Veis, A. (1999) Biochemistry 38 5401-5411 [DOI] [PubMed] [Google Scholar]
- 28.McAlinden, A., Smith, T. A., Sandell, L. J., Ficheux, D., Parry, D. A., and Hulmes, D. J. (2003) J. Biol. Chem. 278 42200-42207 [DOI] [PubMed] [Google Scholar]
- 29.McBride, D. J., Jr., and Shapiro, J. R. (1994) Genomics 20 135-137 [DOI] [PubMed] [Google Scholar]
- 30.Campbell, B. G., Wootton, J. A., Macleod, J. N., and Minor, R. R. (2001) J. Bone Miner. Res. 16 1147-1153 [DOI] [PubMed] [Google Scholar]
- 31.Fitzgerald, J., Lamande, S. R., and Bateman, J. F. (1999) J. Biol. Chem. 274 27392-27398 [DOI] [PubMed] [Google Scholar]
- 32.Doyle, S. A., and Smith, B. D. (1998) J. Cell Biochem. 71 233-242 [PubMed] [Google Scholar]
- 33.Lim, A. L., Doyle, S. A., Balian, G., and Smith, B. D. (1998) J. Cell Biochem. 71 216-232 [PubMed] [Google Scholar]
- 34.Chessler, S. D., Wallis, G. A., and Byers, P. H. (1993) J. Biol. Chem. 268 18218-18225 [PubMed] [Google Scholar]
- 35.Lees, J. F., and Bulleid, N. J. (1994) J. Biol. Chem. 269 24354-24360 [PubMed] [Google Scholar]