

Abstract
SOK1 is a Ste20 protein kinase of the germinal center kinase (GCK) family that has been shown to be activated by oxidant stress and chemical anoxia, a cell culture model of ischemia. More recently, it has been shown to be localized to the Golgi apparatus, where it functions in a signaling pathway required for cell migration and polarization. Herein, we demonstrate that SOK1 regulates cell death after chemical anoxia, as its down-regulation by RNA interference enhances cell survival. Furthermore, expression of SOK1 elicits apoptotic cell death by activating the intrinsic pathway. We also find that a cleaved form of SOK1 translocates from the Golgi to the nucleus after chemical anoxia and that this translocation is dependent on both caspase activity and on amino acids 275-292, located immediately C-terminal to the SOK1 kinase domain. Furthermore, SOK1 entry into the nucleus is important for the cell death response since SOK1 mutants unable to enter the nucleus do not induce cell death. In summary, SOK1 is necessary to induce cell death and can induce death when overexpressed. Furthermore, SOK1 appears to play distinctly different roles in stressed versus non-stressed cells, regulating cell death in the former.
Reactive oxygen species (ROS)6 have been reported to mediate the cell death seen in a variety of pathophysiological conditions including ischemia, ischemia followed by reperfusion, neurodegenerative disorders, and cancer (1-3). Cells react to elevated levels of ROS in various ways depending on the status of the cell itself and the severity of the insult. In general, cells can either activate mechanisms that minimize the damage induced by ROS or undergo programmed cell death or necrotic cell death if the damage is too severe to be repaired. A large number of molecules have been implicated in the stress response to ROS (4), but much remains unknown, particularly about the proximal effectors regulating the cell response to this type of stress.
The Ste20 proteins were first described in yeast as the upstream components of signal transduction pathways (5, 6). In mammals, more than 30 members have been described to date, and they fall into two families (7, 8), the p21-activated protein kinase family and the germinal center kinase (GCK) family. Based on phylogenetic analysis, Dan et al. (8) further classified these two families into two distinct subfamilies for the p21-activated protein kinases and eight subfamilies for the GCKs These proteins are implicated in a wide range of biological responses such as regulation of cell proliferation, cell death, and cytoskeleton rearrangements in response to either extracellular stimuli or various forms of cellular stress. Some family members activate known mitogen-activated protein kinase pathways (9, 10), whereas for others uncertainty remains as to whether they activate these core signal transduction modules (11, 12).
The proteins belonging to the GCK-II and -III subfamilies are closely related and have recently been dubbed mammalian sterile twenty-like (MST) kinases. The Mst1 and Mst2 proteins belong to the GCK-II subfamily, whereas SOK1/YSK1, Mst3, and Mst4 belong to the GCK-III subfamily (8, 13). Enforced overexpression of Mst1, Mst2, and Mst3 has been reported to induce apoptotic cell death (10, 14-17). It has also been shown that these proteins are cleaved by caspases in response to certain types of stress. Mst1 and Mst3 have known nuclear localization signals (NLS) and nuclear export signals (14, 18, 19). After caspase cleavage, the nuclear export signal is separated from the rest of the kinase, which can then enter the nucleus driven by its NLS. Further levels of regulation have been reported for Mst1 and 2 that control their apoptotic potential (20, 21). Physiological substrates and effectors for these kinases have only recently been reported, but a wide variety of them have been proposed in the last years. For instance, Mst1 has been shown to phosphorylate histone H2B, and this correlates with chromatin condensation seen in apoptotic cells, suggesting a possible explanation of how this protein is involved in cell death. More recently, another substrate of Mst1 has been identified. Upon oxidative stress in mammalian neurons, Mst1 phosphorylates the FOXO3 transcription factor, which translocates to the nucleus to play a role in oxidative stress-induced cell death (22). The finding that Mst1/2 and Mst3 phosphorylate the serine/threonine kinases Lats1 and NDR, respectively, places these proteins in yet another signal transduction pathway (23, 24). Although the functional significance of this pathway remains poorly understood in mammals, interestingly, the orthologs in Drosophila (Hippo and Warts) have been shown to coordinate both cell division and cell death (25-29). Further suggesting that these proteins are implicated in apoptotic cell death is the fact that they are activated by stress stimuli and, for Mst1, by the Fas death-inducing receptor (10).
The GCK-III subfamily member SOK1 is activated by oxidative stress as its name implies (Ste-20 oxidant stress response kinase 1) (30, 31). This suggests that it may play a role in the cellular response to this kind of stress. However, the consequences of this activation are unclear (30, 32). Recently SOK1 has been reported to play a role in cytoskeletal rearrangements as its activity is necessary to redirect the Golgi apparatus toward the leading edge of the migrating cell (33). Thus, although the role of SOK1 in regulating specific functions in non-stressed cells is beginning to be understood, what role, if any, the marked activation of this kinase by extreme stress might play in cell response to these stresses is not known.
Herein, we demonstrate that SOK1 drives the apoptotic response to ROS and chemical anoxia, a model of ischemia characterized by marked ROS production and severe ATP depletion. Under these conditions SOK1 dissociates from the Golgi and translocates to the nucleus, a process that depends on caspase activation and on the integrity of amino acids 275-292. Thus, SOK1 has distinctly different functions depending on the redox status and energy status of the cell, regulating cell motility in the non-stressed cell via effects at the Golgi and regulating cell death in the stressed cell via effects in the nucleus.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies—Hydrogen peroxide was purchased from Calbiochem. Protein-G-agarose was purchased from Roche Diagnostics. Caspase inhibitors z-VAD-fmk and Ac-DEVD-CHO were from Sigma and Alexis Biochemicals, respectively. Leptomycin B was purchased from Sigma. The lactate dehydrogenase release detection kit was purchased from Roche Diagnostics. The [γ-32P]ATP and the l-[35S]methionine were purchased from Amersham Biosciences. The transcription/translation system was purchased from Promega. All restriction enzymes were from New England Biolabs. Monoclonal antibody to the FLAG tag, monoclonal anti-Golgi-58K protein/formiminotransferase cyclodeaminase clone 58K-9, and anti-goat AlexaFluor 488 were from Sigma Aldrich. Antibodies for poly(ADP-ribose) polymerase (PARP; sc-7150), SOK1 (sc-6865), and Sp1 (sc-59) were from Santa Cruz Biotechnology. The antibody for cytochrome c and the purified active recombinant human caspase 3 (CPP32) were from BD Biosciences, and the antibody for GAPDH (6C5) was from Calbiochem. AlexaFluor 594 goat anti-mouse IgG (H+L) was from Molecular Probes.
DNA Constructs—All plasmids were constructed using standard molecular biology techniques. Point mutants of SOK1 and SOK1ΔC were constructed using the QuikChange site-directed mutagenesis kit (Stratagene) following the manufacturer's instructions. The oligonucleotides designed for the different mutants were purchased from Sigma Aldrich or Thermo, and their sequence is available on request. The plasmid encoding the small hairpin RNA (shRNA) for SOK1 was constructed ligating a double-stranded oligonucleotide (sequence available on request) to the pSUPER plasmid (34). Sok1-R (Rescue) was generated by creating six silent base pair mutations into the wild-type cDNA encoding Sok1 using the QuikChange site-directed mutagenesis kit (Stratagene). The SOK1-expressing adenovirus was made with the AdTrack system, in which SOK1 is encoded downstream of one CMV promoter, and GFP is encoded downstream of a second CMV promoter. The control virus expresses LacZ and GFP from the two promoters. Viruses were propagated in HEK293 cells.
Cell Culture and Treatments—HeLa, HEK293, SaOS2, and COS-7 cells were cultured under standard conditions. For experiments with caspase inhibitors, cells were cultured in the presence of 50 μm z-VAD-fmk or 50 μm Ac-DEVD-CHO.
For chemical anoxia, cells were treated with sodium cyanide (5 mm) and 2-deoxyglucose (5 mm) in KRH medium (115 mm NaCl, 3.6 mm KCl, 1.3 mm KH2PO4, 25 mm NaHCO3, 1 mm CaCl2, 1 mm MgCl2). Treatment with H2O2 was at a concentration of 500 μm at the times specified in each individual experiment.
Transfections and Infections—HEK293 and SaOS2 cells were transfected using the calcium phosphate protocol. COS7 cells were transfected using the DEAE-dextran technique as described (35). Infection with adenovirus in HeLa cells was performed with standard protocols. To ensure moderate levels of expression of the proteins, the multiplicity of infection was 1.
Determination of Cell Viability—Cell viability was determined by trypan blue exclusion assay. Dead cells (blue) are expressed as a percent of total cells.
Western Blot Analysis—Cells were lysed in radioimmune precipitation assay buffer. For SOK1 or M2 detection, cells were lysed in SAPK buffer (20 mm HEPES, pH 7.4, 50 mm β-glycerophosphate, 1 mm sodium orthovanadate, 1% Triton X-100, 10% glycerol, 1 mm EGTA, 1 mm dithiothreitol, 400 μm phenylmethylsulfonyl fluoride, 2 μm leupeptin, 2 μm pepstatin, 10 units/ml Trasylol) (36). Cell extracts were resolved on SDS-PAGE, and proteins were analyzed by Western blot on nitrocellulose membrane. Antigen-antibody complexes were detected with alkaline phosphatase-conjugated secondary antibody (TROPIX, Applied Biosystems).
Cytochrome c Release—Detection of cytochrome c release was performed as described (37).
Determination of Lactate Dehydrogenase Release—Cell cytotoxicity was measured according to the manufacturer instructions (cytotoxicity detection kit, Roche Diagnostics). Briefly, HEK293 cells transfected either with sh-luci or sh-SOK1 were preincubated for 12 h in assay medium (Dulbecco's modified Eagle's medium + 1% fetal bovine serum). Cells were then left untreated or treated with 1% Triton X-100, 5 mm sodium cyanide and 2-deoxyglucose or 1 μm staurosporine, respectively. After 6 h of treatment, 100 μl of supernatant of each well were removed and transferred into an optically clear 96-well flat-bottom microplate. Then, 100 μl of reaction mixture was added to each well and incubated for up to 30 min at 20 °C. The reaction was stopped by the addition of 50 μl/well of 1 n HCl. Lactate dehydrogenase release was determined using an enzyme-linked immunosorbent assay reader at 455 nm, with 595 nm as the reference wavelength.
Preparation of Nuclear and Cytoplasmic Extracts—Nuclear and cytoplasmic extracts were prepared from HeLa cells essentially as described (38) using NaCl to a final concentration 300 mm to extract nuclear proteins.
Kinase Assays—Extracts were immunoprecipitated with anti-M2 tag monoclonal antibody or anti-SOK1 antibody, and kinase assays were performed as described (30).
Immunofluorescence—HEK293 cells were fixed in 50% methanol, 50% acetone and treated with the appropriate antibodies under standard conditions. Nuclei were stained with Hoechst 33342 (5 μm). The coverslips were mounted in aqueous medium with anti-fading agents (Gel/Mount).
Microscopy—The expression of GFPSOK1 and its mutants in living cells was monitored using an inverted fluorescence microscope (Olympus IX70) with a 60×/0.65 objective. Bright field visualization was performed in the same microscope using a Hoffman modulation contrast system. Immunofluorescence of endogenous SOK1 was visualized using a Leica AF6000 series microscope equipped with an HCX PL APO 63× objective with a NA of 1.40. Photoimages were acquired using an Olympus DP50 digital camera. Adobe Photoshop software was used to cut, resize, and mount the photographs in the figures.
In Vitro Translation and Caspase Cleavage—Mst3 and Sok1 were translated in vitro using a coupled transcription-translation system (Promega) in the presence of l-[35S]methionine (Amersham Biosciences). After translation they were incubated for 2 h al 37 °C with 200 ng of caspase 3 (BD Biosciences) in the presence or absence of 1 μm z-VAD-fmk. Reactions were stopped by the addition of 5× Laemmli sample buffer and subjected to SDS-PAGE before drying and autoradiography.
Statistical Analysis—A non-parametric Mann-Whitney test was done to perform a statistical analysis of the data.
RESULTS
SOK1 Induces Apoptotic Cell Death—SOK1 is activated by ROS and chemical anoxia. However, the effects of SOK1 on the biology of the cell are not known. As a first approach toward understanding the cellular responses elicited by SOK1, we studied the effect of enforced expression of the protein. We transfected either HEK293, SaOS2, or COS-7 cells with full-length SOK1 or infected HeLa cells with recombinant adenoviruses encoding GFP and SOK1 in independent ORFs (Ad-SOK1). In all four cell types, overexpression of SOK1 induced cell death as measured by trypan blue exclusion (Fig. 1A). These results are consistent with a role for SOK1 in cell death in a variety of cell types. Fig. 1A also shows that the degree of cell death in each cell line was different, which correlated with the expression levels of the overexpressed protein in the different cell types (data not shown). To verify a correlation between the level of SOK1 and the degree of cell death, we transfected increasing amounts of full-length SOK1 in HEK293 cells and assessed the number of non-viable cells. As shown in Fig. 1B, the percentage of non-viable cells is higher as the level of the overexpressed SOK1 increases, confirming that the level of SOK1 protein correlates with its capacity to induce cell death.
FIGURE 1.

Overexpression of SOK1 induces apoptotic cell death. A, overexpression of SOK1 induces cell death. HEK293, SaOS2, or COS7 cells were transfected with pCMV5-SOK1 (SOK1) or pCMV5 plasmid (vector), and HeLa cells were infected with either an adenovirus expressing GFP (Ad-GFP), or an adenovirus expressing both SOK1 and GFP (Ad-SOK1). The percentage of dead cells was determined by trypan blue exclusion 72 h after transfection of HEK293, SaOS2, and COS7 cells and 48 h after infection of HeLa cells. Shown is the mean and S.D. of four independent experiments. *, p < 0.05 versus empty control. B, the levels of overexpressed SOK1 correlate with its cell death-inducing capacity. HEK293 cells were transfected with increasing amounts (0, 1.25, 2.5, 3.75, or 5 μg) of pCMV5-M2SOK1 (SOK1). The percentage of cell death was determined by trypan blue exclusion 72 h after transfection. GAPDH is shown as a loading control. C, overexpression of SOK1 induces chromatin condensation and membrane blebbing. HeLa cells were infected with Ad-SOK1, which also expresses GFP, and visualized in vivo. GFP was used to score for successfully infected cells (left panel). Chromatin condensation was visualized by Hoechst 33342 (central panel), and cell morphology was evaluated using Hoffman interference microscopy (right panel). GFP-positive cells with apoptotic features are marked with arrows. D, SOK1 induces cytochrome c release to the cytoplasm. HEK293 cells were transfected with control or a SOK-1 expressing plasmid and either incubated with 500 μm H2O2 or left untreated. Cytoplasmic cytochrome c (cyt. c) was determined by Western blot. E, SOK1-induced cell death is caspase-dependent. HEK293 cells were transfected with empty plasmid (pCMV5) or pCMV5-SOK1 (SOK1) and treated with a caspase inhibitor (z-VAD-fmk or Ac-DEVD-CHO) or vehicle. The percentage of non-viable cells was determined by trypan blue exclusion. Shown is the mean and S.D. of four independent experiments. *, p < 0.05 for SOK1 + caspase inhibitors versus SOK1 alone. F, PARP is cleaved during SOK1-induced cell death. Extracts were prepared from HeLa cells infected with control or SOK1-expressing adenovirus for the times indicated. Western blots were performed for SOK1 and PARP. The arrow marks the predicted position of caspase-cleaved PARP.
To determine the type of cell death induced by SOK1, we examined the morphological and biochemical features of SOK1-overexpressing cells. Cells successfully transduced with Ad-SOK1, which are readily identified by co-expression of GFP, demonstrated condensed chromatin as determined by Hoechst 33342 staining (Fig. 1C, middle panel) and also show membrane blebbing and shrinkage when observed in bright field (right panel). These characteristics are hallmarks of apoptotic cell death and were not observed in cells transfected with control Ad-GFP (data not shown). These data suggest that SOK1 induces an apoptotic form of cell death.
At the molecular level one of the hallmarks of apoptosis is release of cytochrome c from the mitochondrial intermembrane space to the cytoplasm, triggering activation of the intrinsic cell death pathway. We determined whether cytochrome c is released in response to expression of SOK1. In HEK293 cells, expression of SOK1 increased the amount of cytoplasmic cytochrome c (Fig. 1D) to the same magnitude as treatment with H2O2. To further test if SOK1 induces an apoptotic type of cell death, we examined whether caspase activity was required for SOK1-induced death using z-VAD-fmk, a broad-spectrum caspase inhibitor, in SOK1-transfected HEK293 cells. When cell death was measured by trypan blue exclusion assay, the ability of SOK1 to induce cell death was significantly reduced by z-VAD-fmk (Fig. 1E). A different caspase inhibitor, Ac-DEVD-CHO, had similar effects (Fig. 1E). This indicates that a significant fraction of SOK1-induced cell death is caspase-dependent. Consistent with this, PARP, which is a biological substrate of caspases, is cleaved in Ad-SOK1-infected cells (Fig. 1F), confirming that caspases are activated in the course of SOK1-induced death. In summary, expression of SOK1 induces cell death with apoptotic features and is dependent at least in part on activation of caspases.
SOK1 Mediates Cell Death Induced by Different Stress Insults—To confirm a role for SOK1 in cell death, we down-regulated SOK1 levels by RNA interference. Specifically, we silenced SOK1 using a shRNA expressed from the pSUPER vector. As a control we transfected cells with a shRNA directed against firefly luciferase. Five days after transfection of HEK293 cells with SOK1 shRNA, the levels of SOK1 were less than 50% that of those in cells transfected with control shRNA (Fig. 2A). In untreated cells knockdown of SOK1 had no effect on cell viability, but when chemical anoxia was induced in these cells with sodium cyanide and 2-deoxyglucose (CN+DOG), cell death was significantly reduced in cells treated with SOK1 shRNA (Fig. 2B). Despite the extreme stress of prolonged chemical anoxia, knockdown of SOK-1 protected cells for up to 24 h.
FIGURE 2.

Down-regulation of SOK1 protects cells from apoptosis induced by chemical anoxia. A, down-regulation of SOK1 by a hairpin RNA. HEK293 cells were transfected with a pSUPER-derived plasmid encoding a small hairpin RNA directed against SOK1 (sh-SOK1) or a control shRNA directed against firefly luciferase (sh-luci). 5 days later extracts were prepared, and a Western blot was performed to evaluate SOK1 down-regulation. GAPDH Western blot is shown as a loading control. B, inhibition of endogenous SOK1 delays cell death induced by chemical anoxia. HEK293 cells transfected with sh-SOK1 or sh-luci were either left untreated (C) or treated with sodium cyanide and 2-deoxyglucose (CN+2-DG) for the times indicated, and the percentage of non-viable cells was determined by the trypan blue exclusion assay. Shown is the mean ± S.D. of four independent experiments. *, p < 0.05 versus treated cells transfected with control shRNA. C, the SOK1R mutant is refractory to SOK1 shRNA. Cells transfected with sh-SOK1 or sh-luci were transfected again with either SOK1 wild-type or the shRNA-resistant SOK1R mutant. A Western blot was performed to assess the effect of sh-SOK1 on the different mutants of SOK1. D, the effects of sh-SOK1 are mediated by down-regulation of SOK1. Cells were then subjected to chemical anoxia for 6 h, and the percentage of dead cells assessed by trypan blue exclusion. Shown is the mean and S.D. of four independent experiments. p > 0.05 (N.S.) and p < 0.05 (*) versus untreated cells. E, SOK1 knockdown protects from the apoptotic cell death induced by chemical anoxia. HEK293 cells transfected with sh-SOK1 or sh-luci were either left untreated or treated with sodium cyanide and 2-deoxyglucose for 6 h. Cytochrome c (cyt. c) release was determined by Western blotting. GAPDH is shown as a loading control. F, SOK1 knockdown does not protect from the necrotic cell death induced by chemical anoxia. HEK293 cells transfected with sh-SOK1 or sh-luci were either left untreated or treated with sodium cyanide and 2-deoxyglucose for 6 h, and lactate dehydrogenase levels in the medium were measured. Triton X-100 was used as positive control, and staurosporine was used as a negative control of necrotic cell death. N.S., non-significant differences (p > 0.05) of treated cells with SOK1 shRNA versus treated cells with control shRNA.
To demonstrate the specificity of the SOK1 shRNA, we performed a rescue experiment. We generated a mutated form of SOK1 (SOK1-R) that has the same amino acid sequence as SOK1 but is resistant to sh-SOK1 (Fig. 2C). SOK1-R, but not SOK1, significantly reduced the ability of SOK1 shRNA to protect cells from chemical anoxia-induced cell death (Fig. 2D). Thus, SOK1 shRNA inhibits chemical anoxia-induced cell death via specific knockdown of SOK1 rather than off-target effects of the shRNA construct.
The role of SOK1 in cell death is not restricted to that induced by chemical anoxia. Death induced by osmotic stress was clearly diminished in cells transfected with sh-SOK1 (supplemental Fig. S1). This correlates with the delayed activation of SOK1 (2-6 h) after osmotic stress. Osmotic stress has been shown to induce reactive oxygen species (39), and this provides a possible mechanism for activation of SOK1 in this situation. The above results indicate that, besides being able to induce cell death upon overexpression, endogenous SOK1 plays a significant role in cell death induced by at least two different forms of extreme cellular stress.
Enforced expression of SOK1 induces an apoptotic form of cell death, but the knockdown of SOK1 protects cells from types of insults that can cause either necrotic or apoptotic cell death (40). To determine whether the effects of silencing SOK1 protects the cells from one or both death mechanisms, we quantified the magnitude of necrosis elicited by chemical anoxia in cells that either express normal levels of SOK1 or in which the levels of the protein were knocked-down. In our cellular system chemical anoxia resulted in a very modest induction of necrosis, as judged by lactate dehydrogenase release by injured cells (Fig. 2F), and SOK1 down-regulation had no effect on this. In contrast, chemical anoxia resulted in cytochrome c release from the mitochondria (Fig. 2E). This together with the absence of plasma membrane injury under the treatment suggests that at least part of the cell death induced by this stress is through an apoptotic mechanism. Significantly, the release of cytochrome c in response to chemical anoxia is clearly diminished in cells transfected with the SOK1 shRNA (Fig. 2E), showing that SOK1 has a role in this induction.
SOK1 Enters the Nucleus in Response to Cellular Stress—Preisinger et al. (33) recently reported that SOK1 localizes to the Golgi apparatus and is required for cells to reorientate the Golgi toward the direction of forward movement. To reconcile this observation with ours (i.e. that SOK1 induces cell death), we hypothesized that SOK1 may have dual roles depending on the cellular status. We first asked whether this might be reflected in its intracellular location, as described for other Mst kinases. To address this issue we performed immunofluorescent staining of endogenous SOK1 in control cells and upon induction of chemical anoxia. As shown in Fig. 3, SOK1 localizes preferentially to the Golgi apparatus in control cells, as its staining aligns with Golgi-58K, a protein known to localize at the Golgi. In contrast, SOK1 becomes uniformly distributed throughout the cell (pancellular), including in the nucleus, after 6 h of treatment. Interestingly, Golgi-58Kalsobecomes delocalized, possibly reflecting the disassembly of the Golgi upon extreme stresses. However, in contrast to SOK1, Golgi-58K is still located mainly in the cytoplasm. Even GAPDH, a protein that has been shown to translocate to the nucleus in many forms of apoptotic cell death (41), remains in the cytoplasm after CN/2DOG at a time when SOK1 is pancellular and enters the nucleus (supplemental Fig. S2). This suggests that stress-induced nuclear entry of SOK1 is not due to a generalized breakdown of nuclear membrane integrity but is characteristic of this protein and may be important for its biology.
FIGURE 3.

SOK1 locates to the Golgi in control cells and enters the nucleus upon chemical anoxia. HEK293 cells were left untreated (Control) or incubated for 6 h with sodium cyanide and 2-deoxyglucose. They were then processed for immunofluorescence and stained for SOK1 (green), Golgi-58K (red), and DNA (Hoechst, blue). CN+2-DG, sodium cyanide and 2-deoxyglucose.
To study the mechanisms regulating entry of SOK1 into the nucleus, we evaluated the cellular localization of different mutants of the protein (Fig. 4A). We constructed fusion proteins of several mutants of SOK1 with GFP and visualized their cellular distribution in HEK293 cells by direct fluorescence. To avoid cell death induced by transfection of the various mutants of SOK1 (see below), we used lower amounts of SOK1-expressing plasmids and analyzed cells 24 h after transfection (death induced by SOK1 in these cells is maximal at 72 h posttransfection). We found that wild-type SOK1 fused to GFP is predominantly cytoplasmic and largely excluded from the nucleus in control cells (Fig. 4C), although a small percentage of cells shows a nuclear location of GFP-SOK1. Importantly, GFP-SOK1 becomes pancellular with significant nuclear accumulation after treatment with CN+DOG. The percent of cells with pancellular SOK1 begins to increase significantly 90 min after treatment (not shown), and more than 50% of GFP-SOK1-expressing cells have nuclear-localized SOK1 at 3 h and ∼90% at 6 h after treatment (Fig. 4B). Thus, under these conditions, GFP-SOK1 closely reflects the behavior of the endogenous protein and demonstrates nuclear entry in response to stress.
FIGURE 4.

Structural requirements of SOK1 for nuclear entry. HEK293 cells were transfected with GFP-SOK1 fusion proteins and either wild-type (WT) SOK1 or the mutants SOK1K49R, SOK1T174A, and SOK14A. 24 h after transfection cells were incubated with sodium cyanide and 2-deoxyglucose (treated) or left untreated (control). A, schematic representation of SOK1 and the mutants used in this study. B, percentage of cells in which the different mutants of GFP-SOK1 show a nuclear signal. At least 100 GFP-positive cells per mutant and time point were scored for GFP nuclear positivity. Shown is the mean and S.D. of the percentage of cells positive for nuclear GFP from three independent experiments. C, control; T3 and T6, 3 and 6 h after treatment. p > 0.05 (N.S.) and p < 0.05 (*) versus wild-type SOK1. C, images of representative fields of control and treated cells transfected with the different GFP-SOK1 mutants in control cells and cells treated for 6 h. The bar represents 10 μm.
When we constructed two kinase-inactive mutants, GFP-SOK1K49R (with the lysine essential for its catalytic activity mutated) and GFP-SOK1T174A (mutated in the T-loop), we found that both could enter the nucleus with the same efficiency as the wild-type protein (Fig. 4, B and C) despite having no detectable kinase activity (Fig. 5B). Thus, kinase activity is not necessary for the nuclear translocation of SOK1 upon chemical anoxia.
FIGURE 5.

Structural requirements of cell death induction by SOK1. M2-flagged versions of wild-type (WT) SOK1 and the mutants SOK1K49R, SOK1T174A, and SOK14A were transfected into HEK293 cells. A, the levels of expression of the different mutants were determined by Western blotting using anti-M2 antibody. B, the kinase activity of each mutant toward the unspecific substrate myelin basic protein (MBP) was determined after immunoprecipitation with the M2 antibody. C, the ability of each mutant to induce cell death was assessed by determining the percentage of non-viable cells by trypan blue exclusion. Shown is the mean and S.D. of four independent experiments. N.S., non-significant differences versus wild type SOK1 (p = 0.190 for K49R and p = 0.064 for T174A). *, p < 0.01 versus wild type SOK1.
We next explored the role of amino acids 275-302 of SOK1, immediately C-terminal to the kinase domain. Preisinger et al. (33) have shown that this region together with a functional kinase domain, is necessary for binding of SOK1 to the Golgi protein GM130, an interaction necessary for full kinase activity of SOK1. However, amino acids 275-292 of SOK1 are also highly homologous to residues 279-296 of Mst3, a region that has been shown to act as a NLS. Therefore, to determine whether nuclear entry of SOK1 might be dependent on this sequence, we mutated residues 275, 276, 288, and 289 to alanines. This mutant of SOK1 (GFP-SOK14A) has a low but detectable kinase activity, as expected from its inability to bind to GM130 (see Fig. 5B for its kinase activity and supplemental Fig. S3 for binding to GM130). Significantly, it is greatly impaired in its nuclear entry after treatment with CN+DOG, as even after 6 h of treatment, greater than 60% of cells exclude it from the nucleus. Because the kinase activity of SOK1 is not essential for its nuclear entry, we conclude that the sequence in question is important for the nuclear entry of SOK1 independently of its effect on SOK1 kinase activity.
We next transfected the various mutants tagged with M2 epitope into HEK293 cells. Immunoblotting confirmed equivalent levels of expression of each mutant (Fig. 5A). We also immunoprecipitated the transfected protein and assayed kinase activity measured as phosphorylation of myelin basic protein (MBP; Fig. 5B). When we measured the induction of cell death, we found that both of the kinase-inactive SOK1 mutants (SOK1K49R and SOK1T174A) could induce cell death, albeit with a reduced efficiency when compared with the wild-type protein, demonstrating that in the context of a full-length protein and with the expression levels achieved in this experiment, the kinase activity of SOK1 may play a role but is not necessary for induction of cell death. In contrast, induction of cell death was clearly impaired in the case of SOK14A despite the fact that kinase activity of this mutant, although significantly reduced, was greater than SOK1K49R and SOK1T174A. These findings suggest that nuclear entry plays a role in the induction of cell death by SOK1.
Nuclear Entry of SOK1 Is Dependent on Caspases—Taken together, the above results suggest that SOK1 enters the nucleus after treatment with CN+DOG driven by a nuclear localization signal placed C-terminal to its kinase domain. This is reminiscent of other Mst kinases like Mst1, Mst2, or Mst3. In these proteins their C terminus inhibits nuclear translocation of the kinase until phosphorylation or caspase cleavage of the protein drives nuclear entry of the protein. Thus, we hypothesized that caspase-mediated cleavage of SOK1 could allow the kinase to translocate to the nucleus. If so, the nuclear entry of SOK1 after chemical anoxia should be caspase-dependent. We studied the localization of SOK1 in GFP-SOK1-transfected HEK293 cells that had been pretreated with vehicle versus the caspase inhibitor z-VAD-fmk (50 μm for 180 min) and then were treated with vehicle versus CN+DOG. Pretreatment with z-VAD-fmk efficiently inhibited the nuclear entry of SOK1 in CN+DOG-treated cells (Fig. 6, A and B). Thus, we concluded that SOK1 entry is indeed dependent on caspase activity.
FIGURE 6.
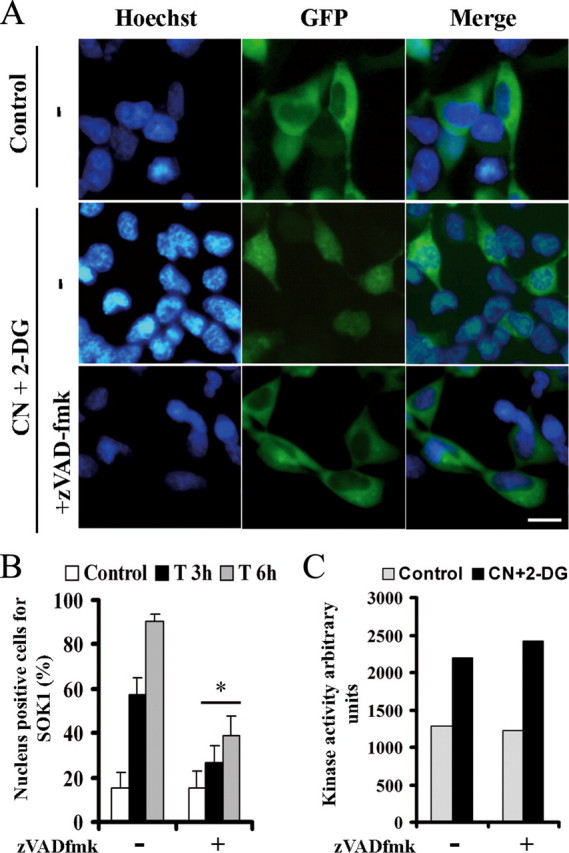
Nuclear entry of SOK1, but not its early activation, is dependent on caspases. A, HEK293 cells were transfected with a plasmid expressing GFP-SOK1. 24 h later they were left untreated (Control) or incubated with sodium cyanide and 2-deoxyglucose for 3 or 6 h either alone (-) or with a preincubation of the caspase inhibitor z-VAD-fmk. DNA staining and GFP of representative fields of cells subject to each treatment for 6 h are shown. B, the percentage of cells with the fusion protein excluding nucleus were counted and represented as the mean and S.D. of four independent experiments. *, p < 0.05 versus cells subject to chemical anoxia without z-VAD-fmk. C, cells were either left untreated (Control) or subjected to chemical anoxia for 40 min sodium cyanide and 2-deoxyglucose either alone or with a preincubation with the z-VAD-fmk caspase inhibitor. Endogenous SOK1 was immunoprecipitated and its kinase activity assayed.
The kinase activity of SOK1 is increased at early times after some forms of extreme stresses such as chemical anoxia or ROS generation (31). We wanted to know whether caspase inhibition could also affect this stimulation. Fig. 6C shows that the kinase activity of SOK1 increased after 40 min of chemical anoxia, and this was not affected by pretreatment with the caspase inhibitor z-VAD-fmk. This suggests that the early stimulation of the kinase activity of SOK1 does not depend on caspase activity and occurs independent of nuclear translocation.
We wanted to confirm the nuclear localization of SOK1 by an alternative technique. To do so, we prepared cytoplasmic and nuclear extracts from control and CN+DOG-treated HeLa cells and determined the localization of the endogenous protein in both extracts by Western blot. In control cells, an immunoreactive band that corresponds in molecular mass to the full-length protein (48 kDa) appeared in the cytoplasm (Fig. 7A, first lane), and no or very little SOK1 appeared in the nucleus. In treated cells, the amount of the 48-kDa band decreased in cytoplasmic extracts compared with control cells. In parallel, a band of ∼35 kDa appeared in the nuclear extracts from treated cells, and this band was not present in the cytoplasm or in the nucleus of non-treated cells. Caspase inhibition reduced the intensity of the nuclear band in treated cells and prevented the reduction in the cytoplasmic band. Again, GAPDH remained in the cytoplasm in all the conditions tested. This is consistent with caspase-dependent nuclear entry of SOK1 under stress conditions but, importantly, suggests that it is a modified form of SOK1 that enters the nucleus. Supporting this conclusion, when we overexpressed M2-SOK1, a 35-kDa band also appeared in the nuclear fraction on Western blotting with anti-M2 antibody, and this band is absent when cells are pretreated with caspase inhibitor (supplemental Fig. S4).
FIGURE 7.

SOK1 enters the nucleus in cells subject to chemical anoxia and is a substrate of caspases. A, a low molecular weight form of SOK1 enters the nucleus upon chemical anoxia. HEK293 cells were either left untreated or treated with sodium cyanide and 2-deoxyglucose for 3 h in the absence or presence of 50 μm z-VAD-fmk caspase inhibitor. Nuclei were separated from cytoplasm, extracts were prepared, and a Western blot against SOK1 was performed. The asterisk marks a band appearing in the nucleus of treated cells in a caspase-dependent manner. Western blots for the nuclear protein SP1 and the cytoplasmic protein GAPDH were performed to ensure proper separation of nucleus and cytoplasm (lower panels). B, SOK1 is a caspase 3 substrate. The caspase substrate Mst3 or SOK1 were in vitro translated in the presence of [35S]methionine and incubated in the absence or presence of caspase 3 and the caspase inhibitor z-VAD-fmk. Reactions were separated by electrophoresis and exposed by autoradiography. FL, full-length.
Given the dependence of SOK1 nuclear entry and cell death induction on caspase activity, we hypothesized that the 35-kDa band might be SOK1 cleaved by caspases. To determine whether SOK1 could be cleaved by caspases, we translated SOK1 in vitro and incubated it with caspase 3. SOK1 was cleaved by this enzyme into a 35-kDa fragment, albeit less efficiently than Mst3, a previously described caspase 3 substrate (Fig. 7B). This shows that SOK1 can be cleaved by caspase 3, generating a fragment the size of the immunoreactive band detected in the nucleus of cells subject to chemical anoxia.
An N-terminal Fragment of SOK1 Is Pancellular in Untreated Cells and Induces Cell Death with High Efficiency—The 35-kDa band in the nucleus of treated cells was detected with an antibody directed against the N terminus of SOK1, suggesting that it represents an N-terminal fragment of the protein. As discussed above, this is similar to other members of the Mst kinase family such as Mst1, Mst2, and Mst3 that are cleaved by caspases into an N-terminal fragment that translocates to the nucleus. We reasoned that if this were the case for SOK1, an N-terminal fragment of the protein should be able to efficiently enter the nucleus even in the absence of a stressor. We used the previously described SOK1ΔC mutant (30) that encompasses the 331 N-terminal amino acids of the protein. When we fused this mutant to GFP, we found that it readily entered the nucleus even in untreated cells and that chemical anoxia did not modify its cellular distribution (Fig. 8). Two kinase-inactive versions of the fragment (SOK1ΔCK49R and SOK1ΔCT174A) behaved identically, consistent with kinase activity not being necessary for nuclear entry. In contrast, the SOK1ΔC4A mutant was excluded from the nucleus, further suggesting that amino acids 275-292 are essential for nuclear entry.
FIGURE 8.

An N-terminal fragment of SOK1 can enter the nucleus in a constitutive manner independently of its kinase activity but dependent on amino acids 275-292. HEK293 cells were transfected with GFP-SOK1ΔC (bearing amino acids 1-331 of SOK1) fusion protein or with the mutants SOK1ΔCK49R, SOK1ΔCT174A, and SOK1ΔC4A. 24 h after transfection cells were incubated with sodium cyanide and 2-deoxyglucose or left untreated (control). A, schematic representation of the mutants used in this study. B, percentage of cells in which the different mutants of GFP-SOK1 show a nuclear signal. At least 100 GFP-positive cells per mutant and time point were scored for GFP nuclear positivity. Shown is the average and S.D. of the percentage of cells positive for nuclear GFP from at least four independent experiments. C, control; T3 and T6, 3 and 6 h after treatment. p > 0.05 (N.S.) and p < 0.05 (*) versus SOK1ΔC. C, images of representative fields of control and treated cells transfected with the different GFP-SOK1 mutants in control cells and cells treated for 6 h. The bar represents 10 μm. CN+2-DG, sodium cyanide and 2-deoxyglucose.
We then transfected the mutants of SOK1 described above tagged with M2 into HEK293 cells. We found that all of them attained levels similar to each other but clearly lower than their full-length versions (Fig. 9A, comparison not shown). Despite this, SOK1ΔC had a very high kinase activity. On the contrary and as expected, SOK1ΔCK49R and SOK1ΔCT174A did not have detectable activity. The kinase activity of SOK1ΔC4A was greatly reduced with respect to SOK1ΔC but clearly higher than wild-type SOK1, even more if we consider the different expression levels (Fig. 9B, comparison with wild-type SOK1 not shown).
FIGURE 9.

The N-terminal fragment of SOK1 can induce cell death with high efficiency, and this is dependent on its kinase activity and amino acids 275-292. M2-flagged versions of SOK1ΔC, SOK1ΔCK49R, SOK1ΔCT174A, and SOK1ΔC4A were transfected into HEK293 cells. A, the levels of expression of the different mutants were determined by Western blotting using anti-M2 antibody. B, the kinase activity of each mutant toward the unspecific substrate myelin basic protein (MBP) was determined after immunoprecipitation with the M2 antibody. C, the ability of each mutant to induce cell death was assessed by determining the percentage of non-viable cells by trypan blue exclusion. Shown is the mean and S.D. of four independent experiments. *, p < 0.05 versus SOK1ΔC. WT, wild-type.
SOK1ΔC induced cell death to the same degree as the full-length protein despite its markedly lower level of expression (Fig. 9, A and B). The number of non-viable cells was significantly lower in the cells transfected with the SOK1ΔC-K49R and SOK1ΔC-T174A when compared with those transfected with SOK1ΔC despite similar levels of expression. Thus, in the context of a truncated protein and at the levels achieved in this experiment, the kinase activity is necessary for cell death induction. Significantly, the induction of cell death is also impaired for the SOK1ΔC4A construct. Because the kinase activity of this mutant is higher than the wild-type protein, this result suggests that a feature in addition to kinase activity is essential for cell death induction by SOK1, and this is likely to be nuclear entry.
DISCUSSION
The biological functions of SOK1, a GCK III subfamily member, are the subject of debate. On one hand, its kinase activity is stimulated by extreme stresses such as reactive oxygen species and anoxia, suggesting that it might play a role in the cellular response to these insults (30, 31). On the other hand, SOK1 is localized to the cis-Golgi network, and its inhibition by RNA interference leads to Golgi disassembly in stationary cells and Golgi misplacement in migrating cells (33). We report here that, whereas SOK1 is normally localized on the Golgi complex, when the cell is subject to extreme stresses and caspases are activated, it plays a role in the apoptotic death of the cell. SOK1 is cleaved and enters the nucleus under these circumstances, and this relocation is likely to contribute to cell death.
SOK1 Is Involved in Cell Death—The ability of overexpressed SOK1 to induce cell death, its characteristic redistribution in response to extreme stresses, and especially the reduction in cell death upon inhibition of its expression all implicate SOK1 in the cell death response to some stresses. The ability of SOK1 to induce death is evident in several cell types, suggesting that it is a general phenomenon. Most importantly, based on RNA-mediated interference-mediated knockdown of SOK1, endogenous SOK1 is critical to the cell death induced by extreme stresses such as chemical anoxia or osmotic stress. These findings suggest that the death-inducing effects of SOK1 expression are not a consequence of nonspecific toxicity but, rather, reflect a physiological property of SOK1.
The two primary pathways to cell death are apoptosis and necrosis (though strict distinctions between the two have recently been challenged (40)). Significantly, the cell death induced by SOK1 is most consistent with apoptosis, as denoted by the membrane blebbing, cytochrome c leakage from the mitochondria, dependence on caspase activity, cleavage of the caspase substrate PARP, and chromatin condensation of cells overexpressing the kinase. The ability of the enforced expression of SOK1 to induce apoptosis is consistent with other kinases of the Mst family such as Mst1, Mst2, and Mst3 (see also Refs. 10, 15-17, 42). However, in this work we not only show that SOK1 expression induces apoptosis but also that its down-regulation by shRNA protects the cell from extreme stresses such as chemical anoxia. This had only been demonstrated for Mst1, the best studied member of the family, in neurons exposed to hydrogen peroxide (22). Our results suggest that regulation of cell death may be a common characteristic of Mst kinases, with the possible exception of Mst4, an issue that deserves a deeper study in the future. While this article was under review, Mst3 was reported to be important in placental cell death subject to oxidative stress (43).
The stimuli that most potently activate SOK1, such as chemical anoxia and ROS, are usually thought to induce necrotic cell death. However, it is important to note that ROS and chemical anoxia can also induce apoptosis, particularly when the insult is not sufficiently intense to induce necrosis (40). This is the case in our system, where a very clear leakage of cytochrome c to the cytoplasm is seen in response to chemical anoxia, whereas there is very little release of lactate dehydrogenase to the medium. Significantly, SOK1 protects against this apoptosis rather than against necrosis. This together with its apoptosis-inducing ability and its relation to caspases places this kinase in the apoptotic response to extreme stresses.
SOK1 and Caspases—During late stages of apoptotic cell death SOK1 could be a target of caspases, and their activity is important for entry of SOK1 into the nucleus (Fig. 5). Our results are consistent with SOK1 being cleaved between amino acids 300 and 340, a region that is highly homologous to Mst3, a protein that is known to be cleaved by caspases. Nevertheless, mutation of the various aspartate residues of this zone alone or in combination does not prevent in vitro cleavage of SOK1 by caspase 3 (supplemental Fig. S5), suggesting that caspases may cleave SOK1 at atypical sites. We are currently pursuing this possibility. Alternatively, the effect of caspases on nuclear entry of SOK1 might be indirect. Nevertheless, this is not consistent with the in vitro cleavage of SOK1 by recombinant caspase 3.
Although our results place SOK1 downstream of caspases, the facts that cell death induced by SOK1 is dependent on their activity and that the early activation of SOK1 by chemical anoxia is independent of caspases also places SOK1 upstream of them. Being both upstream and downstream of caspases is not unusual for kinases regulating apoptosis such as Mst1 (15), protein kinase Cδ (44), and another GCK, SLK (45). In some cases this has been proposed to form part of a positive feedback or auto-amplification loop that ensures that the induction of apoptosis is an all-or-nothing event, and this scenario may also be the case for SOK1.
Nuclear Entry of SOK1 during Cell Death—As described above, our results show that SOK1 participates in the cell death response elicited by ROS and ATP depletion. We also show that in response to this type of stress, SOK1 translocates from the Golgi apparatus to the nucleus in its cleaved form. This is not secondary to an unspecific nuclear permeabilization, as Golgi-58K and, especially, GAPDH do not enter the nucleus after the same stimulus. Our data also suggest that the nuclear entry of SOK1 depends on the integrity of the sequence between amino acids 275 and 292. This together with the compliance of this sequence to a canonical NLS and its homology to the well characterized NLS of Mst3 all suggest that it constitutes a bona fide NLS that is inactive until the C terminus of the protein is separated from the rest of the protein. Furthermore, nuclear entry of SOK1 seems to be necessary for cell death induction by SOK1 since death induction by SOK14A and SOK1ΔC4A is severely impaired. Our data are less informative on the requirement of kinase activity for cell death induction by SOK1. It seems to be partially dispensable in the context of the overexpressed full-length protein but necessary when truncated proteins are used. Whether this difference is due to the different context of sequences or to the different levels of expression of full-length versus ΔC proteins awaits further investigation.
Nuclear entry of SOK1 after the activation of caspases parallels the behavior of another GCKIII family member, Mst3, under the same circumstances, although it is completely different to the biology of the other member of the family, Mst4, which seems to be involved in cell proliferation rather than in cell death. There are even some differences in the biology of SOK1 and Mst3 as substrates of caspases, most prominently the cleavage sequence. SOK1 mutated in the residues that are essential for Mst3 cleavage are still cut by caspase 3. Besides, we have not been able to identify a nuclear export signal in SOK1, contrary to Mst3 (data not shown). Further study of these differences will no doubt shed new light on the biology of these interesting proteins.
SOK1 is activated by chemical anoxia in a caspase-independent manner, something that has not been described for any other of the GCKIII family members (Fig. 6C). We have not tried in this paper to study the effects of this activity, but it is likely to be important in the early stages of induction of cell death. However, this activity can be bypassed in the context of an overexpressed full-length protein. We believe that our data show that SOK1 can induce cell death entering the nucleus, but this is not necessarily the only way it can kill a cell. The related GCK, Mst1, can also enter the nucleus to induce apoptosis after being cleaved by caspases to phosphorylate histone H2B, and also, like SOK1, it is activated in the early stages of stress response independently of caspase activity. In this case the substrate is the FOXO3a transcription factor (46). Future experiments are needed to unveil the substrates that SOK1 phosphorylates in the early activation after extreme stresses.
In summary, we demonstrate here that SOK1 is important in cell death after chemical anoxia, and it translocates from the Golgi apparatus to the nucleus under these circumstances. Nuclear translocation depends on both caspase activity and on a nuclear localization signal. These data add to our understanding of the complex cellular response to this type of insult and open new avenues to explore the pathophysiology of ischemic insults.
Supplementary Material
Acknowledgments
We thank Carlos Diéguez and Anxo Vidal for helpful discussions and comments.
This work was supported, in whole or in part, by National Institutes of Health Grant HL67371 (to T. F.) from the NHLBI. This work was also supported by Ministerio de Educación y Ciencia of Spain Grant BMC2002-03110 (to C. P.) and Xunta de Galicia Grants PGIDIT03PXIC20808PN (to C. P.) and PGIDT06PXIB208061PR (to J. Z.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1-S5.
Footnotes
The abbreviations used are: ROS, reactive oxygen species; GCK, germinal center kinase; MST, mammalian sterile twenty-like; Z-VAD-fmk, benzyloxycarbonyl-VAD-fluoromethyl ketone; NLS, nuclear localization signals; PARP, poly(ADP-ribose) polymerase; shRNA, short hairpin RNA; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; CMV, cytomegalovirus; GFP, green fluorescent protein; Ad, adenovirus; DOG, deoxyglucose.
References
- 1.Klein, J. A., and Ackerman, S. L. (2003) J. Clin. Investig. 111 785-793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benhar, M., Engelberg, D., and Levitzki, A. (2002) EMBO Rep. 3 420-425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Droge, W. (2002) Physiol. Rev. 82 47-95 [DOI] [PubMed] [Google Scholar]
- 4.Martindale, J. L., and Holbrook, N. J. (2002) J. Cell. Physiol. 192 1-15 [DOI] [PubMed] [Google Scholar]
- 5.Leberer, E., Dignard, D., Harcus, D., Thomas, D. Y., and Whiteway, M. (1992) EMBO J. 11 4815-4824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friesen, H., Lunz, R., Doyle, S., and Segall, J. (1994) Genes Dev. 8 2162-2175 [DOI] [PubMed] [Google Scholar]
- 7.Kyriakis, J. M. (1999) J. Biol. Chem. 274 5259-5262 [DOI] [PubMed] [Google Scholar]
- 8.Dan, I., Watanabe, N. M., and Kusumi, A. (2001) Trends Cell Biol. 11 220-230 [DOI] [PubMed] [Google Scholar]
- 9.Lin, J. L., Chen, H. C., Fang, H. I., Robinson, D., Kung, H. J., and Shih, H. M. (2001) Oncogene 20 6559-6569 [DOI] [PubMed] [Google Scholar]
- 10.Graves, J. D., Gotoh, Y., Draves, K. E., Ambrose, D., Han, D. K., Wright, M., Chernoff, J., Clark, E. A., and Krebs, E. G. (1998) EMBO J. 17 2224-2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schinkmann, K., and Blenis, J. (1997) J. Biol. Chem. 272 28695-28703 [DOI] [PubMed] [Google Scholar]
- 12.Zhou, T. H., Ling, K., Guo, J., Zhou, H., Wu, Y. L., Jing, Q., Ma, L., and Pei, G. (2000) J. Biol. Chem. 275 2513-2519 [DOI] [PubMed] [Google Scholar]
- 13.Pombo, C. M., Force, T., Kyriakis, J., Nogueira, E., Fidalgo, M., and Zalvide, J. (2007) Front. Biosci. 12 850-859 [DOI] [PubMed] [Google Scholar]
- 14.Ura, S., Masuyama, N., Graves, J. D., and Gotoh, Y. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 10148-10153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ura, S., Masuyama, N., Graves, J. D., and Gotoh, Y. (2001) Genes Cells 6 519-530 [DOI] [PubMed] [Google Scholar]
- 16.Huang, C. Y., Wu, Y. M., Hsu, C. Y., Lee, W. S., Lai, M. D., Lu, T. J., Huang, C. L., Leu, T. H., Shih, H. M., Fang, H. I., Robinson, D. R., Kung, H. J., and Yuan, C. J. (2002) J. Biol. Chem. 277 34367-34374 [DOI] [PubMed] [Google Scholar]
- 17.Dan, I., Ong, S. E., Watanabe, N. M., Blagoev, B., Nielsen, M. M., Kajikawa, E., Kristiansen, T. Z., Mann, M., and Pandey, A. (2002) J. Biol. Chem. 277 5929-5939 [DOI] [PubMed] [Google Scholar]
- 18.Lee, K. K., and Yonehara, S. (2002) J. Biol. Chem. 277 12351-12358 [DOI] [PubMed] [Google Scholar]
- 19.Lee, W. S., Hsu, C. Y., Wang, P. L., Huang, C. Y., Chang, C. H., and Yuan, C. J. (2004) FEBS Lett. 572 41-45 [DOI] [PubMed] [Google Scholar]
- 20.Praskova, M., Khoklatchev, A., Ortiz-Vega, S., and Avruch, J. (2004) Biochem. J. 381 453-462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khokhlatchev, A., Rabizadeh, S., Xavier, R., Nedwidek, M., Chen, T., Zhang, X. F., Seed, B., and Avruch, J. (2002) Curr. Biol. 12 253-265 [DOI] [PubMed] [Google Scholar]
- 22.Lehtinen, M. K., Yuan, Z., Boag, P. R., Yang, Y., Villen, J., Becker, E. B., DiBacco, S., de, l. I., Gygi, S., Blackwell, T. K., and Bonni, A. (2006) Cell 125 987-1001 [DOI] [PubMed] [Google Scholar]
- 23.Chan, E. H., Nousiainen, M., Chalamalasetty, R. B., Schafer, A., Nigg, E. A., and Sillje, H. H. (2005) Oncogene 24 2076-2086 [DOI] [PubMed] [Google Scholar]
- 24.Stegert, M. R., Hergovich, A., Tamaskovic, R., Bichsel, S. J., and Hemmings, B. A. (2005) Mol. Cell. Biol. 25 11019-11029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jia, J., Zhang, W., Wang, B., Trinko, R., and Jiang, J. (2003) Genes Dev. 17 2514-2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu, S., Huang, J., Dong, J., and Pan, D. (2003) Cell 114 445-456 [DOI] [PubMed] [Google Scholar]
- 27.Harvey, K. F., Pfleger, C. M., and Hariharan, I. K. (2003) Cell 114 457-467 [DOI] [PubMed] [Google Scholar]
- 28.Udan, R. S., Kango-Singh, M., Nolo, R., Tao, C., and Halder, G. (2003) Nat. Cell Biol. 5 914-920 [DOI] [PubMed] [Google Scholar]
- 29.Pantalacci, S., Tapon, N., and Leopold, P. (2003) Nat. Cell Biol. 5 921-927 [DOI] [PubMed] [Google Scholar]
- 30.Pombo, C. M., Bonventre, J. V., Molnar, A., Kyriakis, J., and Force, T. (1996) EMBO J. 15 4537-4546 [PMC free article] [PubMed] [Google Scholar]
- 31.Pombo, C. M., Tsujita, T., Kyriakis, J. M., Bonventre, J. V., and Force, T. (1997) J. Biol. Chem. 272 29372-29379 [DOI] [PubMed] [Google Scholar]
- 32.Osada, S., Izawa, M., Saito, R., Mizuno, K., Suzuki, A., Hirai, S., and Ohno, S. (1997) Oncogene 14 2047-2057 [DOI] [PubMed] [Google Scholar]
- 33.Preisinger, C., Short, B., De, C. V., Bruyneel, E., Haas, A., Kopajtich, R., Gettemans, J., and Barr, F. A. (2004) J. Cell Biol. 164 1009-1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brummelkamp, T. R., Bernards, R., and Agami, R. (2002) Science 296 550-553 [DOI] [PubMed] [Google Scholar]
- 35.Pombo, C. M., Kehrl, J. H., Sanchez, I., Katz, P., Avruch, J., Zon, L. I., Woodgett, J. R., Force, T., and Kyriakis, J. M. (1995) Nature 377 750-754 [DOI] [PubMed] [Google Scholar]
- 36.Pombo, C. M., Bonventre, J. V., Avruch, J., Woodgett, J. R., Kyriakis, J. M., and Force, T. (1994) J. Biol. Chem. 269 26546-26551 [PubMed] [Google Scholar]
- 37.Hatai, T., Matsuzawa, A., Inoshita, S., Mochida, Y., Kuroda, T., Sakamaki, K., Kuida, K., Yonehara, S., Ichijo, H., and Takeda, K. (2000) J. Biol. Chem. 275 26576-26581 [DOI] [PubMed] [Google Scholar]
- 38.Robb, G. B., Brown, K. M., Khurana, J., and Rana, T. M. (2005) Nat. Struct. Mol. Biol. 12 133-137 [DOI] [PubMed] [Google Scholar]
- 39.Kultz, D. (2005) Annu. Rev. Physiol 67 225-257 [DOI] [PubMed] [Google Scholar]
- 40.Zong, W. X., and Thompson, C. B. (2006) Genes Dev. 20 1-15 [DOI] [PubMed] [Google Scholar]
- 41.Chuang, D. M., Hough, C., and Senatorov, V. V. (2005) Annu. Rev. Pharmacol. Toxicol. 45 269-290 [DOI] [PubMed] [Google Scholar]
- 42.Veal, E. A., Findlay, V. J., Day, A. M., Bozonet, S. M., Evans, J. M., Quinn, J., and Morgan, B. A. (2004) Mol. Cell 15 129-139 [DOI] [PubMed] [Google Scholar]
- 43.Wu, H. Y., Lin, C. Y., Lin, T. Y., Chen, T. C., and Yuan, C. J. (2008) Apoptosis 13 283-294 [DOI] [PubMed] [Google Scholar]
- 44.Brodie, C., and Blumberg, P. M. (2003) Apoptosis 8 19-27 [DOI] [PubMed] [Google Scholar]
- 45.Sabourin, L. A., Tamai, K., Seale, P., Wagner, J., and Rudnicki, M. A. (2000) Mol. Cell. Biol. 20 684-696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheung, W. L., Ajiro, K., Samejima, K., Kloc, M., Cheung, P., Mizzen, C. A., Beeser, A., Etkin, L. D., Chernoff, J., Earnshaw, W. C., and Allis, C. D. (2003) Cell 113 507-517 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.