

Abstract
The pathogenesis of Huntington disease (HD) is attributed to the misfolding of huntingtin (htt) caused by an expanded polyglutamine (polyQ) domain. Considerable effort has been devoted to identifying molecules that can prevent or reduce htt misfolding and the associated neuropathology. Although overexpression of chaperones is known to reduce htt cytotoxicity in cellular models, only modest protection is seen with Hsp70 overexpression in HD mouse models. Because the activity of Hsp70 is modulated by co-chaperones, an interesting issue is whether the in vivo effects of chaperones on polyQ protein toxicity are dependent on other modulators. In the present study, we focused on BAG1, a co-chaperone that interacts with Hsp70 and regulates its activity. Of htt mice expressing the N171-82Q mutant, we found that male N171-82Q mice show a greater deficit in rotarod performance than female N171-82Q mice. This sex-dependent motor deficit was improved by crossing N171-82Q mice with transgenic mice overexpressing BAG1 in neurons. Transgenic BAG1 also reduces the levels of mutant htt in synaptosomal fraction of male HD mice. Overexpression of BAG1 augmented the effects of Hsp70 by reducing aggregation of mutant htt in cultured cells and improving neurite outgrowth in htt-transfected PC12 cells. These findings suggest that the effects of chaperones on HD pathology are influenced by both their modulators and sex-dependent factors.
Huntington disease (HD)2 is an inherited neurodegenerative disease caused by a polyglutamine (polyQ) repeat expansion near the N terminus of the protein huntingtin (htt) (HD collaborative research group (49). Patients with HD suffer an array of cognitive, psychiatric, and motor disorders before death, which typically occurs 10-20 years after onset of symptoms. Pathological hallmarks of this disease include selective neuronal loss and htt inclusions or aggregates that are formed in neuronal nuclei and processes (1, 2). Several genetic mouse models of HD have been developed, and they recapitulate various aspects of the disease observed in patients, including age-dependent inclusion formation and behavioral abnormalities (3). Of these mouse models, the transgenic N171-82Q model, which was generated by using the mouse prion promoter to drive neuronal expression of N-terminal mutant htt, exhibits progressive inclusion formation, motor deficits, weight loss, and shortened life-span (4). The well characterized phenotypes of HD transgenic mice have proven valuable for evaluating potential therapeutics.
Misfolding of mutant htt is thought to contribute to abnormal protein-protein interactions, inclusion formation, and neuronal death. Currently, no treatment exists for those afflicted with HD, although considerable attention has been given to molecules that might alleviate the burden of misfolded htt. The major cytosolic chaperones of the Hsp40 and Hsp70 families act in concert to recognize misfolded proteins and refold them. Hsp70 family members interact with mutant N-terminal htt fragments and localize to inclusions in cellular and mouse models of HD (5). Although Hsp70 can effectively reduce polyQ-mediated cytotoxicity in cultured cells, its effect in HD mouse models is modest or not significant (6, 7). The modest benefits of overexpression of Hsp70 in HD mouse models indicate that Hsp70 alone is insufficient to revert HD pathology and that other modulators are likely required for its action.
The activity of Hsp70 is regulated by its co-chaperones. BAG1 is an Hsp70 co-chaperone that interacts directly with both Hsp70 and the proteasome. Therefore, BAG1 acts as a unique link between the two primary systems for handling misfolded proteins (8). The direct interaction between BAG1 and Hsp70 leads to an increase in Hsp70 chaperone activity in neuronal cell lines (9). Consequently, mice overexpressing BAG1 in neurons display significant neuroprotection in an in vivo stroke model and in an in vitro excitotoxicity model (10). In cellular and mouse models of HD, BAG1 interacts with N-terminal htt fragments and localizes to htt inclusions (11). These findings suggest BAG1 is a strong candidate to assist the effect of Hsp70 on the toxicity caused by mutant htt. However, the in vivo role of BAG1 on HD pathology remains unknown.
To test the effect BAG1 on HD mice, we crossed BAG1 transgenic mice with N171-82Q mice and evaluated levels of soluble and aggregated htt and various chaperones. Although several phenotypic parameters in N171-82Q mice remained unchanged in the presence of transgenic BAG1, we identified a gender-specific improvement in motor deficits in N171-82Q mice. The protective benefit of BAG1 on motor dysfunction of HD mice fits with the previously reported phenomenon of an increased Hsp70 response in male animals (12, 13) and with observations that male HD mice show more severe behavioral phenotypes than female HD mice (14, 15). Our findings suggest that HD pathology is influenced by sex-dependent factors and chaperone function. Thus, evaluation of effects of chaperones or experimental therapeutics on HD pathology should consider these factors.
EXPERIMENTAL PROCEDURES
Animals—Mice expressing a 171-amino acid N-terminal fragment htt containing an 82 polyQ stretch under the mouse prion promoter (4) were maintained on a hybrid (B6CBA) background. To generate N171-82Q mice overexpressing BAG1 in neurons, male N171-82Q mice were mated to female FVB mice expressing the p29 isoform of mouse BAG1 with a FLAG tag under the control of the neuron-specific enolase promoter (10). Three separate groups of F1 littermates resulting from these crosses were used for all experiments described below. Genotyping was carried out according to published protocols (4, 10). All mice were bred and maintained in an animal facility at Emory University in accordance with institutional guidelines.
Antibodies—Mouse (mEM48), rabbit (rEM48), and guinea pig (EM73) antibodies to htt were generated in our previous studies (16, 17). Commercially available antibodies included Hsp90 and Hsp60 (Stressgen); BAG1, Hsp70, and Hsp40 (Santa Cruz Biotechnology, Santa Cruz, CA); α-tubulin (Sigma-Aldrich); and synaptophysin (Roche Applied Science).
Immunoblotting—To quantify protein expression levels of mutant htt and chaperones in mouse forebrain tissue (aged three or five months) or transfected cells, soluble lysates were normalized for total protein content and analyzed by immunoblotting, according to standard procedures. Blots were initially probed for htt and then reprobed for chaperones or the loading control synaptophysin. Relative protein expression levels were determined using ImageQuaNT software (Molecular Dynamics) with synaptophysin levels used as the reference for each sample.
Immunohistochemistry—Histochemical labeling of mutant htt in perfusion-fixed mouse brain sections (aged 4 months) with EM48 was performed as previously described (18).
Rotarod Test—Motor performance was evaluated using an AccuRotor rotarod apparatus (Accuscan Instruments). During the week prior to testing, mice were trained on two consecutive days for two 5-min trials at 5 rpm. Testing began at 15 weeks of age and continued twice weekly for up to 10 weeks. Because of the shortened lifespan of mice expressing mutant htt, we analyzed data collected between 15 and 22 weeks for statistical differences. During testing, the rotating rod was set to accelerate from 0 to 40 rpm at a rate of 0.1 rpm/s. Each mouse performed three trials on each testing day with 5-min resting periods between each trial. The time to fall from the rotating rod was measured and averaged for the three trials. A maximum score of 400 s was recorded for mice that remained on the rod for the duration of the trial.
Limb Clasping—N171-82Q mice exhibit a limb clasping phenotype when suspended from their tails (4). To test the effect of BAG1 overexpression on this phenotype, two parameters were quantified: age of initial onset of the clasping phenotype and the duration to clasping at 145 days of age. Mice were monitored for onset at least twice weekly by suspending from the tail for up to 3 min. Positive onset was noted as the first day during which a mouse sustained clasping of the forelimbs or hind limbs for longer than 3 s. At 145 days of age, all mice expressing mutant htt were positive for the clasping phenotype. At this age, all mice were suspended from their tails for up to 3 min, and the time elapsed before the start of sustained limb clasping was recorded.
Body Weight—Mice were weighed twice weekly starting at 7 weeks of age. Percent change in weight was calculated from the starting baseline.
Survival—Significant decreases in weight and/or rotarod performance were indicators of end stage disease in both N171-82Q and DbTg mice. End stage mice were monitored daily and euthanized when necessary. Survival curves and average age of death were used to evaluate the effect of BAG1 overexpression on N171-82Q mouse survival.
Expression of htt and Chaperones in PC12 Cells—Transfection of PC12 cells was performed as described previously (17). To assess the protective effects of chaperones on neurite out-growth of PC12 cells, BAG1 (pcDNA3 vector) and Hsp70 (pRK vector) (16) were co-expressed with N208-htt containing 120Q. After transfection for 5 h, the cells were treated with nerve growth factor (100 ng/ml) for 40 h. For single transfection of htt, anti-tubulin was used to visualize neurites. For co-transfected cells, mouse (mEM48) or guinea pig (EM73) antibodies to htt and antibodies to BAG1 and Hsp70 were used for immunofluorescent staining. The percentage of transfected cells (71-232 cells each group in three transfection experiments) containing neurites longer than two cell bodies was counted.
Knockdown of BAG1—Plasmids encoding BAG1 siRNA (siBAG1A and siBAG1C) (25) were co-transfected with N208-htt containing 120Q into HEK 293 cells. After 72 h, cells were fixed and immunostained with mEM48 to examine the aggregation of mutant htt. Whole cell lysates were also immunoblotted according to standard procedures to examine the expression of BAG1 and the accumulation and aggregation of mutant htt.
Synaptosomal Fractionation—Synaptosomal fractions were prepared from brains of 3- to 4-month-old mice following a previously described method (19) with minor changes. The protocol was scaled down to allow isolation of synaptosomes from individual mouse brains. After ultracentrifugation, isolated synaptosomes were rinsed twice with homogenization buffer prior to immunoblotting.
Statistics—Student's t test, and repeated measure and factorial analysis of variance with Tukey post-test were used to determine statistical significance between groups. p < 0.05 was considered significant in all cases.
RESULTS
Expression of Chaperones in N171-82Q Mice—As transgenic expression of the Hsp70 co-chaperone BAG1 can protect against stroke (10), we examined the effect of BAG1 on HD pathogenesis by crossing the BAG1 transgenic mice with N171-82Q mice, which express N-terminal mutant htt (171 amino acids) containing an 82-glutamine repeat (4). Using immunoblot analysis of brain cortical lysates from mice at 5 months of age, we first verified the expression of transgenic BAG1 in double transgenic mice (DbTg) that also expressed transgenic htt (Fig. 1A). Mutant htt was present as both soluble and aggregated forms, with the latter remaining in the stacking gel (inner bracket). Overexpression of BAG1 did not appear to alter the levels of these two forms of mutant htt. Hsp70 levels were decreased in N171-82Q mice, confirming previous reports of reduced Hsp70 expression in HD mouse models (6, 7, 20). However, transgenic BAG1 was unable to reverse this deficit. Next, we examined the expression of other chaperones (Hsp90 and Hsp60) (Fig. 1A) and quantified the relative amounts of aggregated or soluble htt and chaperones by measuring their ratios relative to the synaptic protein synaptophysin (Fig. 1B). Interestingly, Hsp90 levels were increased in the N171-82Q brain compared with WT mouse brain, and BAG1 overexpression seemed to prevent this increase in Hsp90 in DbTg animals. No difference in Hsp60 levels was seen between any of the groups tested. These results suggest that HD transgenic mice have selective alterations in their chaperone protein levels or activities.
FIGURE 1.

Expression of chaperones and htt in transgenic mice. A, immunoblot analysis of brain cortical lysates from wild-type (WT), BAG1 transgenic, N171-82Q (Tg), and BAG1/N171-82Q double transgenic (DbTg) mice at 5 months of age. The blots were probed with rabbit EM48 to detect aggregated (agg) htt (inner bracket) in the stacking gel and soluble (sol) htt in the resolving gel. Blots were subsequently probed with antibodies to Hsp70, Hsp90, Hsp60, and BAG1. Transgenic BAG1 with a FLAG tag and endogenous BAG1 are indicated. The samples were also probed for the neuronal protein synaptophysin to reveal the loading amounts of proteins. B, densitometric quantification of the relative levels of htt and chaperones in transgenic mice. The ratios of htt (aggregated and soluble) or chaperones (Hsp70, Hsp60, and Hsp90) to synaptophysin are presented. *, p < 0.05 compared with wild type (WT).
htt Aggregates—Chaperones are important for preventing protein misfolding and aggregation. In HD mouse brains, mutant htt forms aggregates in neuronal nuclei and neuropil (4, 21, 22). To evaluate the effect of transgenic BAG1 on aggregate formation in vivo, we performed EM48 immunostaining on 4-month-old N171-82Q and DbTg mouse brains (Fig. 2). High magnification images revealed that the size and density of nuclear htt inclusions were similar in N171-82Q and DbTg mouse striatum and cortex. Neuropil aggregates, which are much smaller than nuclear inclusions and located outside of cell bodies, did not appear to be significantly changed in DbTg mouse brains as compared with N171-82Q mouse brains. Thus, using immunohistochemistry (Fig. 2) and Western blotting of whole cell lysates (Fig. 1), we were unable to find significant changes of aggregated htt in the presence of transgenic BAG1.
FIGURE 2.

Nuclear htt inclusions and neuropil aggregates in N171-82Q and N171-82Q/BAG1 double transgenic mice. EM48 staining of the cortex (left panel) and striatum (right panel) of N171-82Q and double transgenic mice (DbTg) at 4 months of age. Note that mutant htt forms nuclear inclusions and neuropil aggregates.
Neurological Symptoms—N171-82Q mice display significant neurological symptoms, including limb clasping, motor deficits, body weight loss, and early death at the age of 5-6 months. These mice failed to gain weight after 12 weeks of age and weighed significantly less than wild-type littermates by 20 weeks of age (Fig. 3A). BAG1 transgenic mice increased in weight at the same rate as wild-type mice. However, no significant difference was observed in weight between N171-82Q and DbTg mice at any age (Fig. 3A). N171-82Q mice showed an increased mortality compared with both wild-type and BAG1 mice (Fig. 3B), but transgenic BAG1 was unable to prolong the survival of DbTg mice or to delay the age at death (Fig. 3, B and C). Thus, like transgenic Hsp70 (6, 7), transgenic BAG1 is unable to influence the age of onset of symptoms, body weight loss, and early death of HD transgenic mice. In addition, neither age of onset nor duration to onset of the hind limb clasping phenotype was significantly different between N171-82Q and DbTg mice (Fig. 3, D and E).
FIGURE 3.

Neurological phenotypes of N171-82Q (Tg) and N171-82Q/BAG1 (DbTg) double transgenic mice. Body weight (A), survival (B), age at death (C), and the age of onset (D) and duration to onset (E) of the clasping phenotype of N171-82Q and DbTg mice were examined. The data are mean ± S.E. from three separate groups of F1 littermates.
We then focused on the motor function of N171-82Q mice, which is more likely to reflect neuronal dysfunction in HD. We observed a significant gender-dependent deficit in rotarod performance in N171-82Q mice. Although male wild-type or BAG transgenic mice showed slightly better performance than female littermates of the same genotype (Fig. 4, A and B), male N171-82Q mice performed significantly worse than female N171-82Q mice at all ages examined (Fig. 4C). In DbTg mice expressing both BAG1 and mutant htt, both males and females performed equally well on the rotarod test (Fig. 4D). To verify that the sex-dependent difference on rotarod performance is dependent on the expression of transgenic BAG1, we compared the rotarod performance among N171-82Q and DbTg mice. Interestingly, male DbTg mice performed significantly better than male N171-82Q mice (Fig. 4E), whereas no significant difference was observed in rotarod performance between female N171-82Q and DbTg mice (Fig. 4F).
FIGURE 4.

Alleviation of motor deficits in male HD transgenic mice by transgenic BAG1. The rotarod test was performed to examine latency to fall (seconds) of transgenic mice. A and B, no significant difference was observed in rotarod performance between male and female wild-type (A) or BAG1 transgenic (B) mice. C, male N171-82Q (Tg) mice show a significant decrease in latency to fall compared with female N171-82Q mice. D, male and female double transgenic (DbTg) mice performed equally well on the rotarod test. E, comparison of male N171-82Q and DbTg mice revealed that mice overexpressing BAG1 performed significantly better than N171-82Q mice. *, p < 0.05. F, no significant difference in rotarod performance was seen between female N171-82Q and DbTg mice. The data are presented as mean ± S.E. from three separate groups of F1 littermates. *, p < 0.05; **, p < 0.01.
Gender-related Expression of Hsp70—The above findings suggest that the motor deficit of N171-82Q mice is influenced by gender and that this gender-related deficit can be reversed or delayed by transgenic BAG1. These findings led us to examine more closely whether gender-dependent differences in expression of htt and Hsp70 occur in brain. Consistent with the results in Fig. 1, transgenic HD mouse brains displayed a decrease in Hsp70 expression (Fig. 5A). We did not find any significant difference in the expression of endogenous and transgenic BAG1 between male and female mice (supplemental Fig. S2). Analysis of whole cell lysates did not reveal a significant effect of BAG1 on the levels of mutant htt (Fig. 5A). As mutant htt accumulates in nerve terminals to form aggregates and to affect synaptic function in HD mice, we isolated synaptosomal fractions to examine the expression of mutant htt and Hsp70, an assay that would be more quantitative than immunohistochemistry to reveal soluble htt. Unlike the whole cell extracts (Fig. 5A), synaptosomal fractions show a clearer doublet of transgenic htt (Fig. 5B). The lower band of this doublet in N171-82Q mouse brain tissue is likely a degraded htt product as reported previously (4). The increased level of the degraded htt product in synaptosomal fractions could be due to increased synaptic proteolysis or decreased activity of ubiquitin-proteasomal system seen in the synapses of HD mouse brains (23). Thus, we chose the intact transgenic htt (the upper band) to assess the expression of transgenic htt in synaptosomal fractions. By quantifying the ratio of this band to the synaptic protein syntaxin, we verified that the relative level of mutant htt in synaptosomes from male transgenic mice is significantly decreased in the presence of transgenic BAG1 (Fig. 5C). Synaptosomal mutant htt from female HD mice was also reduced, although not statistical significantly, when transgenic BAG1 is present. Analysis of Hsp70 expression revealed that Hsp70 is indeed decreased in N171-82Q mouse brain as compared with wild-type mice, and transgenic BAG1 does not seem to significantly revert this decrease. This result suggests that overexpression of BAG1 may increase chaperone activity, rather than its expression, to reduce the accumulation of mutant htt. Interestingly, the levels of Hsp70 in whole cell lysates and synaptosomes appeared to be lower in male HD mice than in female HD mice (Fig. 5C). The gender-related difference in the level of Hsp70 in HD mouse brains could contribute to the sex-dependent effect of mutant htt on motor function of transgenic N171-82Q mice. Accordingly, BAG1 overexpression-induced chaperone activity may be more pronounced in male mice when Hsp70 level or activity is reduced by sex-dependent factors.
FIGURE 5.
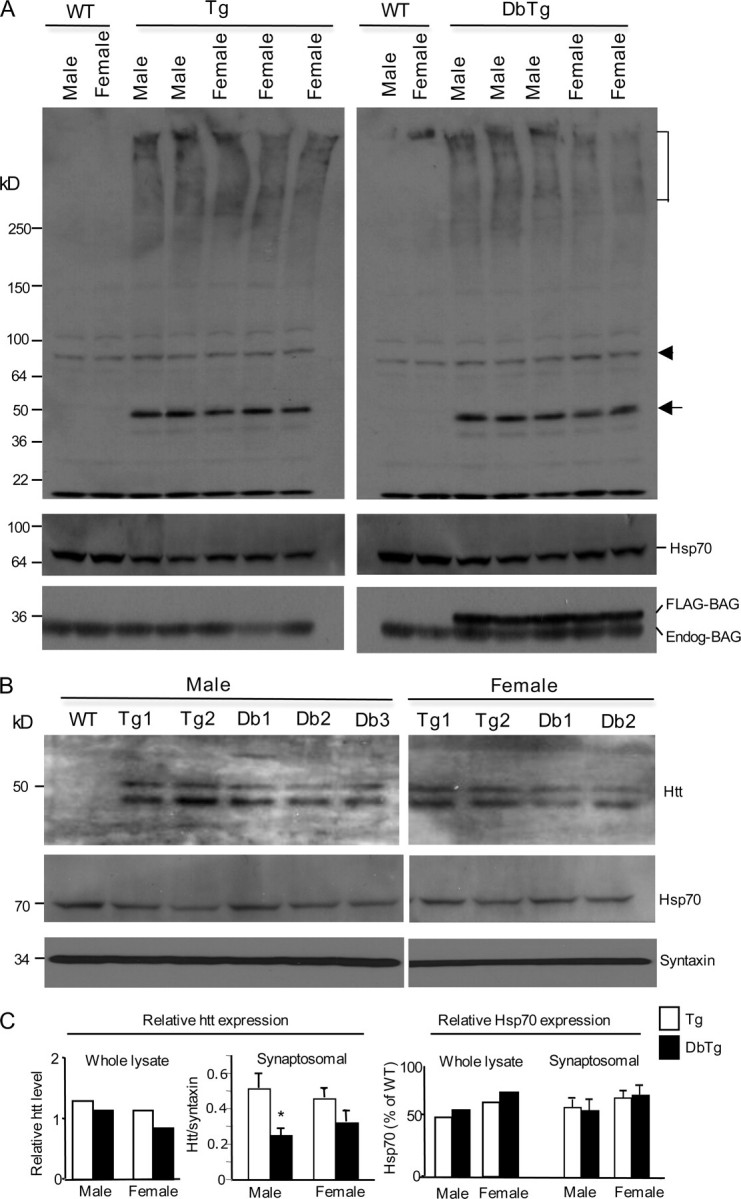
Expression of Hsp70 in male and female HD mice. A, immunoblots showing the expression of mutant htt, Hsp70, transgenic BAG1 (FLAG-BAG), and endogenous BAG (Endog-BAG) in the soluble lysates from the forebrain tissue of wild type (WT), N171-82Q (Tg), and double transgenic (DbTg) mice. The bracket indicates the stacking gel. The arrow indicates soluble transgenic htt, and the arrowhead indicates a nonspecific product that served as a loading control. The samples were probed with antibodies to htt (rabbit EM48, upper blot), Hsp70, and BAG1. Two to three male and female HD mice of each genotype were examined. B, Western blotting of synaptosomal fractions from 3- to 4-month-old male and female N171-82Q (Tg) or double transgenic (Db) mice expressing N171-82Q and BAG1. The blots were probed with mEM48 for transgenic htt (upper panel) and antibodies to Hsp70 (middle panel) and the synaptic protein synatxin (lower panel). Note that a smaller band or degraded product of mutant htt is more pronounced in synaptosomal fractions. C, densitometry analysis of the ratio of transgenic htt to the loading control in whole cell lysates or syntaxin in the synaptosomal fraction in N171-82Q (Tg) or double transgenic (DbTg) mice. The intact form or upper band of transgenic htt was used for quantification. The relative level of Hsp70 represents the percentage of Hsp70 level relative to the Hsp70 level in the wild-type mouse sample. The data (mean ± S.E.) were obtained from five to six samples of each group of two Western blotting experiments. *, p < 0.05.
Synergistic Effect of BAG1 and Hsp70—If cofactors of Hsp70 are important for restoring the activity of Hsp70 when its levels are reduced in vivo, we may see their synergistic effects in vitro. Because BAG1 is both a co-chaperone and regulator of Hsp70 (9), we next asked whether BAG1 can promote the protective effects of Hsp70 on mutant htt toxicity in cellular models of HD. We transfected Hsp40 alone or BAG1 with Hsp70 into HEK293 cells expressing an N-terminal fragment of mutant htt similar to the fragment expressed in N171-82Q mice. Confirming previous findings (24), overexpression of Hsp40, but not Hsp70, alone inhibited the formation of mutant htt aggregates (Fig. 6A). Immunofluorescence studies suggested that expression of BAG1 alone led to a slight reduction in the size of mutant htt aggregates and that co-expression of BAG1 with Hsp70 appeared to reduce aggregate size and aggregate number further (Fig. 6A). We also used immunoblotting to reveal aggregated proteins that remained in the stacking gel. As expected, Hsp40 overexpression markedly reduced the aggregation of mutant htt, which is reflected by the reduced levels of htt immunoreactive products in the stacking gel and the increased amount of soluble htt in the resolving gel compared with 120Q-htt alone (Fig. 6B). BAG1 transfection reduced htt aggregation. Although Hsp70 transfection alone did not significantly affect htt aggregates as compared with vector transfection, its co-expression with BAG1 also reduced htt aggregates (Fig. 6B). Further, suppressing BAG1 expression via siRNA (25) in HEK293 cells increased the accumulation and aggregation of transfected mutant htt (Fig. 7).
FIGURE 6.
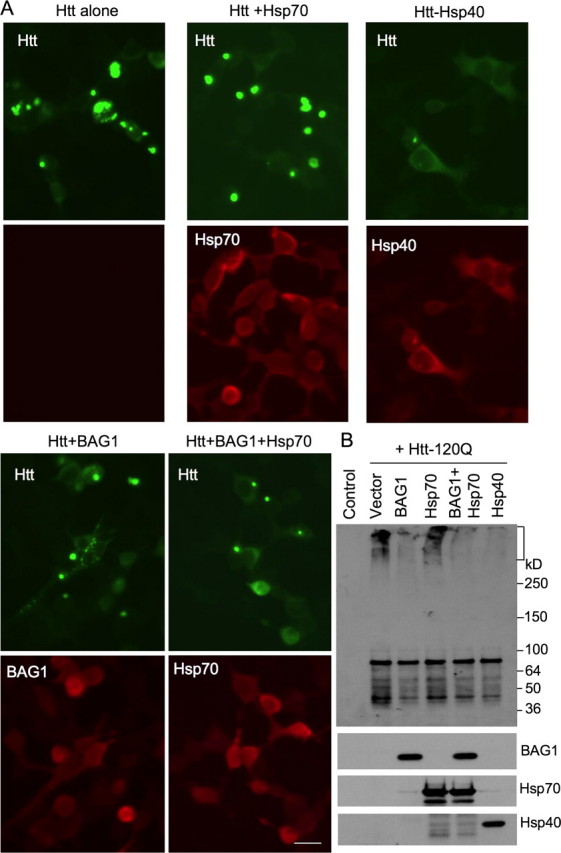
Co-expression of chaperones and mutant htt in HEK293 cells. A, HEK293 cells were transfected with N208-120Q htt alone or together with Hsp70, Hsp40, or BAG1. Cells were stained with antibodies to htt and chaperones and visualized via fluorescent microscopy. B, transfected HEK293 cells were analyzed by immunoblotting. The samples were probed with antibodies to htt (mEM48, upper blot), BAG1, Hsp70, and Hsp40.
FIGURE 7.

Increased htt aggregation in HEK293 cells treated with BAG1 siRNA. A, immunofluorescent staining of HEK293 cells that were transfected with N208-120Q (control) or N208-120Q with plasmids encoding BAG1 siRNA. The cells were examined 3 days after transfection. Note that co-transfection of BAG1 siRNA increased the size and number of htt aggregates. B, Western blot analysis of the expression of transfected htt and endogenous BAG1 in HEK293 cells. BAG1 siRNA transfection reduced the level of endogenous BAG1 and increased the aggregation of transfected htt seen in the stacking gel (bracket). The blots were probed with antibodies to htt (mEM48, upper panel), BAG1 (middle panel), and tubulin (lower panel).
To determine the effect of chaperone expression on cell function, we measured neurite extension in PC12 cells after stimulation by nerve growth factor. Mutant htt is known to affect this neuron-like property via nerve growth factor signaling impairment (17, 26). Misfolded htt could also impair overall health of PC12 cells, leading to reduced neurite extension. Also, BAG1 overexpression induces differentiation of the neuronal cell line CSM14.1 (27). When PC12 cells were transfected with N-terminal mutant htt (208 amino acids with 120Q), a significant decrease was observed in the number of cells extending their neurites longer than two cell bodies compared with PC12 cells transfected with normal htt (208 amino acids with 23Q; Fig. 8, A and B, and supplemental Fig. S1). Expression of either BAG1 or Hsp70 alone increased the number of mutant htt-transfected PC12 cells with long neurites, whereas co-expression of BAG1 and Hsp70 together potentiated the improvement of the number of HD cells with neurites (Fig. 8B). These results indicate that BAG1 and Hsp70 act synergistically to protect against toxicity of mutant htt in neuronal cells.
FIGURE 8.
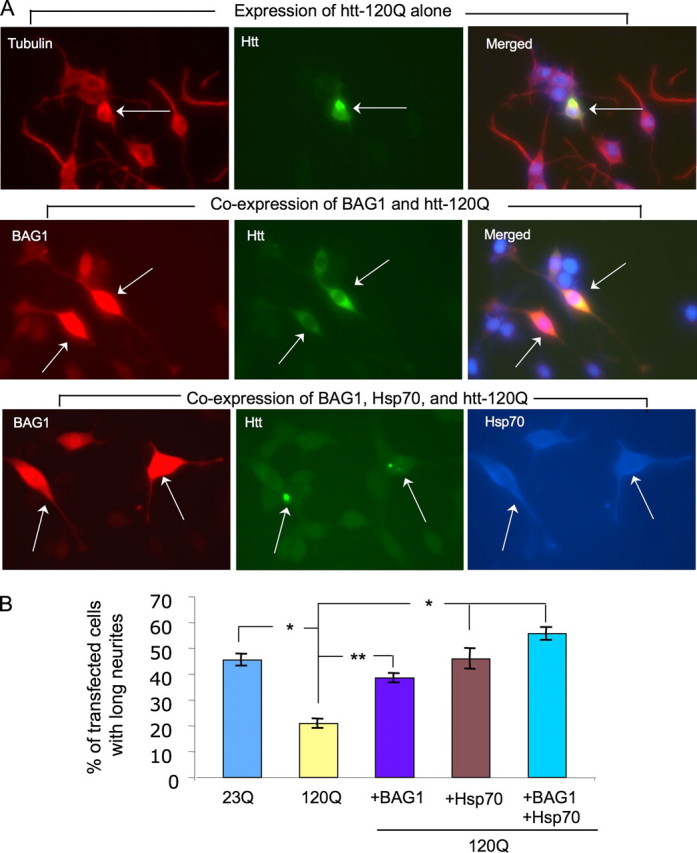
Chaperone protection against mutant htt toxicity in PC12 cells. A, transfection of mutant htt (N208-120Q) into PC12 cells (arrow) reduces nerve growth factor-induced neurite outgrowth (upper panel). Cells co-expressing BAG1 and mutant htt develop neurites (middle panel). Long neurites are also seen in triple transfected cells that expressed BAG1 (red), mutant htt (green), and Hsp70 (blue) (bottom panel). Arrows indicate transfected PC12 cells. Images for expression of normal htt (N208-23Q) or BAG1 alone and Hsp70 with mutant htt are presented in supplemental Fig. S1. B, quantitative assessment of PC12 cells with neurites longer than two cell bodies. The percentage of transfected PC12 cells showing long neurites was calculated for each group. The data are presented as mean ± S.E. and were obtained from three transfections. *, p < 0.05; **, p < 0.01 compared with mutant htt-transfected cells (120Q).
DISCUSSION
The expansion of the polyQ repeat causes htt misfolding and leads to abnormal protein interactions and neuronal death. In support of this notion, the length of the polyQ domain correlates inversely with the age of onset of disease symptoms (28). In the majority of HD patients, however, neurological symptoms occur in mid-life. It is possible that the late onset of symptoms results from the age-dependent accumulation of toxic htt products due to the decline in the protein handling capacity of aging neurons (16). Consistently, human HD patients and mouse models of HD show age-dependent accumulation of short N-terminal mutant htt fragments in the form of insoluble proteins or inclusions in neuronal nuclei and neuritic processes (1, 2, 29). Although inclusion formation is thought to be a cellular self-defense mechanism for handling overwhelming amounts of misfolded and toxic fragments (30), large inclusions could be harmful if they disrupt cellular cytoskeleton structure and intracellular trafficking (31). Thus, a more efficient way to prevent toxicity of misfolded proteins is to reduce or prevent protein misfolding. Chaperones are able to refold proteins and prevent protein misfolding. The decreased levels of chaperones observed in patients and animal models of HD (6, 7, 20) suggest that this important protective ability is impaired in HD. Although studies in cell models of HD provide compelling evidence that overexpressed chaperones can protect against mutant htt toxicity (24, 32-35), overexpression of Hsp70 alone in HD mouse models yields only modest benefit or no significant protection (6). It is possible that other chaperone modulators are also required for chaperone protective function when chaperones are expressed at the endogenous level. For example, the C terminus of Hsc70 (heat shock cognate protein 70)-interacting protein, a ubiquitin ligase, has been shown to protect against toxicity caused by different polyglutamine proteins (11, 36-38). BAG1 is an Hsp70 co-chaperone that physically links members of the Hsp70 chaperone family to proteins of the ubiquitin-proteasome degradation complex (8). Transgenic mice overexpressing BAG1 in neurons are resistant to stroke-induced neurotoxicity in vivo as well as excitotoxins in vitro (10). Although little is known about the role of BAG1 in HD pathogenesis, a recent report suggests that BAG1 co-localizes with mutant htt inclusions (11).
By crossing BAG1 transgenic mice with N171-82Q mice, we anticipated that BAG1 overexpression would reduce the amount of misfolded htt or its aggregates in the mouse brain. However, immunoblots and immunocytochemistry of brain tissue from HD mice showed no significant alteration of the levels of soluble htt fragments and htt aggregates. Similarly, overexpression of Hsp70 in brain tissue does not affect neuronal htt aggregates in R6/2 mice (6). It was reported that N171-82Q mice crossed with mice overexpressing yeast Hsp104 displayed a reduced aggregate load despite no change in the diminished levels of Hsp70 in these mice (20). These results suggest that yeast chaperones may have different protective functions on mutant htt aggregation than Hsp70 and its co-chaperones. The lack of obvious effects of BAG1 and Hsp70 on htt aggregation in the brain, which occurs over days or weeks, suggests that these chaperones may only act on soluble mutant htt but not aggregated htt in vivo. Soluble htt is probably more toxic than aggregated htt, because it is able to bind various partners while aggregated htt forms a compact structure that can reduce the ability of the sequestered mutant htt to interact with other partners. However, significant protective effects of chaperones on htt misfolding were observed in our cellular models of HD. This difference from the in vivo situation is perhaps because the chaperones were overexpressed in transfected cells in which the transfected mutant htt formed aggregates rapidly in vitro. Further, transgenic chaperone levels in the brain are lower than that of transfected chaperones in cultured cells, and their major impact is likely on the soluble form of mutant htt. In addition, the in vivo protective effect of transgenic BAG1 is determined by its expression driven by the neuron-specific enolase promoter that may not equally express transgene in various types of neuronal cells. The modest protective effect of BAG1 could also implicate that BAG1 alone is not sufficient to markedly alleviate neuronal toxicity of mutant htt. Indeed, the sex-dependent effect of BAG1 in HD mice suggests that the protective function of BAG1 is at least modulated by sex-related factors.
Our evaluation of motor function in N171-82Q mice revealed a gender-specific deficit in which the male N171-82Q mice were more severely impaired than their female counterparts (Fig. 4C). Importantly, this impairment was reversed by neuronal expression of transgenic BAG1 in male DbTg mice (Fig. 4E). Previous characterization of motor function of N171-82Q mice did not look at the influence of gender (4). To our knowledge, our finding is the first report of a gender-specific effect on motor function of HD mouse models.
In light of the significant benefits seen with transgenic BAG1 in other models of neurodegeneration (10), it is interesting that BAG1 did not offer protection against other phenotypes such as the loss of body weight and shortened survival of N171-82Q mice. Further, no effect of gender was found on these severe symptoms. Thus, the specific gender difference for BAG1 protection against motor deficits has several implications. First, a recent finding shows that N171-82Q mice have severe metabolic defects that could be caused by the peripheral effects of mutant htt (39), which may not be influenced by overexpressing BAG1 and chaperones in the brain. Second, the protective effects of chaperones may be particularly effective against neuronal dysfunction caused by certain forms of misfolded mutant htt. In support of this idea, overexpression of Hsp70 only improves motor function of HD mice without any effect on other severe symptoms (6, 7). Third, the effect of chaperones is also dependent on other modulators such as sex-related cellular function and modulation.
The gender-specific difference on motor performance in N171-82Q mice underscores the importance in identifying modifiers other than polyglutamine expansion. Also, transgenic BAG1 can selectively alleviate motor deficits in male HD mice, suggesting that chaperone function is dependent on gender as well. In this regard, various chaperones, including BAG1 and Hsp70, interact with and modulate the activity of steroid receptors (40). Recent studies also demonstrate gender differences in both basal and inducible levels of various chaperones (41-44) and that such gender-specific differences can be modulated by sex steroids (45, 46). For example, estradiol increases chaperone expression in brain tissue (47), whereas testosterone inhibits the up-regulation of Hsp70 and Hsp90 in brain tissue following ischemia/reperfusion in rats (48). It should be noted that the neuroprotective effects of BAG1 on stroke were observed in male mice (10). Although we did not find significantly different levels of Hsp70 between male and female wild-type mice, it is possible that a cell-type-specific difference exists in chaperone expression or activity between male and females, which could not be revealed by immunoblotting of whole cell extracts. Because the brain level of Hsp70 appeared to be lower in male HD mice than in female HD mice, the level of Hsp70 is also affected by other sex-dependent factors. When the level of Hsp70 is decreased, its co-chaperones may have a greater impact on its function. This possibility may explain the sex-dependent degree of motor deficit in HD mice and gender-related improvement of this deficit by BAG1. Accordingly, sex-dependent modulators should be considered when evaluating the protective effects of chaperones and other therapeutics against the neuronal toxicity caused by mutant polyQ proteins.
Supplementary Material
Acknowledgments
We thank C. L. Kress and Z. H. Fang for technical assistance and R. Zhou and H. K. Manji at the National Institutes of Health for BAG1 siRNA plasmids.
This work was supported, in whole or in part, by National Institutes of Health Grants AG019206, NS045106, CA6739, AG15393, and NS041669. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
Footnotes
The abbreviations used are: HD, Huntington disease; polyQ, polyglutamine; htt, huntingtin; siRNA, small interference RNA; DbTg, double transgenic.
References
- 1.DiFiglia, M., Sapp, E., Chase, K. O., Davies, S. W., Bates, G. P., Vonsattel, J. P., and Aronin, N. (1997) Science 2771990 -1993 [DOI] [PubMed] [Google Scholar]
- 2.Gutekunst, C. A., Li, S. H., Yi, H., Mulroy, J. S., Kuemmerle, S., Jones, R., Rye, D., Ferrante, R. J., Hersch, S. M., and Li, X. J. (1999) J. Neurosci. 192522 -2534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rubinsztein, D. C. (2002) Trends Genet. 18202 -209 [DOI] [PubMed] [Google Scholar]
- 4.Schilling, G., Becher, M. W., Sharp, A. H., Jinnah, H. A., Duan, K., Kotzuk, J. A., Slunt, H. H., Ratovitski, T., Cooper, J. K., Jenkins, N. A., Copeland, N. G., Price, D. L., Ross, C. A., and Borchelt, D. R. (1999) Hum. Mol. Genet. 8 397-407 [DOI] [PubMed] [Google Scholar]
- 5.Jana, N. R., Tanaka, M., Wang, G., and Nukina, N. (2000) Hum. Mol. Genet. 92009 -2018 [DOI] [PubMed] [Google Scholar]
- 6.Hansson, O., Nylandsted, J., Castilho, R. F., Leist, M., Jaattela, M., and Brundin, P. (2003) Brain Res. 970 47-57 [DOI] [PubMed] [Google Scholar]
- 7.Hay, D. G., Sathasivam, K., Tobaben, S., Stahl, B., Marber, M., Mestril, R., Mahal, A., Smith, D. L., Woodman, B., and Bates, G. P. (2004) Hum. Mol. Genet. 131389 -1405 [DOI] [PubMed] [Google Scholar]
- 8.Alberti, S., Esser, C., and Hohfeld, J. (2003) Cell Stress Chaperones 8 225-231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liman, J., Ganesan, S., Dohm, C. P., Krajewski, S., Reed, J. C., Bahr, M., Wouters, F. S., and Kermer, P. (2005) Mol. Cell. Biol. 253715 -3725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kermer, P., Digicaylioglu, M. H., Kaul, M., Zapata, J. M., Krajewska, M., Stenner-Liewen, F., Takayama, S., Krajewski, S., Lipton, S. A., and Reed, J. C. (2003) Brain Pathol. 13 495-506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jana, N. R., and Nukina, N. (2005) Neurosci. Lett. 378171 -175 [DOI] [PubMed] [Google Scholar]
- 12.Paroo, Z., Haist, J. V., Karmazyn, M., and Noble, E. G. (2002) Circ. Res. 90 911-917 [DOI] [PubMed] [Google Scholar]
- 13.Thorp, D. B., Haist, J. V., Leppard, J., Milne, K. J., Karmazyn, M., and Noble, E. G. (2007) Am. J. Physiol. 293R363 -R371 [DOI] [PubMed] [Google Scholar]
- 14.Van Raamsdonk, J. M., Pearson, J., Rogers, D. A., Bissada, N., Vogl, A. W., Hayden, M. R., and Leavitt, B. R. (2005) Hum. Mol. Genet. 141379 -1392 [DOI] [PubMed] [Google Scholar]
- 15.Dorner, J. L., Miller, B. R., Barton, S. J., Brock, T. J., and Rebec, G. V. (2007) Behav. Brain Res. 178 90-97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou, H., Cao, F., Wang, Z., Yu, Z. X., Nguyen, H. P., Evans, J., Li, S. H., and Li, X. J. (2003) J. Cell Biol. 163109 -118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rong, J., McGuire, J. R., Fang, Z. H., Sheng, G., Shin, J. Y., Li, S. H., and Li, X. J. (2006) J. Neurosci. 266019 -6030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li, S. H., Yu, Z. X., Li, C. L., Nguyen, H. P., Zhou, Y. X., Deng, C., and Li, X. J. (2003) J. Neurosci. 236956 -6964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Phillips, G. R., Huang, J. K., Wang, Y., Tanaka, H., Shapiro, L., Zhang, W., Shan, W. S., Arndt, K., Frank, M., Gordon, R. E., Gawinowicz, M. A., Zhao, Y., and Colman, D. R. (2001) Neuron 3263 -77 [DOI] [PubMed] [Google Scholar]
- 20.Vacher, C., Garcia-Oroz, L., and Rubinsztein, D. C. (2005) Hum. Mol. Genet. 143425 -3433 [DOI] [PubMed] [Google Scholar]
- 21.Li, S. H., and Li, X. J. (1998) Hum. Mol. Genet. 7777 -782 [DOI] [PubMed] [Google Scholar]
- 22.Li, S. H., Li, H., Torre, E. R., and Li, X. J. (2000) Mol. Cell Neurosci. 16168 -183 [DOI] [PubMed] [Google Scholar]
- 23.Wang, J., Wang, C. E., Orr, A., Tydlacka, S., Li, S. H., and Li, X. J. (2008) J. Cell Biol. 1801177 -1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou, H., Li, S. H., and Li, X. J. (2001) J. Biol. Chem. 27648417 -48424 [DOI] [PubMed] [Google Scholar]
- 25.Zhou, R., Gray, N. A., Yuan, P., Li, X., Chen, J., Chen, G., Damschroder-Williams, P., Du, J., Zhang, L., and Manji, H. K. (2005) J. Neurosci. 254493 -4502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wyttenbach, A., Sauvageot, O., Carmichael, J., Diaz-Latoud, C., Arrigo, A. P., and Rubinsztein, D. C. (2002) Hum. Mol. Genet. 111137 -1151 [DOI] [PubMed] [Google Scholar]
- 27.Kermer, P., Krajewska, M., Zapata, J. M., Takayama, S., Mai, J., Krajewski, S., and Reed, J. C. (2002) Cell Death Differ 9405 -413 [DOI] [PubMed] [Google Scholar]
- 28.Duyao, M., Ambrose, C., Myers, R., Novelletto, A., Persichetti, F., Frontali, M., Folstein, S., Ross, C., Franz, M., Abbott, M., Gray, J., Conneally, P., Young, A., Penney, J., Hollingsworth, Z., Shoulson, I., Lazzarini, A., Falek, A., Koroshetz, W., Sax, D., Bird, E., Vonsattel, J., Bonilla, E., Alvir, J., Bickham Conde, J., Cha, J. H., Dure, L., Gomez, F., Ramos, M., Sanchez-Ramos, J., Snodgrass, S., de Young, M., Wexler, N., Moscowitz, C., Penshaszadeh, G., MacFarlane, H., Anderson, M., Jenkins, B., Srinidhi, J., Barnes, G., Gusella, J., Macdonald, M. (1993) Nat. Genet. 4387 -392 [DOI] [PubMed] [Google Scholar]
- 29.Lin, C. H., Tallaksen-Greene, S., Chien, W. M., Cearley, J. A., Jackson, W. S., Crouse, A. B., Ren, S., Li, X. J., Albin, R. L., and Detloff, P. J. (2001) Hum. Mol. Genet. 10 137-144 [DOI] [PubMed] [Google Scholar]
- 30.Arrasate, M., Mitra, S., Schweitzer, E. S., Segal, M. R., and Finkbeiner, S. (2004) Nature 431805 -810 [DOI] [PubMed] [Google Scholar]
- 31.Chang, D. T., Rintoul, G. L., Pandipati, S., and Reynolds, I. J. (2006) Neurobiol. Dis. 22 388-400 [DOI] [PubMed] [Google Scholar]
- 32.Bonini, N. M. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 Suppl. 4, 16407-16411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan, H. Y., Warrick, J. M., Gray-Board, G. L., Paulson, H. L., and Bonini, N. M. (2000) Hum. Mol. Genet. 92811 -2820 [DOI] [PubMed] [Google Scholar]
- 34.Paulson, H. L. (1999) Am. J. Hum. Genet. 64339 -345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sakahira, H., Breuer, P., Hayer-Hartl, M. K., and Hartl, F. U. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 Suppl. 4,16412 -16418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adachi, H., Waza, M., Katsuno, M., Tanaka, F., Doyu, M., and Sobue, G. (2007) Neuropathol. Appl. Neurobiol. 33 135-151 [DOI] [PubMed] [Google Scholar]
- 37.Al-Ramahi, I., Lam, Y. C., Chen, H. K., de Gouyon, B., Zhang, M., Perez, A. M., Branco, J., de Haro, M., Patterson, C., Zoghbi, H. Y., and Botas, J. (2006) J. Biol. Chem. 28126714 -26724 [DOI] [PubMed] [Google Scholar]
- 38.Miller, V. M., Nelson, R. F., Gouvion, C. M., Williams, A., Rodriguez-Lebron, E., Harper, S. Q., Davidson, B. L., Rebagliati, M. R., and Paulson, H. L. (2005) J. Neurosci. 259152 -9161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weydt, P., Pineda, V. V., Torrence, A. E., Libby, R. T., Satterfield, T. F., Lazarowski, E. R., Gilbert, M. L., Morton, G. J., Bammler, T. K., Strand, A. D., Cui, L., Beyer, R. P., Easley, C. N., Smith, A. C., Krainc, D., Luquet, S., Sweet, I. R., Schwartz, M. W., and La Spada, A. R. (2006) Cell Metab. 4 349-362 [DOI] [PubMed] [Google Scholar]
- 40.Cheung, J., and Smith, D. F. (2000) Mol. Endocrinol. 14939 -946 [DOI] [PubMed] [Google Scholar]
- 41.Voss, M. R., Stallone, J. N., Li, M., Cornelussen, R. N., Knuefermann, P., and Knowlton, A. A. (2003) Am. J. Physiol. 285H687 -H692 [DOI] [PubMed] [Google Scholar]
- 42.Fekete, A., Vannay, A., Ver, A., Rusai, K., Muller, V., Reusz, G., Tulassay, T., and Szabo, A. J. (2006) Am. J. Physiol. 291F806 -F811 [DOI] [PubMed] [Google Scholar]
- 43.Nickerson, M., Kennedy, S. L., Johnson, J. D., and Fleshner, M. (2006) J. Appl. Physiol. 101566 -575 [DOI] [PubMed] [Google Scholar]
- 44.Shinohara, T., Takahashi, N., Ooie, T., Ichinose, M., Hara, M., Yonemochi, H., Saikawa, T., and Yoshimatsu, H. (2004) J. Mol. Cell Cardiol. 371053 -1061 [DOI] [PubMed] [Google Scholar]
- 45.Olazabal, U. E., Pfaff, D. W., and Mobbs, C. V. (1992) Brain Res. 596311 -314 [DOI] [PubMed] [Google Scholar]
- 46.Krebs, C. J., Jarvis, E. D., and Pfaff, D. W. (1999) Proc. Natl. Acad. Sci. U. S. A. 961686 -1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu, A., Ran, R. Q., Clark, J., Reilly, M., Nee, A., and Sharp, F. R. (2002) J. Cereb. Blood Flow Metab. 22 183-195 [DOI] [PubMed] [Google Scholar]
- 48.Yang, S. H., Liu, R., Wen, Y., Perez, E., Cutright, J., Brun-Zinkernagel, A. M., Singh, M., Day, A. L., and Simpkins, J. W. (2005) J. Neurobiol. 62 341-351 [DOI] [PubMed] [Google Scholar]
- 49.HD Collaborative Research Group (1993) Cell 26971 -983 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.