

Abstract
The rapamycin-sensitive mammalian target of rapamycin (mTOR) complex 1 (mTORC1) contains mTOR, raptor, mLST8, and PRAS40 (proline-rich Akt substrate of 40 kDa). PRAS40 functions as a negative regulator when bound to mTORC1, and it dissociates from mTORC1 in response to insulin. PRAS40 has been demonstrated to be a substrate of mTORC1, and one phosphorylation site, Ser-183, has been identified. In this study, we used two-dimensional phosphopeptide mapping in conjunction with mutational analysis to show that in addition to Ser-183, mTORC1 also phosphorylates Ser-212 and Ser-221 in PRAS40 when assayed in vitro. Mutation of all three residues to Ala markedly reduces mTORC1-mediated phosphorylation of PRAS40 in vitro. All three sites were confirmed to be phosphorylated in vivo by [32P]orthophosphate labeling and peptide mapping. Phosphorylation of Ser-221 and Ser-183 but not Ser-212 is sensitive to rapamycin treatment. Furthermore, we demonstrate that mutation of Ser-221 to Ala reduces the interaction with 14-3-3 to the same extent as mutation of Thr-246, the Akt/protein kinase B-phosphorylated site. We also find that mutation of Ser-221 to Ala increases the inhibitory activity of PRAS40 toward mTORC1. We propose that after mTORC1 kinase activation by upstream regulators, PRAS40 is phosphorylated directly by mTOR, thus contributing to the relief of PRAS40-mediated substrate competition.
Mammalian target of rapamycin (mTOR)2 has been demonstrated as a key element in signaling pathways controlling cell size, proliferation, and metabolism (1, 2). Two mTOR signaling complexes, mTORC1 and mTORC2, have been discovered. The rapamycin-sensitive mTORC1 consists of the catalytic subunit mTOR, the substrate-binding subunit raptor (regulatory associated protein of mTOR), mLST8 (also known as GβL), and PRAS40 (1, 2) and controls protein translation (1, 2). mTORC2, the rapamycin-insensitive form, contains mTOR, rictor, SIN1, mLST8, and PRR5 (1, 2) and functions as a PDK2 (phosphoinositide-dependent protein kinase 2) to phosphorylate Akt/protein kinase B at Ser-473 and regulates the actin cytoskeleton (1, 2). The best characterized downstream effectors of mTORC1 are S6K1 and 4E-BP1 (also known as PHAS-I), both of which are phosphorylated by mTORC1 at multiple sites and are involved in the control of mRNA translation (1, 2). The nature of the phosphorylation sites in these two mTORC1 substrates is surprisingly different: either (S/T)P (3) or h(S/T)h (where h represents hydrophobic) (4). Thus, the surrounding amino acids appear not to be the major determinant for phosphorylation. A critical motif called the TOR signaling (TOS) motif has been discovered in the NH2 terminus of S6K1 (FDIDL) and COOH terminus of 4E-BP1 (FEMDI) (5, 6). Mutation of the TOS motif not only decreases the rate of phosphorylation of S6K1 and 4E-BP1 by mTOR but also disrupts interaction between these substrates and raptor (5–8).
PRAS40 has been recently identified as a protein associated with mTORC1 (9, 10). PRAS40 predominantly interacts with raptor, although it may also interact with mTOR (9, 10). Importantly, a TOS motif (FVMDE) is found in amino acids 129–133 of PRAS40 and is required to mediate the interaction of PRAS40 and raptor (11–13). This finding leads to the possibility that PRAS40 is a direct substrate of mTORC1, as is S6K1 and 4E-BP1. Indeed, Oshiro et al. discovered that PRAS40 was phosphorylated by mTORC1, and one phosphorylation site, Ser-183, was identified (12). In response to insulin and nutrients, PRAS40 disassociates from mTORC1, and recombinant PRAS40 inhibits mTORC1 activity toward S6K1 and 4E-BP1 in vivo and in vitro (9–13). The inhibitory activity of PRAS40 is suggested to occur by direct competition with 4E-BP1 and S6K1 for binding to raptor (11, 12). PRAS40 was originally isolated with a 14-3-3 affinity column, and its binding with 14-3-3 depends on Akt/protein kinase B-mediated phosphorylation at Thr-246 (14). PRAS40 binding to 14-3-3 also depends on nutrient sufficiency and is inhibited by treatment with rapamycin, the specific inhibitor against mTORC1 (13, 15). This suggests that mTORC1-mediated phosphorylation of PRAS40 may regulate interaction of PRAS40 and 14-3-3.
The cellular function of PRAS40 is poorly understood. It has been shown that overexpression of PRAS40 inhibits apoptotic neuronal cell death after transient focal cerebral ischemia (16), decreased phosphorylation of PRAS40 at Thr-246 is correlated with increased tumor cell apoptosis (17), and phosphorylation of PRAS40 at Thr-246 is reduced in rats fed a high fat diet, which induces insulin resistance (18). However, decreased expression of PRAS40 has also been shown to protect against induction of apoptosis by tumor necrosis factor-α and cycloheximide in HeLa cells, suggesting that PRAS40 may be a proapoptotic agent (19). Presumably, the function of PRAS40 is regulated by its phosphorylation. In this study, we investigate phosphorylation sites in PRAS40 catalyzed by mTORC1. Two sites, Ser-212 and Ser-221, were identified by two-dimensional phosphopeptide mapping, and the regulation of phosphorylation at these sites upon mTORC1 activity and PRAS40 binding to 14-3-3 was determined.
EXPERIMENTAL PROCEDURES
Materials—Antibodies to raptor (20), 4E-BP1 (21), Akt1 (22), S6K1 (23), PRAS40 (14), HA (20), and phosphospecific antibodies to the Thr-36 and Thr-45 sites (24) have been described previously. Phosphospecific antibodies to the Thr-389 site in S6K1 were from Cell Signaling Technology, Inc. FLAG antibodies were from Sigma. Recombinant human insulin (Novolin R) was from Novo Nordisk. Rapamycin was from Calbiochem-Novabiochem. Tween 20 was from Fischer, and Triton X-100 was from Sigma. TPCK-treated trypsin was from Worthington. Glutathione-Sepharose 4B was from GE Healthcare Biosciences AB. Cellulose TLC plates were from EMD Chemicals Inc.
Cell Culture, Treatment, and Extract Preparation—3T3-L1 fibroblasts were grown in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) containing 10% newborn calf (Invitrogen) serum. Fibroblasts were converted to adipocytes by using differentiation medium as described previously (25) and then were cultured in DMEM containing 10% fetal bovine serum (Invitrogen) for 10–12 days. 3T3-L1 adipocytes were used to study insulin-stimulated mTOR kinase activity in vitro. HEK293, HEK293E, and HEK293T cells were cultured in DMEM containing 5% fetal bovine serum and used for transfection experiments. Compared with HEK293 and HEK293T cells, there are low basal levels of phosphorylation of mTOR targets in HEK293E cells, and thus HEK293E cells were used for insulin signal study. For experiments, cells were serum-starved with DMEM overnight and then incubated at 37 °C without or with insulin and/or other additions. To terminate the incubation, cells were rinsed once with chilled phosphate-buffered saline and then homogenized with a syringe with a 20-gauge needle in lysis buffer as described previously (20). Homogenates were centrifuged at 12,000 × g for 10 min, and the supernatants were retained for analyses.
Mutagenesis and Purification of Recombinant Proteins—PRAS40 cDNAs having point mutations were generated by oligonucleotide-directed mutagenesis of HA-tagged PRAS40 in pcDNA3 by using a QuikChange II kit (Stratagene) according to the manual instructions. All mutations were confirmed by sequencing.
The pGEX-4T-1 constructs encoding NH2-terminal GST-tagged PRAS40 and PRAS point mutations were expressed in bacteria (BL21) and purified as described previously (26). Samples were subjected to SDS-PAGE and stained with Coomassie Blue to assess purity and confirm concentrations of the recombinant proteins. The pGEX-4T-1 constructs of 14-3-3 and FKBP12 with GST tag were expressed and purified with glutathione-Sepharose 4B. The amount of recombinant proteins retained on glutathione beads was assessed by Coomassie Blue staining after SDS-PAGE with bovine serum albumin as a standard.
Transfection, Immunoprecipitation, and GST Pulldown Assay—HEK293 or HEK293E cells were seeded in a 10- or 6-cm dish or 6-well plate. Twenty-four hours later, plasmids were transfected using Lipofectamine 2000 (Invitrogen) at a 1:1 ratio (w/v). The cells were harvested and analyzed at 36 h after transfection. Cell extracts were incubated with antibodies (2 μg) bound to protein A- or G-agarose beads or GST-tagged recombinant proteins (10 μg) bound to glutathione beads at 4 °C for 2 h with constant mixing. The beads were then washed four times.
In Vitro Kinase Assay—As described previously (20), immune complex beads were rinsed with 1 ml of kinase buffer (50 mm NaCl, 0.1 mm EGTA, 1 mm dithiothreitol, 0.5 μm microcystin LR, 10 mm HEPES, and 50 mm β-glycerophosphate, pH 7.4) and suspended in 60 μl of kinase buffer. After removing a sample for immunoblots, the kinase reactions were initiated by adding to 20 μl of the suspension 5 μl of kinase buffer supplemented with 0.5 mm [γ-32P]ATP (1000 mCi/mmol; PerkinElmer Life Sciences), 50 mm MnCl2, and 1 μg of PRAS40 wild type, mutants, or 4E-BP1 as substrates. Reactions were terminated after 30 min at 30 °C by adding SDS sample buffer. The relative amounts of 32P incorporated into the PRAS40 and 4E-BP1 proteins were determined by phosphorimaging after SDS-PAGE.
In Vivo 32P Radiolabeling—Prior to radiolabeling, HEK293 cells cultured in 10-cm dishes were serum-starved overnight. The medium was then replaced by phosphate-free DMEM (Invitrogen), and cells were labeled with [32P]orthophosphate (0.4 mCi/ml) (PerkinElmer Life Sciences) for 3 h at 37°C, followed by incubation with rapamycin (20 nm) and insulin (200 nm) for 30 min. The cells were harvested and immunoprecipitated by HA antibody described as above. The samples were subject to SDS-PAGE and transferred to polyvinylidene difluoride membrane, and the radioactivity was analyzed by autoradiography.
Two-dimensional Phosphopeptide Mapping—Two-dimensional phosphopeptide mapping was performed according to the manual instructions of the Hunter Thin Layer Peptide Mapping Electrophoresis System. Briefly, 32P-labeled PRAS40 wild type and mutant proteins either phosphorylated in vitro by mTORC1 isolated from insulin-stimulated HEK293 cells or immunoprecipitated from radiolabeled and insulin-stimulated HEK293 cells were excised from polyvinylidene difluoride membrane and digested with 10 μg of TPCK-treated trypsin in buffer containing 50 mm NH4HCO3 (pH 8.0) twice for 12 and 3 h, respectively, at 37 °C. Digests were lyophilized and resuspended in 50 μl of hydrogen peroxide/formic acid (1:9) for oxidation, incubating on ice for 30 min. After lyophilization, samples were resuspended in the first dimension running buffer and loaded onto cellulose TLC plate and subject to two-dimensional peptide mapping. The first dimension was run for 30 min at 1000 V at pH 1.9 (formic acid/acetic acid/water (50:156:1794)), and second dimension chromatography was performed in phosphochromo buffer (butanol/pyridine/acetic acid/water (750:500:150:600)) for 16 h. 32P-Labeled peptides were subsequently visualized by phosphorimaging chromatography plates.
RESULTS
mTORC1 Phosphorylates Ser-212 and Ser-221 in Vitro in Addition to Ser-183—To investigate whether mTORC1 phosphorylates PRAS40 at sites other than Ser-183, recombinant proteins of PRAS40 wild type and mutants of Ser-183 to Ala (S183A) and Thr-246 to Ala (T246A) were generated and utilized in phosphorylation assays in vitro. mTORC1, Akt1, and S6K1 were immunoprecipitated from extracts of 3T3-L1 adipocytes by using antibodies to raptor, Akt1, and S6K1. Intact mTORC1 was recovered, and PRAS40 disassociation was induced by insulin treatment as reported previously (11) (Fig. 1A). Akt1 and S6K1 immune complexes did not contain mTORC1 components. To measure mTORC1 kinase activity toward PRAS40, immune complexes were incubated with [γ-32P]ATP and recombinant PRAS40. As shown by 32P incorporation, PRAS40 was phosphorylated by mTORC1, and treating adipocytes with insulin increased PRAS40 phosphorylation more than 2-fold (Fig. 1A). mTORC1 phosphorylated the PRAS40 mutant T246A to the same extent as wild type, whereas the S183A mutation decreased but did not eliminate the phosphorylation of PRAS40 compared with wild type (Fig. 1A). In contrast, Akt1 phosphorylated wild type PRAS40 and the S183A mutant equally well, whereas the T246A mutation completely eliminated phosphorylation by Akt1 (Fig. 1A), indicating that Thr-246 is the only site phosphorylated by Akt1, as previously reported (14). As a control, in an S6K1 kinase assay, 32P incorporation into PRAS40 showed only background level. Thus, mTORC1 specifically phosphorylates PRAS40 in vitro at site(s) in addition to Ser-183. The specificity of mTORC1-catalyzed phosphorylation of PRAS40 was confirmed by using mTOR kinase-dead (S2338A, KD) and constitutive active (deleting 2433–2451, ΔRD) mutants in HEK293 cells. The mTOR kinase-dead mutant abolished kinase activity toward PRAS40, whereas mTOR ΔRD substantially enhanced phosphorylation of PRAS40 (Fig. 1B).
FIGURE 1.
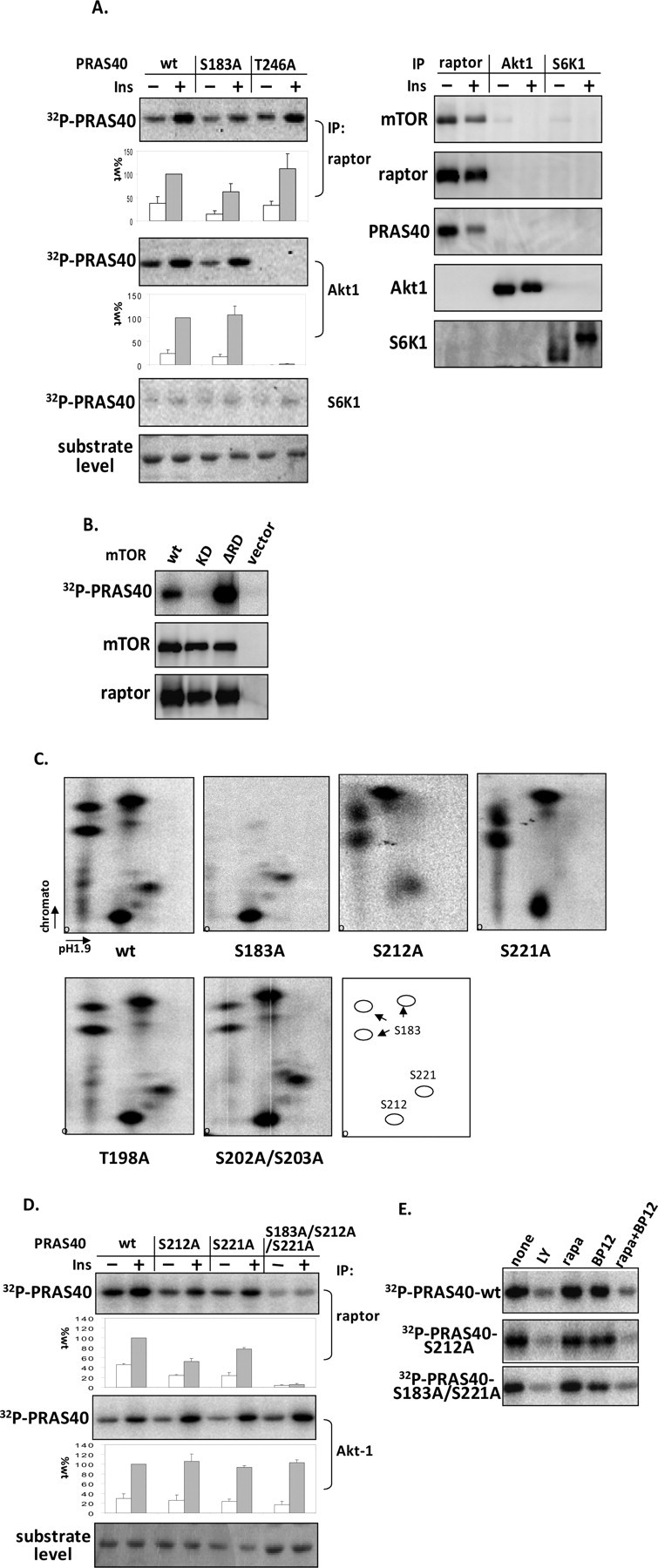
Identification of Ser-212 and Ser-221 as in vitro mTORC1-catalyzed phosphorylation sites in PRAS40. 3T3-L1 adipocytes were incubated with insulin (60 nm) for 30 min (A and D). A, immune complex kinase assays were performed with [γ-32P]ATP and PRAS40 wild type (wt) and the indicated mutative proteins as substrates after conducting immunoprecipitations (IP) of raptor, Akt1, and S6K1. After SDS-PAGE, a phosphor image of 32P-labeled PRAS40 (left) and immunoblots of mTOR, raptor, PRAS40, Akt1, and S6K1 (right) were prepared, and substrates were stained with 0.5% Ponceau S, as shown with substrate level. The effect on 32P incorporation into PRAS40 (corrected for substrate level) is expressed as a percentage relative to wild type (mean ± S.E. from three experiments). B, mTOR wild type, kinase-dead (S2338A, KD), and constitutive active (deleting 2433–2451, ΔRD) mutants were co-expressed with raptor in HEK293 cells. mTOR complexes were isolated with antibodies toward FLAG-tagged mTOR, and then mTOR kinase assays were performed with PRAS40 as substrate. C, PRAS40 wild type and mutants as indicated were phosphorylated in vitro by mTORC1 immune complexes (raptor IP) from insulin-stimulated HEK293 cells. After SDS-PAGE and transferring, 32P-incorporated PRAS40 was excised from the polyvinylidene difluoride membrane, digested with trypsin, and then subjected to two-dimensional phosphopeptide mapping. The results were visualized by a PhosphorImager. The pattern of migration for the three identified sites is shown in the last panel. D, phosphorylation of the indicated PRAS40 mutants in kinase assays with mTORC1 and Akt1 immune complexes were assessed by 32P incorporation. The effect on 32P incorporation into PRAS40 (corrected for substrate level) is expressed as a percentage relative to wild type (mean ± S.E. from three experiments). E, washed raptor antibody immune complexes isolated from HEK293 cells were incubated in kinase reaction mixtures containing no additions (None) for 20 min or the following: 10 μm LY294002 (LY), 5 μm rapamycin (rapa), 5 μm GST-FKBP12 (BP12), or 5 μm GST-FKBP12 plus 5 μm rapamycin (rapa + BP12). Phosphor images of 32P-labeled PRAS40 wild type and mutants of S212A and S183A/S221A are presented.
We next used two-dimensional phosphopeptide mapping in conjunction with point site mutagenesis to identify sites in PRAS40 phosphorylated by mTORC1. Preliminary results showed that the truncation of PRAS40 with amino acid residues from 1 to 181 substantially reduced the phosphorylation of PRAS40 by mTORC1, whereas the truncation with amino acid residues from 97 to 256 was phosphorylated by mTORC1 to the same extent as wild type (data not shown). This suggests that the mTORC1 phosphorylation sites are predominantly located in the COOH terminus of PRAS40. Therefore, PRAS40 recombinant proteins with point mutations in the COOH terminus (S183A, T198A, S202A/S203A, S211A, S212A, S221A, S232A, T246A, and S247A) were generated. PRAS40 wild type and mutant proteins were phosphorylated by mTORC1 in vitro, and after SDS-PAGE, 32P-labeled PRAS40 was digested by trypsin and subjected to phosphopeptide mapping analysis. The tryptic digestion pattern of PRAS40 extracted directly from the gel was compared with the pattern of PRAS40 digested after transfer to polyvinylidene difluoride membrane and was found to be identical (data not shown); therefore, the latter method was used for subsequent analyses. As illustrated in Fig. 1C (first panel) and summarized in Fig. 1C (last panel), five major phosphopeptides were obtained after digestion of 32P-incorporated PRAS40 wild type phosphorylated by mTORC1 in vitro. The cluster of three spots in the upper left were confirmed to arise from incomplete digestion of the peptide that contains Ser-183, since mutation of Ser-183 to Ala eliminated these spots (Fig. 1C). The other two sites phosphorylated by mTORC1 were Ser-212 and Ser-221, demonstrated by alanine substitution abolishing the two remaining spots (Fig. 1C). T198A and S202A/S203A and the rest of the mutants (S211A, S232A, T246A, and S247A; data not shown) did not change the phosphopeptide mapping pattern.
To confirm that these identified residues are the major sites phosphorylated by mTORC1 in vitro, PRAS40 mutants S212A, S221A, and the combined S183A/S212A/S221A mutations were used in an mTORC1 kinase assay. S212A and S221A modestly reduced the mTORC1-catalyzed phosphorylation of PRAS40, and mutations in Ser-212, -221, and -183 together essentially eliminated the phosphorylation of PRAS40 (Fig. 1D). Taken together, these results demonstrate that Ser-183, -212, and -221 account for the essential sites in PRAS40 phosphorylated by mTORC1 in vitro. To investigate further whether PRAS40 phosphorylation catalyzed by mTORC1 is rapamycinsensitive in vitro, the immune complex isolated by raptor antibody was incubated with the addition of rapamycin and FKBP12. Incubating mTORC1 with rapamycin or FKBP12 alone was without effect on PRAS40 phosphorylation; however, the combination of rapamycin and FKBP12 abolished the phosphorylation of PRAS40 (Fig. 1E). As predicted, LY294002, which is previously shown to inhibit mTOR (20), abolished PRAS40 phosphorylation as well. PRAS40 phosphorylation was also inhibited by rapamycin-FKBP12 in the S212A and S183AS221A mutants (Fig. 1E), demonstrating that phosphorylation at individual sites catalyzed by mTORC1 in vitro, such as Ser-183 and Ser-212, were rapamycin-sensitive.
Phosphorylation of PRAS40 by mTORC1 in Vivo—To investigate whether Ser-212 and Ser-221 are phosphorylated by mTORC1 in vivo, HEK293 cells were transfected with PRAS40 and labeled by [32P]orthophosphate. Recombinant PRAS40 was isolated by HA immunoprecipitation. Two-dimensional phosphopeptide mapping shows six major 32P-labeled peptides (a–f) after digestion of wild-type PRAS40 (Fig. 2A). In comparison with the in vitro mapping pattern, spots a, c, and f run in similar positions with the peptides shown to contain Ser-183, -221, and -212 individually (Fig. 2A). Indeed, the S183A mutant eliminated spot a, confirming that Ser-183 is phosphorylated in vivo (Fig. 2A). Spot c represents the highly phosphorylated peptide containing Thr-246, as demonstrated by its elimination in the T246A mutant (Fig. 2A). In contrast, mutants S212A and S221A did not alter the in vivo phosphopeptide pattern (Fig. 2A). Importantly, mutation of Thr-246 to Ala uncovered a previously obscured spot (spot g), which was eliminated by the combined mutation of Thr-246 and Ser-221 (Fig. 2A). This indicates that Ser-221 is also phosphorylated in vivo (spot g). Inspecting the predicted peptides arising from trypsin digestion revealed that Ser-212 is located in the same peptide as Ser-202 and Ser-203. The combined mutations of S202A/S203A and S212A substantially eliminated spot f, whereas mutation of S202A/S203A or S212A alone was not able to abolish spot f (Fig. 2A). This suggests that both Ser-202/203 and Ser-212 are phosphorylated in vivo. Of note, in PRAS40 wild type, there was a long tailed spot below spot g, but this spot disappeared in the S202A/S203A mutant (Fig. 2A). It is possible that phosphorylation at Ser-202/203 causes incomplete digestion of trypsin between Arg-201 and Ser-202 and produces a longer and more hydrophilic peptide with four extra amino acids of Thr-Glu-Ala-Arg. We also consistently observed peptides represented in spots b, d, and e that were phosphorylated in vivo (Fig. 2A), but what phosphorylation sites these spots represent is yet to be identified. Compared with HEK293 cells, in HEK293E cells, the spots of d and e showed very little phosphorylation (data not shown). Therefore, as indicated in the last panel of Fig. 2A, Ser-183, Ser-221, Ser-212, Ser-202/203, and Thr-246 in PRAS40 are phosphorylated in vivo.
FIGURE 2.
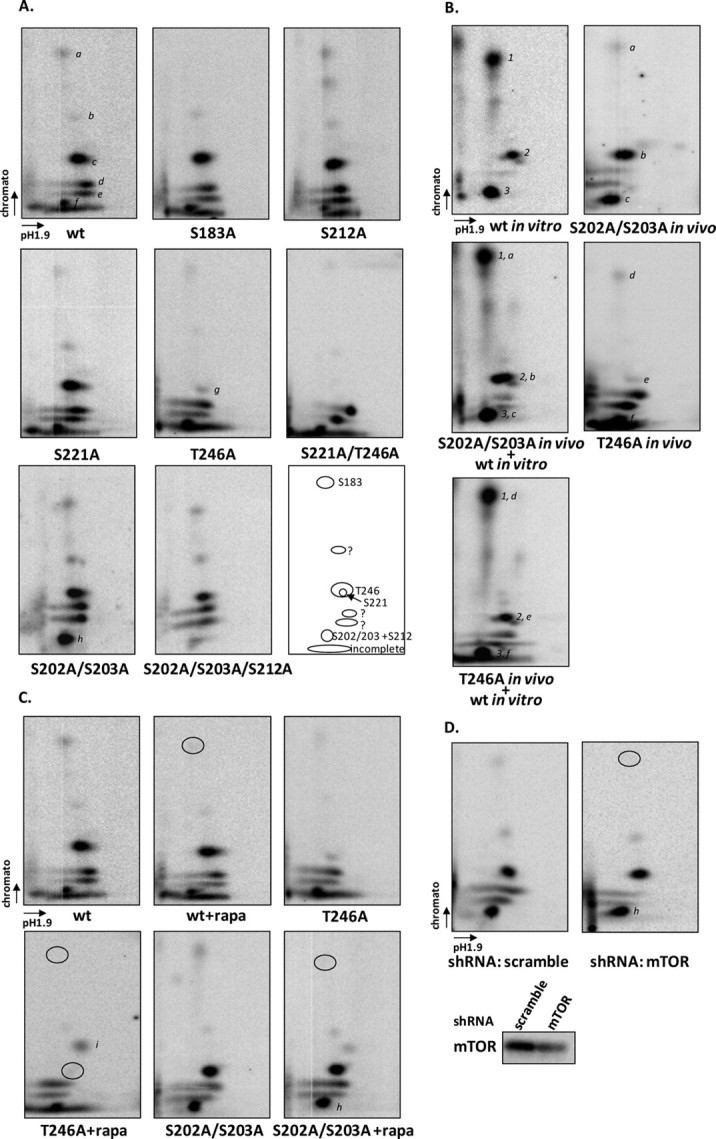
Phosphorylation of PRAS40 by mTORC1 in vivo. PRAS40 and mutants were transfected in HEK293 cells (A and B), and cells were labeled with [32P]orthophosphate for 3 h followed by insulin treatment (200 nm) for 30 min. 32P-Labeled PRAS40 was immunoprecipitated by HA antibody, and trypsin-digested peptides were resolved by two-dimensional phosphopeptide mapping. A, a variety of PRAS40 mutants were used to assess phosphopeptides. Major peptide spots are indicated by a–g, and phosphorylation sites are indicated in the last panel. B, in vitro and in vivo phosphorylated PRAS40 were mixed after trypsin digestion. 1, 2, and 3 indicate phosphopeptides obtained from in vitro phosphorylation by mTORC1 as described in Fig. 1C. The phosphopeptides in vivo are indicated by a–c in S202A/S203A (SS202,3AA) mutants and d–f in the T246A mutant. C, HEK293 cells were treated without or with rapamycin (rapa; 20 nm) for 20 min before insulin treatment. The circles indicate the spots that were altered after rapamycin treatment. D, HEK293T cells with lentivirus expressing either scramble or mTOR short hairpin RNA (shRNA) were transfected with PRAS40 mutant S202A/S203A. The circles indicate the spots that were altered in mTOR knockdown cells. wt, wild type.
To compare the sites phosphorylated in PRAS40 in vivo and in vitro by mTORC1, we mixed PRAS40 peptides phosphorylated by mTORC1 in vitro with in vivo phosphorylated T246A and S202A/S203A peptides, respectively, after trypsin digestion, followed by two-dimensional peptide mapping. The peptides in vitro phosphorylated by mTORC1 represented Ser-183 (spot 1), Ser-221 (spot 2), and Ser-212 (spot 3) comigrated with phosphopeptides in vivo containing Ser-183 (spot a in S202A/S203A and spot d in T246A), Thr-246 (spot b in S202A/S203A), Ser-221 (spot e in T246A), and Ser-212 (spot c in S202A/S203A and spot f in T246A), respectively (Fig. 2B).
We further investigated whether the phosphorylations of Ser-212 and Ser-221 are sensitive in vivo to rapamycin, a specific inhibitor of mTORC1. As predicted, phosphorylation of Ser-183 was reduced by rapamycin treatment in PRAS40 wild type and mutants (T246A and S202A/S203A) (Fig. 2C). In mutant T246A, which is used to show the Ser-221-containing phosphopeptide, rapamycin treatment decreased phosphorylation at Ser-221, as shown in Fig. 2C. However, in mutant S202A/S203A, the Ser-212-containing phosphopeptide (spot h) was not altered after rapamycin treatment (Fig. 2C). This result indicates that phosphorylation of Ser-212 is not affected by rapamycin treatment in cells, despite the rapamycin-FKBP12 complex inhibiting the mTORC1-catalyzed phosphorylation of Ser-212 in vitro (Fig. 1E). In order to investigate whether phosphorylation of Ser-212 in vivo is mediated by mTOR, we performed phosphopeptide mapping of PRAS40 mutant S202A/S203A in HEK293T cells, where mTOR was reduced by lentivirus-mediated RNA interference. Compared with the phosphopeptide map in the cell with scrambled short hairpin RNA, phosphorylation of Ser-183 was obviously reduced, but phosphorylation of Ser-212 (spot h) was not affected in mTOR knockdown cells (Fig. 2D). Together with a lack of sensitivity to rapamycin, the phosphorylation of Ser-212 is clearly mediated by a kinase other than mTORC1 in vivo. In addition, we found that rapamycin treatment resulted in the presence of one new phosphopeptide (indicated as spot i) (Fig. 2C), but this was not consistently observed. Determination of whether it is from PRAS40 requires further investigation.
Disassociation of mTOR and Raptor by Triton X-100 and Disruption of the TOS Motif in PRAS40 Are Unable to Eliminate Phosphorylation of PRAS40—S6K1 and 4E-BP1 are the two best characterized mTORC1 substrates, and their phosphorylation depends on mTOR and raptor association. Raptor binds substrates for mTOR phosphorylation (27), and disruption of the mTOR and raptor interaction by nonionic detergents, such as Triton X-100 and Nonidet P-40, abolishes mTOR activity toward S6K1 and 4E-BP1 (20). Interestingly, adding 0.2% Triton X-100 to the washed mTORC1 immune complex only decreased mTOR activity toward PRAS40 in vitro by 60%, whereas the same concentration of this detergent completely abolished phosphorylation of 4E-BP1 in vitro (Fig. 3A). The detergent did not affect phosphorylation of PRAS40 by Akt1 in vitro. This indicates that mTOR alone, even when disassociated from raptor, is able to phosphorylate PRAS40, suggesting that PRAS40 could have a direct binding site with mTOR. In fact, unlike S6K1 and 4E-BP1, in which the TOS motif is indispensable for phosphorylation by mTORC1 (5–8), disruption of the TOS motif (FVMDE) in PRAS40 by mutating Phe to Ala (F129A) was unable to eliminate phosphorylation of PRAS40 by mTORC1 (Fig. 3B). Therefore, although mutation in the TOS motif impairs PRAS40 interaction with raptor, the binding of PRAS40 to mTORC1 directly via other sites seems to be sufficient to allow phosphorylation of PRAS40 by mTOR. Further reduction of the mTORC1-PRAS40 interaction by combining the mutation (F129A/P185A) in the TOS motif and KSLP region (amino acid residues 182–185), which is also important for interaction of mTORC1 and PRAS40 (11), markedly decreased mTORC1 activity toward PRAS40 (Fig. 3B). It is not surprising that PRAS40 appears to have a stronger substrate-binding affinity with mTORC1 than S6K1 and 4E-BP1, since in addition to being a substrate PRAS40 is a partner tightly bound with mTORC1. PRAS40 phosphorylation by mTORC1 could thus be mediated by multiple interactions with raptor and mTOR.
FIGURE 3.
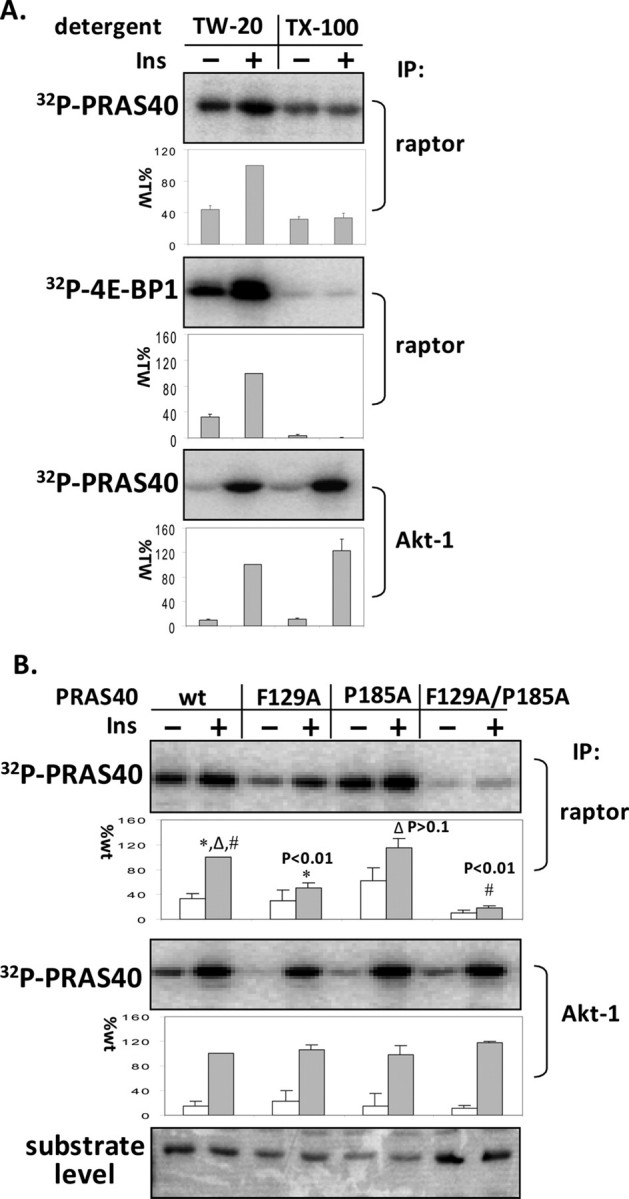
Disassociation of mTOR and raptor by Triton X-100 and disruption of the TOS motif in PRAS40 are unable to eliminate phosphorylation of PRAS40. 3T3-L1 adipocytes were incubated with insulin (60 nm) for 30 min. A, mTORC1 and Akt1 were isolated from adipocyte extracts in the presence of 0.2% Tween 20 by using raptor and Akt1 antibodies. The indicated detergents (0.2% final concentration) were added to the washed complexes, which were then incubated with [γ-32P]ATP and PRAS40 or 4E-BP1. 32P-Labeled PRAS40 and 4E-BP1 were detected by phosphorimaging. The amounts of incorporated 32P in PRAS40 and 4E-BP1 relative to the level of phosphorylation in the presence of Tween 20 with insulin treatment (%TW) were determined (mean ± S.E. from three experiments). TW-20, Tween 20; TX-100, Triton X-100. B, mTORC1 and Akt1 kinase assays were conducted using PRAS40 wild type (wt) and the indicated mutative proteins as substrates. The effect on 32P incorporation into PRAS40 (corrected for substrate level) is expressed as a percentage relative to wild type (mean ± S.E. from three experiments), and a t test was conducted between wild type and mutants. IP, immunoprecipitation.
Phosphorylation at Ser-221 Regulates the Affinity of PRAS40 Binding to 14-3-3—PRAS40 binding to 14-3-3 is insulin-inducible (14) and requires the presence of amino acids (13, 15). We used GST-fused 14-3-3 recombinant proteins coupled to glutathione beads to retain PRAS40 from lysates of HEK293E cells overexpressing PRAS40 wild type and point mutants. As a negative control, GST-fused FKBP12 did not bind PRAS40. Insulin induced an increase in PRAS40 binding to 14-3-3, and rapamycin reversed the effect of insulin (Fig. 4A). This is consistent with the results reported previously (13) and suggests that mTORC1 may regulate association between PRAS40 and 14-3-3. Interestingly, mutation of Ser-221 decreased the interaction to an extent similar to that of Thr-246 (Fig. 4B), indicating that in addition to Thr-246, phosphorylation of Ser-221 regulates insulin-induced PRAS40 binding to 14-3-3. Of note, there is an Arg at the –4-position of Ser-221, which conforms to the features of the 14-3-3 binding sequence (28). Thus, 14-3-3 binding appears to require phosphorylation of PRAS40 by both Akt and mTORC1, including at Ser-221 and Thr-246. Although mutating Ser-183 to Ala resulted in the disruption of the PRAS40 and 14-3-3 interaction, as previously described (13), mutating Pro-185 to Ala had a similar effect (Fig. 4B). In addition, mutating Ser-183 or Pro-185 to Ala reduced the phosphorylation of Thr-246 (Fig. 4B) and decreased interaction of mTORC1 and PRAS40, as shown in Fig. 5A and as previously reported (11). Why mutations at either Ser-183 or Pro-185 cause these actions is yet unclear, and a mutative effect in addition to dephosphorylation should be considered.
FIGURE 4.

In addition to Thr-246, phosphorylation at Ser-221 regulates the affinity of PRAS40 binding to 14-3-3. HEK293E cells were transfected with 3 μg of HA-tagged PRAS40 plasmid or the indicated mutants. 36 h later, serum-starved cells were treated with or without rapamycin (20 nm)(A) followed by insulin (200 nm) for 30 min (A and B). Cells were lysed in 0.2% Triton X-100. A, the extracts were incubated with glutathione beads coupled with GST-tagged 14-3-3 or FKBP12. Immunoblots of HA-PRAS40 and phospho-Thr-389 of S6K1 were prepared, and the coupled proteins were stained with 0.5% Ponceau S. B, one-half of the extracts containing PRAS40 wild type and mutants were subjected to GST-14-3-3 pulldown assays, and in parallel the other half were subjected to HA immunoprecipitations. Immunoblots of HA-PRAS40 and phospho-Thr-246 of PRAS40 are presented.
FIGURE 5.

Phosphorylation at Ser-221 modulates PRAS40 association with mTORC1 and its inhibitory effect on mTORC1 activity. A, PRAS40 wild type (wt) and various mutants as indicated were transfected into HEK293E cells. Cell extracts were prepared and immunoprecipitated (IP) with anti-HA antibodies. Endogenous mTOR and raptor co-immunoprecipitated with HA-PRAS40 were identified by immunoblotting. Co-immunoprecipitated mTOR and raptor (corrected by HA-PRAS40) from three experiments were calculated by densitometry and statistically analyzed by t test (mean ± S.E.). HEK293E cells were co-transfected with mTOR rapamycin-resistant mutant S2035W (SW), HA-S6K1 (B), or myc-4E-BP1 (C) without or with PRAS40 wild type or the indicated mutant with amounts of 0.5, 0.5, 1, and 2 μg. 36 h later, cells were treated with rapamycin (20 nm) for 20 min and then insulin (200 nm) for 30 min. B, HA-S6K1 was isolated by HA immunoprecipitation and phospho-Thr-389 of S6K1, total HA-S6K1, and HA-PRAS40 were detected by immunoblotting. C, phospho-Thr-36/45 of 4E-BP1, total 4E-BP1, and HA-PRAS40 were analyzed by immunoblotting.
Phosphorylation at Ser-221 Modulates PRAS40 Association with mTORC1 and Its Inhibitory Effect on mTORC1 Activity—PRAS40 has been shown to inhibit mTORC1 activity in vivo and in vitro, and phosphorylation of PRAS40 at Thr 246 by Akt is thought to relieve its inhibitory activity toward mTORC1 (9, 10). When overexpressing PRAS40 in HEK293E cells, we observed that mutations at Thr-246 and Ser-221 but not Ser-212 resulted in an increased association of overexpressed PRAS40 with endogenous mTORC1 (Fig. 5A). This increase in mTORC1 association is likely to result in an increased inhibitory activity toward mTORC1 kinase activity. To examine whether phosphorylation of Ser-221 regulates the inhibitory function of PRAS40 toward mTORC1 activity, we co-expressed PRAS40 wild type and mutants (S212A, S221A, and T246A) together with S6K1 or 4E-BP1 in HEK293E cells. As predicted, overexpressed PRAS40 reduced the insulin-induced phosphorylation of S6K1 at Thr-389 and 4E-BP1 at Thr-36/45, and the PRAS40 mutant T246A increased this inhibitory effect (Fig. 5, B and C). This supports the finding that phosphorylation of Thr-246 represses PRAS40 inhibitory function (9, 10). Similarly, mutation of Ser-221 to Ala also enhanced PRAS40 inhibition of the phosphorylation of S6K1 at Thr-389 and 4E-BP1 at Thr-36/45 induced by insulin (Fig. 5, B and C). In contrast, mutation at Ser-212 did not alter phosphorylation of S6K1 and 4E-BP1 compared with PRAS40 wild type. Therefore, together with Akt-mediated phosphorylation at Thr-246, phosphorylation at Ser-221 due to mTOR activation promotes mTORC1 activity by impairing PRAS40 inhibition action on mTORC1.
DISCUSSION
In this study, we identified two novel sites in PRAS40 phosphorylated by mTORC1: Ser-212 and Ser-221. Like Ser-183, Ser-221 is directly phosphorylated by mTORC1 in vitro, and its phosphorylation is mediated by mTORC1 in vivo, since it was inhibited by rapamycin treatment. Phosphorylations of Ser-221 (Fig. 2A, spot g) and Ser-183 (Fig. 2A, spot a) have similar stoichiometries, but both are phosphorylated much less than Thr-246 (Fig. 2A, spot c) and Ser-212 (Fig. 2A, spot h). It is possible that the observed differences in stoichiometries between the different sites are due to differences in the rate of phosphorylation and dephosphorylation, intracellular specific activity of the ATP available for the kinases, or phosphorylation-targeted protein degradation. Alternatively, the lower amount of phosphorylation at the mTORC1-targeted residues may reflect the relatively small fraction of overexpressed PRAS40 that is associated with mTORC1 and available to be phosphorylated as compared with the amount of PRAS40 that is presumably available for phosphorylation by Akt. Phosphorylation of Ser-221 by mTORC1 appears to be as important as phosphorylation at Thr-246 mediated by Akt in regulating mTORC1 activity and interaction with 14-3-3. PRAS40 is suggested to negatively regulate raptor binding to substrates, such as S6K1 and 4E-BP1 (11, 13). Insulin stimulation activates Akt, which directly phosphorylates PRAS40 at Thr-246 and relieves its inhibition to mTORC1, thus increasing mTORC1 kinase activity. Meanwhile, Akt also activates mTORC1 through the inhibition of TSC2 (tuberous sclerosis complex 2), an upstream inhibitor of mTORC1 (29, 30). Subsequent phosphorylation of PRAS40 at Ser-221 after mTOR activation further relieves the negative effect of PRAS40 on mTORC1 activity. Therefore, phosphorylation of PRAS40 at Ser-221 might contribute to the activation of mTORC1 by integrating mTOR kinase activation by the TSC/Rheb pathway with the release of PRAS40 from raptor, thus allowing mTORC1 access to substrates such as 4E-BP1 and S6K1.
Ser-212 is also phosphorylated by mTORC1 in vitro, and this mTORC1-catalyzed phosphorylation is inhibited by the rapamycin-FKBP12 complex. However, phosphorylation of Ser-212 in vivo is predominantly mediated by a kinase other than mTORC1, because rapamycin treatment and mTOR knockdown did not lead to a decrease in phosphorylation at this site. The phosphorylation of Ser-212 in cells was much stronger than the phosphorylation of Ser-183 and Ser-221, and this also supports the possibility that Ser-212 is phosphorylated by other kinase than mTORC1 in cells. Ser-212 might still be phosphorylated by mTORC1 in vivo, but if we assume that the mTORC1-mediated phosphorylation at Ser-212 is similar to that of Ser-183 and Ser-221, then mTORC1 phosphorylation at Ser-212 is responsible for only a small fraction compared with phosphorylation by another kinase. Phosphorylation of Ser-212 does not appear to regulate mTORC1 activity like Ser-221. However, phosphorylation of Ser-212, predominantly mediated by another kinase, may be important for other cellular functions of PRAS40, such as regulating cell growth and apoptosis.
As shown previously, the amino acid sequence of “KSLP” from 182 to 185 in PRAS40 has similarity with the “RAIP” motif in 4E-BP1 (31) and is important for PRAS40 association with mTORC1 (11). Hypothetically, phosphorylation at Ser-183 could play a role in PRAS40 function and association with mTORC1. However, mutating Ser-183 to other residues, such as Ala, Cys, Asn, and Glu, causes a decrease in the interaction of PRAS40 and mTORC1,3 which seems opposite to the effect of dephosphorylation, since nutrient deficiency leads to increase of PRAS40 and mTORC1 interaction (9, 12). A reasonable explanation could be that mutations in this region (“KSLP”) might alter the normal folding of PRAS40. This effect complicates the study of phosphorylation at Ser-183.
In response to insulin treatment, endogenous PRAS40 is released from mTORC1 (9–12). However, overexpressed PRAS40 has not been observed to disassociate from mTORC1, and in some cases, overexpressed PRAS40 binds more mTORC1 after insulin treatment. This could indicate that overexpressed PRAS40 reassociates with mTORC1 when endogenous PRAS40 disassociates from mTORC1 in response to insulin stimulation. But why is overexpressed PRAS40 unable to release from mTORC1 like endogenous PRAS40 under insulin-activated conditions? Perhaps when overexpressed, there are saturating levels of PRAS40 that can continue to bind mTORC1 even during insulin stimulation. Alternatively, the process of PRAS40 disassociation requires involvement of additional molecules that are not present enough with overexpressed PRAS40. 14-3-3 could be a candidate to participate in this process, since 14-3-3 binding to PRAS40 depends on phosphorylation of PRAS40 at Ser-221 and Thr-246, and overexpression of 14-3-3 relieves PRAS40 inhibition on mTORC1 activity (9). However, we have not observed dissociation of overexpressed PRAS40 from mTORC1 when 14-3-3 is co-overexpressed. The mechanism of PRAS40 release from mTORC1 after insulin treatment requires further investigation.
Acknowledgments
We appreciate Dr. James Garrison for administration of the funding. We thank Dr. Richard Roth, Dr. Pierre Roger, and Dr. Thomas Sturgill for comments on the manuscript. We acknowledge Gina Devasahayam for technical help in peptide mapping.
This work was supported, in whole or in part, by National Institutes of Health Grants DK52753 and DK28312 (to J. C. L.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: mTOR, mammalian target of rapamycin; GST, glutathione S-transferase; TOS, TOR signaling; HA, hemagglutinin; TPCK, l-1-tosylamido-2-phenylethyl chloromethyl ketone; DMEM, Dulbecco's modified Eagle's medium.
L. Wang, unpublished observations.
References
- 1.Wullschleger, S., Loewith, R., and Hall, M. N. (2006) Cell 124 471–484 [DOI] [PubMed] [Google Scholar]
- 2.Yang, Q., and Guan, K. L. (2007) Cell Res. 17 666–681 [DOI] [PubMed] [Google Scholar]
- 3.Brunn, G. J., Fadden, P., Haystead, T. A., and Lawrence, J. C., Jr. (1997) J. Biol. Chem. 272 32547–32550 [DOI] [PubMed] [Google Scholar]
- 4.Burnett, P. E., Barrow, R. K., Cohen, N. A., Snyder, S. H., and Sabatini, D. M. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 1432–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schalm, S. S., and Blenis, J. (2002) Curr. Biol. 12 632–639 [DOI] [PubMed] [Google Scholar]
- 6.Schalm, S. S., Fingar, D. C., Sabatini, D. M., and Blenis, J. (2003) Curr. Biol. 13 797–806 [DOI] [PubMed] [Google Scholar]
- 7.Nojima, H., Tokunaga, C., Eguchi, S., Oshiro, N., Hidayat, S., Yoshino, K., Hara, K., Tanaka, N., Avruch, J., and Yonezawa, K. (2003) J. Biol. Chem. 278 15461–15464 [DOI] [PubMed] [Google Scholar]
- 8.Choi, K. M., McMahon, L. P., and Lawrence, J. C., Jr. (2003) J. Biol. Chem. 278 19667–19673 [DOI] [PubMed] [Google Scholar]
- 9.Vander Haar, E., Lee, S. I., Bandhakavi, S., Griffin, T. J., and Kim, D. H. (2007) Nat. Cell Biol. 9 316–323 [DOI] [PubMed] [Google Scholar]
- 10.Sancak, Y., Thoreen, C. C., Peterson, T. R., Lindquist, R. A., Kang, S. A., Spooner, E., Carr, S. A., and Sabatini, D. M. (2007) Mol. Cell 25 903–915 [DOI] [PubMed] [Google Scholar]
- 11.Wang, L., Harris, T. E., Roth, R. A., and Lawrence, J. C., Jr. (2007) J. Biol. Chem. 282 20036–20044 [DOI] [PubMed] [Google Scholar]
- 12.Oshiro, N., Takahashi, R., Yoshino, K., Tanimura, K., Nakashima, A., Eguchi, S., Miyamoto, T., Hara, K., Takehana, K., Avruch, J., Kikkawa, U., and Yonezawa, K. (2007) J. Biol. Chem. 282 20329–20339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fonseca, B. D., Smith, E. M., Lee, V. H., MacKintosh, C., and Proud, C. G. (2007) J. Biol. Chem. 282 24514–24524 [DOI] [PubMed] [Google Scholar]
- 14.Kovacina, K. S., Park, G. Y., Bae, S. S., Guzzetta, A. W., Schaefer, E., Birnbaum, M. J., and Roth, R. A. (2003) J. Biol. Chem. 278 10189–10194 [DOI] [PubMed] [Google Scholar]
- 15.Harthill, J. E., Pozuelo Rubio, M., Milne, F. C., and MacKintosh, C. (2002) Biochem. J. 368 565–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saito, A., Narasimhan, P., Hayashi, T., Okuno, S., Ferrand-Drake, M., and Chan, P. H. (2004) J. Neurosci. 24 1584–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Madhunapantula, S. V., Sharma, A., and Robertson, G. P. (2007) Cancer Res. 67 3626–3636 [DOI] [PubMed] [Google Scholar]
- 18.Nascimento, E. B., Fodor, M., van der Zon, G. C., Jazet, I. M., Meinders, A. E., Voshol, P. J., Vlasblom, R., Baan, B., Eckel, J., Maassen, J. A., Diamant, M., and Ouwens, D. M. (2006) Diabetes 55 3221–3228 [DOI] [PubMed] [Google Scholar]
- 19.Thedieck, K., Polak, P., Kim, M. L., Molle, K. D., Cohen, A., Jeno, P., Arrieumerlou, C., and Hall, M. N. (2007) PLoS ONE 2 e1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang, L., Rhodes, C. J., and Lawrence, J. C., Jr. (2006) J. Biol. Chem. 281 24293–24303 [DOI] [PubMed] [Google Scholar]
- 21.Hu, C., Pang, S., Kong, X., Velleca, M., and Lawrence, J. C., Jr. (1994) Proc. Natl. Acad. Sci. U. S. A. 91 3730–3734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scott, P. H., and Lawrence, J. C., Jr. (1998) J. Biol. Chem. 273 34496–34501 [DOI] [PubMed] [Google Scholar]
- 23.Lin, T. A., and Lawrence, J. C., Jr. (1994) J. Biol. Chem. 269 21255–21261 [PubMed] [Google Scholar]
- 24.Mothe-Satney, I., Brunn, G. J., McMahon, L. P., Capaldo, C. T., Abraham, R. T., and Lawrence, J. C., Jr. (2000) J. Biol. Chem. 275 33836–33843 [DOI] [PubMed] [Google Scholar]
- 25.Lin, T. A., Kong, X., Saltiel, A. R., Blackshear, P. J., and Lawrence, J. C., Jr. (1995) J. Biol. Chem. 270 18531–18538 [DOI] [PubMed] [Google Scholar]
- 26.Sabers, C. J., Martin, M. M., Brunn, G. J., Williams, J. M., Dumont, F. J., Wiederrecht, G., and Abraham, R. T. (1995) J. Biol. Chem. 270 815–822 [DOI] [PubMed] [Google Scholar]
- 27.Hara, K., Maruki, Y., Long, X., Yoshino, K., Oshiro, N., Hidayat, S., Tokunaga, C., Avruch, J., and Yonezawa, K. (2002) Cell 110 177–189 [DOI] [PubMed] [Google Scholar]
- 28.Mackintosh, C. (2004) Biochem. J. 381 329–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Inoki, K., Li, Y., Zhu, T. Q., Wu, J., and Guan, K. L. (2002) Nat. Cell Biol. 4 648–657 [DOI] [PubMed] [Google Scholar]
- 30.Manning, B. D., Tee, A. R., Logsdon, M. N., Blenis, J., and Cantley, L. C. (2002) Mol. Cell 10 151–162 [DOI] [PubMed] [Google Scholar]
- 31.Tee, A. R., and Proud, C. G. (2002) Mol. Cell. Biol. 22 1674–1683 [DOI] [PMC free article] [PubMed] [Google Scholar]