

Abstract
Humans have two major high density lipoprotein (HDL) sub-fractions, HDL2 and HDL3, whereas mice have a monodisperse HDL profile. Epidemiological evidence has suggested that HDL2 is more atheroprotective; however, currently there is no direct experimental evidence to support this postulate. The amino acid sequence of apoA-I is a primary determinant of HDL subclass formation. The majority of the α-helical repeats in human apoA-I are proline-punctuated. A notable exception is the boundary between helices 7 and 8, which is located in the transitional segment between the stable N-terminal domain and the C-terminal hydrophobic domain. In this study we ask whether the substitution of a proline-containing sequence (PCS) separating other helices in human apoA-I for the non-proline-containing sequence (NPCS) between helices 7 and 8 (residues 184–190) influences HDL subclass association. The human apoA-I mutant with PCS2 replacing NPCS preferentially bound to HDL2. In contrast, the mutant where PCS3 replaced NPCS preferentially associated with HDL3. Thus, the specific amino acid sequence between helices 7 and 8 influences HDL subclass association. The wild-type and mutant proteins exhibited similar physicochemical properties except that the two mutants displayed greater lipid-associated stability versus wild-type human apoA-I. These results focus new attention on the influence of the boundary between helices 7 and 8 on the properties of apoA-I. The expression of these mutants in mice may result in the preferential generation of HDL2 or HDL3 and allow us to examine experimentally the anti-atherogenicity of the HDL subclasses.
HDL3 cholesterol has an inverse relationship with the incidence of coronary artery disease (1, 2). The cardioprotective role of HDL is, in part, related to the ability of apoA-I (the major protein component of HDL) to promote reverse cholesterol transport (3–6) involving ATP-binding cassette transporters and scavenger receptor B, class I (3–9) and to activate lecithin-cholesterol acyl transferase, an enzyme involved in cholesterol esterification (10). The anti-inflammatory and anti-oxidant properties (3, 11, 12) of HDL as well as its ability to stimulate nitric oxide production by endothelial cells (3, 13) may also contribute to its atheroprotective properties.
Most species, including mice, generate monodisperse HDL particles, whereas primates and humans make distinct HDL subclasses, the two major subclasses being HDL2 and HDL3 (14). HDL2 are larger and more buoyant and have a higher apoA-I:apoA-II ratio than HDL3. In addition, apoE is found almost exclusively on HDL2. The molecular and physiological basis for these HDL subclasses in humans are unknown; however, some distinct functional capacities of these HDL subclasses have been identified (15–17). Although epidemiological data suggest that HDL2 is more atheroprotective than HDL3 (1), there is no clear experimental support for this postulate. Studies in apoA-I transgenic (18), adenoviral gene transfer in mice (19), and transfected hepatocytes (20) revealed that the ability to form distinct HDL subclasses is inherent in the sequence of human apoA-I.
The primary structure of apoA-I consists of a series of 11- or 22-amino acid repeats capable of forming amphipathic α-helices that can form helix bundles in the lipid-free state (21, 22). Several structure/function relationships have been identified for specific helical repeats within human apoA-I. Helices 1, 9, and 10 are essential for initial lipid binding (23, 24), and helices 6 and partly 7 are critical for maximal activation of lecithin-cholesterol acyl transferase (10). The seven proline residues in human apoA-I are located between the helical repeats. The sequences between putative helices 2 and 3 and putative helices 7 and 8 in human apoA-I lack proline residues. Several nonprimate species contain a proline residue in the latter position (21). Two crystal structures of lipid-free human apoA-I have been reported. The Δ1–43 crystal structure forms a horseshoe shape, with the proline-containing sequences (PCS) between helices adopting an average angle of 39.9° (25). In contrast, the non-proline-containing sequence (NPCS) between putative helices 7 and 8 adopts a 12–18° angle. The crystal structure of the whole protein forms a four-helix bundle N-terminal domain (NTD) and smaller C-terminal domain (CTD) (22). Recent experimental evidence suggests that the NTD of lipid-free apoA-I forms a stable four-helix bundle, and the CTD forms a hydrophobic random coil (24). The transition between the NTD and CTD involves the NPCS between putative helices 7 and 8. Although the structure of apoA-I on discoidal particles has been extensively studied (24, 26–30), high resolution structural information about apoA-I on spherical HDL particles is not available.
In in vitro binding assays mouse apoA-I associates preferentially with HDL2, whereas the human protein associates equally well with HDL2 and HDL3 (14). The affinities for the HDL subclasses parallel the size and density of the HDL found in human and mouse plasma. When the human sequence from residues 166–209 (putative helices 7 and 8) were substituted for the homologous sequence in murine apoA-I, a chimeric molecule was produced that resembled human apoA-I in its lipoprotein association properties. Inspection of the human and mouse sequences between putative helices 7 and 8 shows that in contrast to the human protein, this region in the mouse protein contains a proline residue. In this study, we focused on the potential role of proline residues in this region of human apoA-I on HDL subclass association.
The lipidation of apoA-I is suggested to occur in two steps (24). apoA-I initially binds to a lipid surface through the random coil in the CTD; this is accompanied by an increase in α-helicity in the CTD (24, 31). Subsequently, the helix bundle of the NTD undergoes a conformational change, converting hydrophobic helix-helix interactions to helix-lipid interactions. In this step, the extra helicity in the NTD appears to come from residues 123–142, as well as residues 1–43 (24). This supports the contention that the majority of apoA-I is α-helical in structure when lipid-associated. This includes the region of the protein where we have focused our attention, residues 184–190.
We hypothesize that it is the segment between putative helices 7 and 8 that contributes to the propensity of the apoprotein to associate with the different HDL subclasses each having a different radius of curvature. This is based upon the following points. The transition between the α-helical NTD and the random coiled CTD spans putative helices 7 and 8 (24). Second, the mouse sequence between these putative helices contains a proline, whereas this is not the case for the human sequence, which instead contains an alanine residue at this position (21). Third, apoA-I encompasses spherical HDL with the hydrophobic face associated with lipid, expressing a concavity toward the lipid surface. We believe that the concave curvature of the protein is facilitated by the PCS. This hypothesis is further supported by Lazar et al. (32) where specific human PCS (particularly those between helices 3/4, 4/5, and 9/10) when placed between an 18-mer canonical apoA-I mimetic sequence drive the structure of the protein.
Here we ask whether the nature of the sequence between putative helices 7 and 8 influences the ability of human apoA-I to associate with HDL2 or HDL3.
EXPERIMENTAL PROCEDURES
Elements Common to All Recombinant apoA-I cDNAs—The restriction enzymes, Vent polymerase, and T4 DNA ligase were purchased from New England Biolabs. The QuikChange site-directed mutagenesis kits and Pfu DNA polymerase were from Stratagene. The oligonucleotide primers were made by Integrated DNA Technologies, Inc. The pET28 vector from Novagen was used for bacterial expression of apoA-I.
Generation of Recombinant apoA-I—The wild-type human apoA-I cDNA was subcloned into the bacterial expression vector pET28c as described previously (14). The recombinant protein contains a poly-His sequence and a T7 tag at the N terminus of the protein. The PCR-based QuikChange site-directed mutagenesis kit was used to replace the natural NPCS (NGGARLA) between putative helices 7 and 8 of human apoA-I (residues 184–190) with the PCS present between other helices of the human protein (i.e. PCS1-QLGPVTQ, residues 63–69; PCS2-KVQPYLD, residues 96–102; PCS3-KVEPLRA, residues 118–124; PCS4-KLSPLGE, residues 140–146; PCS5-HLAPYSD, residues 162–168; PCS6-KAKPALE, residues 206–212; and PCS7-GLLPVLE, residues 217–224). Each plasmid was transformed into protease-minus BL21(DE3)pLys Escherichia coli strain (Novagen) and the protein purification was carried out as described earlier (14). The concentration of recombinant human apoA-I was determined by Bradford protein assay (Bio-Rad).
Cross-linking Experiments—Lipid-free or lipid-bound wild-type/mutant apoA-I (1 mg/ml) in 50 mm sodium phosphate and 50 mm NaCl at pH 7.2 were incubated for 30 min with bis(sulfosuccinimidyl)suberate (Pierce; final concentration, 0.5 mm). The reaction mixture was quenched by adding 1 m Tris-HCl and 5× SDS-PAGE sample buffer and then loaded on a 6–12% Tricine/PAGE gel. The populations of different oligomers were estimated using the band intensities of the oligomers as quantified using the software of Kodak Gel Logic 100 imaging system (33).
Circular Dichroism Spectroscopy—CD measurements of lipid-free and lipid-bound wild-type/mutant apoA-I were carried out on an Aviv model 62DS CD spectrometer (Aviv Instruments, Inc., Lakewood NJ) with a variable temperature capability under computer control within ±0.2 °C. Lyophilized protein samples were suspended in 50 mm sodium phosphate and 50 mm NaCl at pH 7.2 and were adjusted to a concentration of 0.1 mg/ml using the same buffer. The measurements were made at room temperature using a 0.1-cm quartz cell. Five scans between 200 and 250 nm were acquired and averaged. A baseline scan was subtracted to produce the final average scan. The percentage of α-helix content was calculated from the molar ellipticity at 222 nm using the mean residue weight for each of the apoA-I proteins (33).
Preparation and Characterization of apoA-I:rHDL Particles—The rHDL particles were prepared using the sodium cholate dialysis method with phosphatidylcholine (POPC) (Avanti Polar Lipids), wild-type/mutant apoA-I, and sodium cholate in a molar ratio of 80:1:108 (33). POPC was dissolved in CHCl3, dried under nitrogen, and then resuspended in 50 mm sodium phosphate and 50 mm NaCl, pH 7.2. After being vortexed thoroughly, sodium cholate was added into the mixture followed by vortexing for another 3 min. The solution was incubated at 37 °C and vortexed every 15 min until completely clear. Human apoA-I protein was added, and the protein/lipid mixture was incubated for 1 h at 37°C. Sodium cholate was removed by the Bio-bead method (33). Size was examined by nondenaturing gradient gel electrophoresis as described (14).
Lipid-associated (Isothermal) Guanidine HCl Denaturation Studies—Guanidine HCl denaturation was performed as described (33) and monitored by CD at 222 nm in a 0.1-cm path length cuvette at 20 °C.
DMPC (Lipid Binding) Clearance Assay—Ten milligrams of DMPC (Avanti Polar Lipids Inc.) were dissolved in a mixture of chloroform and methanol (3:1 v/v), dried using N2, and placed under vacuum for at least 10 h. 1 ml of prewarmed buffer was added (10 mm Tris-HCl, pH 7.2, 150 mm NaCl, and 0.5 mm EDTA) for a final lipid concentration of 10 mg/ml and vortexed several times (30 s each). Using a 200-nm filter, unilamellar vesicles (∼200 nm in diameter) were prepared by extrusion. The protein-induced transformation of DMPC vesicles into protein/DMPC discoidal complexes were monitored as a function of time (33). Two different ratios of apoA-I and DMPC (1:1 and 1:2.5, w/w) were used in this assay. apoA-I/DMPC vesicles in buffer were added into a 1-ml thermostat cuvette and mixed for 5–10 s at 24 °C. The clearance of solution was monitored using a PerkinElmer spectrophotometer (model Lambda 3B) at 490 nm. All of the solutions were preincubated at 24 °C before being used in the reaction.
Isolation of Human HDL2 and HDL3—HDL2 (ρ = 1.063–1.098 g/ml) and HDL3 (ρ = 1.13–1.21 g/ml) were isolated from human plasma obtained from female donors by sequential flotation as described (14).
apoA-I Quantitation by Immunoturbidometry—The endogenous apoA-I in the isolated HDL was determined by immunoturbidometry using kits from Roche Applied Science (14). The human apoA-I standards were purchased from Sigma or Northwest Lipid Laboratory (Seattle, WA).
In Vitro Association of Recombinant Human apoA-I with Isolated Human HDL—The recombinant human apoA-I proteins were incubated with different proportions of HDL2 and HDL3 (i.e. 1:1, 2:1 or 1:2) according to their endogenous apoA-I content. In a typical 545-μl assay, 10 μg of wild-type/mutant recombinant human apoA-I was incubated with an HDL preparation containing 625 μg of total endogenous apoA-I for 30 min at 4 °C. Following incubation, the final volume of the assay mixture was adjusted to 2.0 ml with Tris-sodium chloride-EDTA (TSE) buffer (10 mm Tris, 140 mm NaCl, 0.25 mm EDTA, and 0.15 mm sodium azide), pH 7.4, and subjected to a 10–20% equilibrium density gradient separation (14). 0.4-ml fractions were collected using an ISCO gradient collector. The gradient fractions corresponding to HDL2 and HDL3 were determined from the A280 tracings.
To detect the recombinant and endogenous apoA-I in these fractions, alternate fractions were electrophoresed on 4–20% SDS-PAGE gels, proteins transferred to Immobilon-P, and immunodecorated with mouse monoclonal anti-T7 antibody horseradish peroxidase conjugate (1:5000 dilution; Novagen) to detect recombinant apoA-I or with rabbit anti-huA-I antibodies (1:25000 dilution)/goat anti-rabbit-IgG-horseradish peroxidase secondary antibody (1:10,000 dilution) for endogenous and recombinant huA-I detection. The immunodecorated proteins were visualized with enhanced chemiluminescence (ECL; Amersham Biosciences/Molecular Dynamics). The protein bands were quantitated by Alpha-Imager (ImageQuant, Genomic Solutions Inc). To compare data between experiments the recombinant apoA-I distributions were normalized to the HDL2:HDL3 proportions by dividing them by the corresponding endogenous apoA-I distributions. Endogenous apoA-I is an appropriate surrogate for starting HDL proportions.
Concentration-dependent Binding of Recombinant apoA-I to HDL Subclasses—To determine the binding curves of wild-type/mutant human apoA-I proteins for HDL subclasses, increasing amounts of recombinant apoA-I (1.25–51 μg) were added to isolated HDL2 or HDL3 (total protein, 200 μg) in 545 μl of TSE buffer. The reaction was incubated at 4 °C for 30 min. An aliquot from each assay was electrophoresed on 0.7% agarose gels to separate HDL bound from unbound protein. Following transfer to Immobilon-P, specifically probing with the mouse monoclonal anti-T7 antibody, as described above, recombinant human apoA-I bound to HDL and unbound were calculated and used to determine the Kd of association. The blots were also immunoblotted for apoE and apoA-IV to assess remodeling of the HDL during the incubation. Previous studies in our laboratory have demonstrated little remodeling at the concentrations of HDL and recombinant proteins used in these studies.
Statistical Analysis—The results are expressed as the means ± S.D. Group differences were tested by analysis of variance and Student's t tests. The significance level was set at p < 0.05.
RESULTS
Because we previously showed that apoA-I recombinant protein HDL subclass association in vitro parallels HDL generation in vivo (14), this study is designed to examine the capacity of the sequence between putative helices 7 and 8 of human apoA-I to influence its ability to associate with human HDL subclasses. The mutant proteins are designated by the specific PCS replacing the NPCS between residues 184 and 190. Thus, when the PCS between helices 3 and 4 (i.e. PCS2) is inserted in place of the NPCS at residues 184–190 of human apoA-I, the mutant is designated NPCS→ PCS2, as illustrated in Fig. 1.
FIGURE 1.

Strategy for generating human apoA-I PCS mutants. A, illustration of the location and amino acid sequence for the PCSs between the putative helices in wild-type human apoA-I and the NPCS between the putative helices 7 and 8. B, human apoA-I mutants were generated by replacing the NPCS between helices 7 and 8 with each of the PCS. The mutants contain two copies of the PCS: one at its native position and one between helices 7 and 8 as illustrated for HuA-I (NPCS→ PCS2).
HDL Preferential Association Assay—Our strategy involves the synthesis of recombinant apoA-I constructs each containing N-terminal tags consisting of a His tag for purification and a T7 tag to specifically detect the recombinant proteins after associating with human HDL. We have previously shown by comparing recombinant wild-type apoA-I (T7-HuA-I) protein with purified plasma apoA-I that the presence of the N-terminal tag did not significantly affect the properties of the protein (14). The recombinant proteins displayed the expected molecular weights determined by SDS-PAGE (data not shown). From densitometric scanning of the gel, the purity of the recombinant proteins was ≥95%. We examined the ability of the various recombinant proteins to associate with mature HDL subclasses. In this association assay, lipid-free recombinant apoA-I was incubated with different ratios of human HDL2 and HDL3 (i.e. 1:1, 1:2, and 2:1) at an endogenous to recombinant apoA-I weight ratio of 62.5:1. At this ratio the added apoA-I “tags” the HDL without significantly altering the HDL size or density. Assuming two to four molecules of apoA-I per HDL, there are about 20 HDL particles (HDL2 + HDL3) per recombinant apoA-I molecule in the association assays. With this excess of HDL, the recombinant apoA-I binds to the HDL subclass for which it has the highest affinity.
The HDL subclasses after incubation with the recombinant apoA-I were separated on NaBr equilibrium density gradients, and the fractions were analyzed for the presence of endogenous apoA-I and recombinant proteins by immunoblotting. To compare data among experiments, the recombinant apoA-I distributions (anti-T7 tag) were normalized to the starting HDL2:HDL3 proportions by dividing the recombinant apoA-I distributions by the corresponding endogenous apoA-I distribution. We found that endogenous apoA-I is an appropriate surrogate for starting HDL subclass proportions, as demonstrated by the similar HDL subclass ratio calculated from A280 tracings and from the densitometric scanning of the Western blot for endogenous apoA-I.
T7-HuA-I distributed to HDL subclasses in a pattern that followed closely that of endogenous apoA-I (Fig. 2). On average about 50% of T7-HuA-I distributed to HDL2 and 50% to HDL3, showing that T7-HuA-I bound with approximately equal affinity to HDL2 and HDL3. The human apoA-I mutants in which PCS2 replaced NPCS (i.e. T7-HuA-I (NPCS→ PCS2)) preferentially associated with HDL2 (75–80%), whereas only 20–25% associated with HDL3. On the other hand, the human apoA-I mutant in which PCS3 replaced NPCS (i.e. T7-HuA-I (NPCS→ PCS3)) preferentially associated with HDL3 (70–75%), with only 25–30% associating with HDL2. The substitution of the other PCS of human apoA-I for NPCS had no statistically significant effect on the HDL subclass distribution of recombinant proteins. These data suggest that the nature of the sequence between putative helices 7 and 8 influences the binding specificity of human apoA-I for HDL subclasses. For this reason we examined the physical properties of the two mutants with different HDL subclass distribution.
FIGURE 2.

Recombinant apoA-I distributions to HDL2 or HDL3. Wild-type or the indicated PCS mutants were incubated with HDL (HDL2 + HDL3), the lipoproteins were separated on equilibrium density gradients, and the fractions were probed for the T7-tagged human apoA-I. The recombinant apoA-I distributions to HDL2 and HDL3 were normalized by dividing by endogenous apoA-I distribution to the HDL subclasses. These values for HDL2 (open bars) and HDL3 (black bars) are expressed as percentages (n = 6). a, HDL2, p < 0.05; b, HDL2, p < 0.05; c, HDL3, p < 0.05; d, HDL3, p < 0.05 compared with T7-HuA-I.
Self-association of Lipid-free apoA-I Proteins—The self-association properties of the recombinant apoA-I proteins in the lipid-free state were examined by cross-linking with bis(sulfosuccinimidyl)suberate. Our laboratory has previously shown that T7-HuA-I self-association properties were similar to those of isolated human plasma apoA-I (14). T7-HuA-I (NPCS→ PCS3) and T7-HuA-I (NPCS→ PCS2) had differing self-association properties (Fig. 3). T7-HuA-I and the T7-HuA-I (NPCS→ PCS3) mutant generated a higher percentage of trimer, tetramer, and pentamers than the T7-HuA-I (NPCS→ PCS2) mutant. These findings suggest that the specific sequence within the segment between helices 7 and 8 affects the self-association properties of the lipid-free protein.
FIGURE 3.

Self-association behavior of lipid-free recombinant apoA-I proteins. Lipid-free apoA-I (1 mg/ml) was cross-linked with bis(sulfosuccinimidyl)suberate and electrophoresed on a 6–12% Tricine gel. Lane M, high molecular weight standard; lane 1, non-cross-linked T7-HuA-I; lane 2, cross-linked T7-HuA-I (NPCS→ PCS3); lane 3, cross-linked T7-HuA-I (NPCS→ PCS2); lane 4, cross-linked T7-HuA-I.
DMPC (Lipid Binding) Clearance Assay—The lipid affinity of the recombinant mutant apoA-I proteins were measured using the DMPC clearance assay. Fig. 4 illustrates that wild-type and mutant recombinant apoA-I proteins display a similar but not identical ability to clear DMPC vesicles. This trend was observed when using a ratio of apoA-I:DMPC (w/w) of either 1:1 (data not shown) or 1:2.5. The clearance by T7-HuA-I and T7-HuA-I (NPCS→ PCS3) mutant were essentially identical, whereas the clearance by the T7-HuA-I (NPCS→ PCS2) mutant was somewhat slower though reaching the same maximum.
FIGURE 4.

DMPC clearance (lipid binding affinity) of recombinant apoA-I proteins. DMPC vesicles were incubated (1:2.5 w/w) apoA-I:DMPC at 24 °C in the absence of protein (solid black diamond) and presence of T7-HuA-I (NPCS→ PCS3) (open circle), T7-HuA-I (NPCS→ PCS2) (solid black triangle), or T7-HuA-I (solid black square).
Physicochemical Properties of rHDL Particles—One of the primary functions of apoA-I is to assemble HDL particles. A surrogate for this is represented by the ability to generate HDL-like particles with a physiologically relevant lipid. Recombinant apoA-I was reconstituted in particles containing POPC and cholate in a molar ratio of 80:1:108 of POPC: apoA-I:cholate. Recombinant wild-type apoA-I:rHDL had a hydrodynamic diameter of 10.3 nm (Table 1) and contained two apoA-I molecules per rHDL. This is not different from plasma-derived apoA-I:rHDL (14). The size and composition of rHDL particles containing each of the two mutants were very close to one another and not different from the wild-type protein (Table 1).
TABLE 1.
Characterization of recombinant apoA-I proteins in rHDL and lipid-free forms
|
Protein component |
rHDL diameter of
NDGGEa |
Ratio of
apoA-I/rHDLb |
α-Helix content |
|
|---|---|---|---|---|
| Lipid-freec | rHDLd | |||
| nm | ||||
| T7-HuA-I (NPCS→PCS3) | 10.3 | 2 | 48.9 ± 3.1 | 67.8 |
| T7-HuA-I (NPCS→PCS2) | 10.3 | 2 | 57.0 ± 4.3 | 70.8 |
| T7-HuA-I | 10.3 | 2 | 50.0 ± 4.6 | 70.4 |
Particle diameters were determined from native polyacrylamide gel electrophoresis using reference globular proteins (Fig. 5; n = 2). NDGGE indicates nondenaturing gradient gel electrophoresis.
The number of proteins/particle was determined by SDS-PAGE of rHDL cross-linked with BS3 (n = 2).
The percentage of α-helical content of lipid-free recombinant and plasma-derived proteins were estimated from molar ellipticity at 222 nm (n = 3).
The percentage of α-helical content of recombinant and plasma apoA-I:rHDL (n = 2).
Recombinant apoA-I Secondary Structure—To ascertain the secondary structure, we examined the α-helical content of lipid-free and lipid-bound recombinant apoA-I proteins by circular dichroism. The α-helical contents for lipid-free T7-HuA-I and T7-HuA-I (NPCS→ PCS3) mutant were similar, whereas the T7-HuA-I (NPCS→ PCS2) mutant appeared to exhibit a higher degree of lipid-free α-helicity (Table 1). The lipid-associated α-helicity of the two mutant proteins closely resembled that of T7-HuA-I.
Lipid-free/Lipid-associated Isothermal Stability of Recombinant apoA-I Proteins—Investigations in several laboratories have shown that the denaturation of lipid-free apoA-I is a reversible process that has a midpoint of denaturation (D½) close to 1.0 m guanidine HCl (24). Furthermore, the effectiveness of guanidine HCl in unfolding apoA-I is significantly decreased when apoA-I is complexed with DMPC. The D½ of lipid-freeT7-HuA-I was similar to published values using plasma human apoA-I (24). The denaturation profiles for lipid-free wild-type and mutant recombinant apoA-I proteins were similar (Fig. 5A). Whereas the D½ for lipid-associated T7-HuA-I was similar to reported values for lipid-associated plasma human apoA-I, both mutants when lipid-associated appeared to be more stable than T7-HuA-I to guanidine denaturation (Fig. 5B).
FIGURE 5.
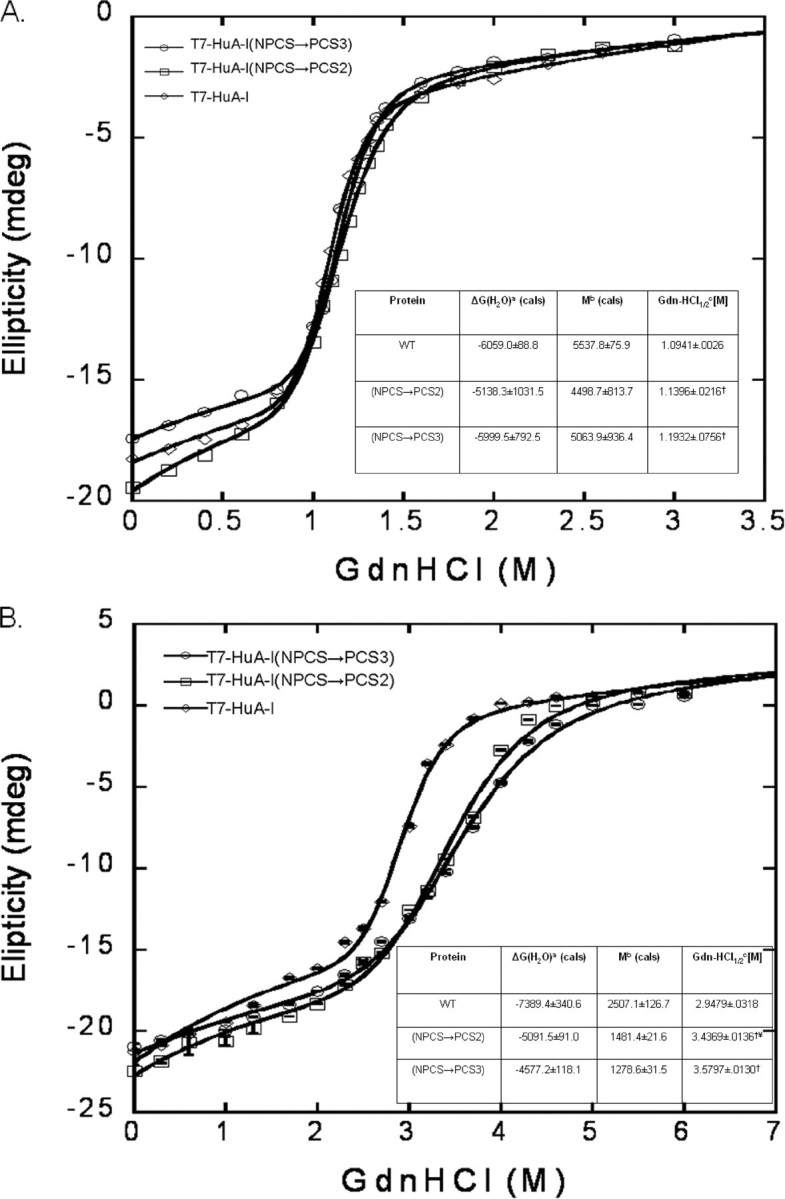
Lipid-free and lipid-associated (isothermal) guanidine HCl denaturation stability. A, lipid-free (n = 3). a, the y intercept of the denaturation curve, represented in calories; b, the slope of the denaturation curve, representedincalories;c, the midpoint concentration of guanidine HCl where half of lipid-free recombinant is folded and unfolded (†, p < 0.05) compared with T7-HuA-I. B, lipid-associated (n = 3). a, the y intercept of the denaturation curve, represented in calories; b, the slope of the denaturation curve, represented in calories; c, the midpoint concentration of guanidine HCl where half of lipid-associated recombinant is folded and unfolded (†, p < 0.0001) compared with T7-HuA-I. ¥, p < 0.0001, compared with T7-HuA-I (NPCS→PCS3); wild-type and recombinant apoA-I proteins. The line represents the best fit curve, which includes both a sigmoid and hyperbola.
Binding Curves of Recombinant apoA-I to HDL—In the association assays presented in Fig. 2, a small amount of recombinant apoA-I was incubated with a mixture of HDL2 and HDL3 so that these two HDL subclasses competed for the added recombinant apoA-I. The results from these experiments suggested that the affinity of some of these recombinants differ for the two HDL subclasses. Having confirmed that overall there are only modest differences, if any, in the physicochemical properties between the two PCS mutants exhibiting preferential binding to HDL subclasses and wild-type human apoA-I, we sought to examine the affinity of our recombinant apoA-I proteins for each HDL subclass. Because added apoA-I did not cause remodeling of HDL or significant desorption of endogenous apoA-I (see below), the binding curves of apoA-I for HDL subclasses could be measured directly using Scatchard analysis. For this analysis we employed nondenaturing agarose electrophoresis to separate free and bound recombinant apoA-I. The Scatchard analysis was based on reversible binding of apoA-I to a site on an HDL particle as shown in Equation 1.
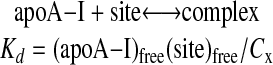 |
(Eq. 1) |
Because (Site)free = (Sites)total–(Sites)occupied = (Sites)total– (apoA-I)bound and Cx = (apoA-I)bound, the value of Kd and (Sites)total could be assessed as parameters using nonlinear least square analysis.
For determination of these binding curves, between 1.25 and 51 μg of recombinant apoA-I was incubated with 200 μg of isolated HDL2 or HDL3, corresponding to 130 or 150 μg of endogenous apoA-I. Scatchard analysis (Fig. 6) demonstrated that T7-HuA-I binding to HDL2 (Kd = 3.2 μm) and HDL3 (Kd = 1.6 μm) are similar. The HuA-I (NPCS→ PCS2) mutant displayed higher affinity for HDL2 (Kd = 5.3 μm) than HDL3 (Kd > 1M), consistent with the association assay (Fig. 2). On the other hand, the HuA-I (NPCS→ PCS3) mutant displayed a higher affinity for HDL3 (Kd = 6.7 μm) versus HDL2 (Kd > 1M). Thus, the two assays used for determining the association of recombinant mutant apoA-I proteins with HDL revealed similar preferences for HDL subclasses.
FIGURE 6.

Scatchard analysis of association of T7-HuA-I, T7-HuA-I (NPCS→PCS2), and T7-HuA-I (NPCS→PCS3) with HDL2 or HDL3. Increasing amounts of recombinant apoA-I proteins were incubated with 200 μg of HDL2 or HDL3. HDL bound apoA-I was separated from lipid-free apoA-I by agarose gel electrophoresis. Bound and free recombinant apoA-I were detected by immunoblotting with horseradish peroxidase-conjugated T7 antibody. The results from three separate experiments are plotted. The line represents the best fit curve.
HDL Remodeling in Binding Assays—To ensure that we are measuring wild-type or mutant apoA-I affinity for an HDL subclass and not an artifact of HDL remodeling, we examined the HDL size following the addition of the highest concentration (51 μg) of apoA-I used in our binding assays. When 51 μg of recombinant apoA-I was incubated with either HDL subclass in the binding assays, the Stokes diameter did not change, nor was apoE or apoA-I released from the particle as examined by Western blotting (data not shown), indicating that by this parameter major HDL remodeling did not occur.
DISCUSSION
In this study we have generated human apoA-I mutants that exhibit a notable preference for association with either HDL2 or HDL3 subclasses. We achieved this by replacing the native NPCS (NGGARLA) (residues 184–190) between putative helices 7 and 8 with a PCS derived from elsewhere in the apoA-I molecule. The native NPCS lacks the canonical proline residue that is thought to center the sequence that separates the repeating amphipathic α-helices. This applies also to each of the engineered proteins here described. When PCS2 (KVQPYLD) is in the NPCS position between putative helices 7 and 8, the protein shows a strong preference and higher affinity for HDL2 than HDL3, whereas when PCS3 (KVEPLRA) is in that position the recombinant protein favorably binds to HDL3 in contrast to HDL2. These affinities have been compared with those of the wild-type protein, which bound equally well to HDL2 and HDL3. The T7-HuA-I (NPCS→ PCS2) mutant bound with a higher affinity to HDL2 than HDL3 (Kd = 5.3 μm and Kd > 1 m, respectively). In contrast the T7-HuA-I (NPCS→ PCS3) mutant displayed a higher affinity for HDL3 than HDL2 (Kd = 6.7 μm and Kd > 1 m, respectively).
DMPC clearance, the size of reconstituted HDL-like particles, and the denaturation profile of lipid-free apoproteins were similar for wild-type and mutant proteins. On the other hand, the T7-HuA-I (NPCS→ PCS3) and T7-HuA-I (NPCS→ PCS2) had differing self-association properties, with T7-HuA-I and the T7-HuA-I (NPCS→ PCS3) mutant generating a higher percentage of trimer, tetramer, and pentamers than the T7-HuA-I (NPCS→ PCS2) mutant (Fig. 3). The self-association of a protein such as apoA-I is probably a reflection of its hydrophobicity, which in this protein is largely determined by helices 9 and 10 (14). However, as can be seen in Fig. 3, seven amino acid substitutions between putative helices 7 and 8 slightly changes the self-association of the resulting proteins versus wild-type apoA-I, with PCS2 forming fewer higher order oligomers. This may allow the monomer to be more readily available for lipoprotein association.
Furthermore, the lipid-free α-helicity was modestly higher for T7-HuA-I (NPCS→ PCS2). In addition, when lipid-associated both mutants were modestly more stable than T7-HuA-I. Overall, the apparent differences in physical properties of the PCS mutant proteins and wild-type apoA-I are small. All of these data suggest that the marked differences in the affinity of the apoA-I mutant for preformed HDL subsets cannot be attributed to deviations in the gross physical properties of these mutants.
Our previous studies highlighted helices 7 and 8 as a likely site contributing to the differences in HDL association of mouse, human, and chimeric apoA-I proteins (14). There is much more sequence divergence within putative helices 7 and 8 in the mouse and human proteins than is the case for any of the other helices of these proteins (with the possible exception of the C-terminal 22 amino acids). Helices 7 and 8 of the human protein bridges the most flexible portion of the apoprotein (24). We focused our attention on the short sequence between putative helices 7 and 8, which is very diverse between human (NGGARLA) and mouse (N--PLTN) proteins. In the case of the human protein, this sequence, unlike that separating most of the other helices in the human protein, does not contain a proline residue.
We substituted PCS separating other helices in human apoA-I for the NPCS between putative helices 7 and 8. Each PCS contained seven residues with the proline in the center. This was chosen because it represents two turns of an α-helix and would be expected to involve little disruption of phase relationships of the surrounding helices. Only two of the PCS mutants displayed statistically significant preferential HDL subclass association, with the mutant containing PCS2 preferentially associating with HDL2 and the mutant containing PCS3 preferentially associating with HDL3.
There are three possible explanations for the preferential association of our PCS mutants with HDL subclasses. In this discussion we take the proline residue as 0; we take the downstream residues as –3, –2, and –1 and the residues upstream from proline as +1, +2, and +3. On the helical wheels illustrated in Fig. 7, these residues correspond respectively to positions 9, 10, 11, 2, 3, and 4. A comparison of the sequence of the two most informative PCS and their surrounding residues reveals interesting differences. The PCS2 contains one negatively charged residue at +3 (position 4 on the wheel) and then a second negative charged residue just outside of the substituted PCS2 at +4 (position 5). Thus, the first possibility is that within the substituted PCS2 the adjacent negative charges are repelling one another and in turn enabling more of the nearby hydrophobic face to become available for lipid surface association. However, this explanation is not sufficient because other PCS mutants contain an acidic residue at +3(e.g. PCS4 and 5), and yet they do not exhibit an HDL2 preferential association (Fig. 2). A second possible explanation for the HuA-I (NPCS→ PCS3) mutant HDL3 preferential association may relate to the possible internal salt bridges between the Glu and either Lys or Arg in the PCS3. Such internal salt bridges have been postulated to account for the properties of lipid-free tandem helices separated by such a PCS (32). However, this possibility is also unlikely because we find the same preferential association behavior when the lipoproteins with associated mutant apoA-I are separated by high salt (NaBr) density gradients, which can be expected to disrupt salt bridges. The third and most likely explanation for our PCS mutant HDL preferential association relates to the change in the size or subtle qualities of the hydrophobic face of the modified putative helices 7 and 8 in each of the mutants.
FIGURE 7.
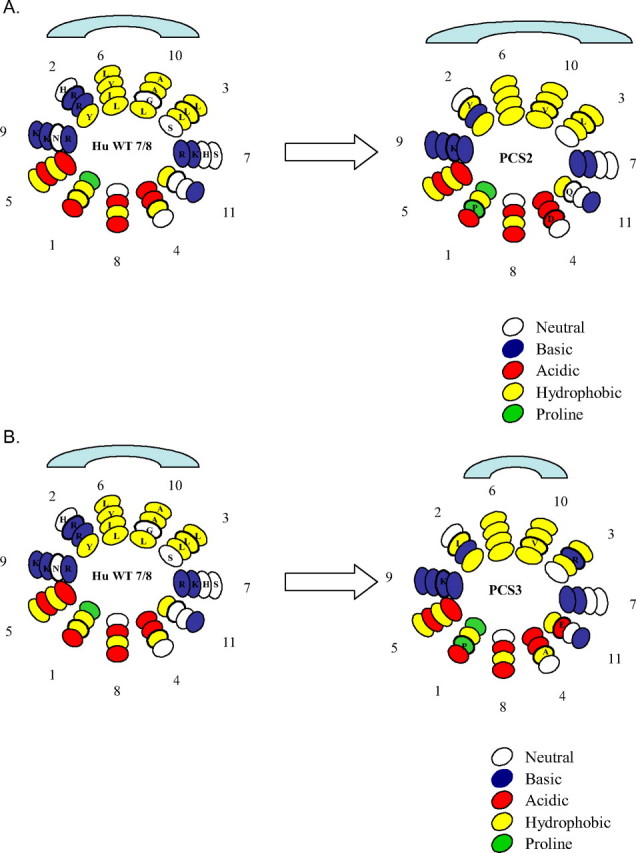
Helical wheel distribution of helices 7 and 8 in the wild-type human apoA-I and NPCS→PCS2 (A) and NPCS→PCS3 (B) illustrating the difference in the overall size of the hydrophobic faces. The loss of a positive charge at position 2 and an increase in hydrophobic residues in PCS2 may increase the hydrophobic face exposure leading to preferential association with HDL2. On the other hand, the insertion of a positive charge at position 3 may decrease the hydrophobic face exposure leading to preferential association with HDL3.
Using the helical wheel diagrams (Fig. 7), we illustrate the effect of substitution of the PCS for the NPCS between helices 7 and 8 on the distribution of hydrophobic and positively charged residues. The illustrations in Fig. 7 are based on model formulations shown in a review by Frank and Marcel (21). The association of amphipathic helices with lipid surfaces will depend upon a balance between the hydrophobicity and hydrophilicity of the two faces of the helices. Both are changed in our mutants. Comparing the hydrophobic faces of helices 7 and 8 in the wild-type protein and the PCS2 mutant, the latter has a Tyr residue substituting for Arg at +1 (position 2 on the wheel) and a Val for Gly at –2 (position 10), which can be expected to increase the hydrophobicity of the extended face, allowing for a higher affinity for the lipid surface of the low radius of curvature HDL2. On the other hand in PCS3, there is a Leu for Arg substitution at +1 (position 2) and Val for Gly at –2 (position 10) but an Arg for Leu at +2 (position 3) on the hydrophobic face. This change could promote its association with the smaller HDL3 particle with a higher radius of curvature. Support for our hypothesis of amino acid distribution affecting potential hydrophobic exposure has been described (34).
The altered conformation induced by the amino acid substitutions in the transition segment in our mutants may not only influence the affinity of the CTD for the lipid surface but may also influence the relationship between the two domains of the protein, affecting the rate and/or extent at which the NTD unfolds onto the lipid surface. This reasoning is derived from the studies of Phillips and co-workers (24, 27). Further mutagenesis studies are required to provide additional information on these issues.
The selectivity of substituting the PCS for the NPCS of putative helices 7/8 is emphasized by further preliminary mutagenesis. The generation of a mutant human apoA-I containing three copies of either PCS2 or PCS3, one at its native location, one substituted for NPCS between putative helices 7 and 8, and another PCS elsewhere in the human protein, does not further alter the association properties of the resultant mutant protein (data not shown). The amino acid substitutions in this transition segment appear to increase the binding of the CTD to lipid as manifest by the increased stability of the two mutants to chemical denaturation in the presence of lipids (Fig. 5B).
We are in the process of examining the ability of these two mutants to preferentially generate HDL subclasses in vivo. Preliminary experiments examining the expression of the PCS mutants in the rat hepatoma cell line McA74 generate nascent HDL particle with densities and sizes that are distinct from those produced by wild-type apoA-I (data not shown). These studies reported here represent an initial platform for the generation of HDL subclasses in intact animals. Success in this endeavor will allow for the direct experimental test of the role of HDL subclasses HDL2 and HDL3 in atheroprotection. This in turn could lead to a capacity (using intact protein or viral vector) to boost HDL2 levels, assuming it to be the most atheroprotective species of HDL, in humans with low HDL2 levels.
This work was supported, in whole or in part, by National Institutes of Health Grants 5T32 HL07237 (a Cardiovascular Pathophysiology and Biochemistry Training Program grant to R. C.), R01 HL68661 (to G. S. G.), R01 NS042852 (to S. C. M.), and R01 HL076620 (to J. W.). This work was also supported by Alzheimer's Association Grant IIRG-06-27794 (to S. C. M.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: HDL, high density lipoprotein; apo, apolipoprotein; PCS, proline-containing sequence; NPCS, non-proline-containing sequence; NTD, N-terminal domain; CTD, C-terminal domain; rHDL, reconstituted HDL; POPC, sn-1-palmitoyl-sn-2-oleoyl-phosphatidylcholine; DMPC, dimyristoylphosphatidylcholine; Tricine, N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine.
References
- 1.Miller, N. E. (1987) Am. Heart J. 113589 –597 [DOI] [PubMed] [Google Scholar]
- 2.Castelli, W. P., Garrison, R. J., Wilson, P. W., Abbott, R. D., Kalousdian, S., and Kannel, W. B. (1986) J. Am. Med. Assoc. 2562835 –2838 [PubMed] [Google Scholar]
- 3.Assmann, G., and Gotto, A. M. (2004) Circulation 109III8 –III14 [DOI] [PubMed] [Google Scholar]
- 4.Singh, I. M., Shishehbor, M. H., and Ansell, B. J. (2007) J. Am. Med. Assoc. 298786 –798 [DOI] [PubMed] [Google Scholar]
- 5.Lewis, G. F. (2006) Curr. Opin. Cardiol. 21345 –352 [DOI] [PubMed] [Google Scholar]
- 6.Rader, D. J. (2006) J. Clin. Investig. 1163090 –3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang, N., Silver, D. L., Thiele, C., and Tall, A. R. (2001) J. Biol. Chem. 27623742 –23747 [DOI] [PubMed] [Google Scholar]
- 8.Kozarsky, K. F., Donahee, M. H., Rigotti, A., Iqbal, S. N., Edelman, E. R., and Krieger, M. (1997) Nature 387414 –417 [DOI] [PubMed] [Google Scholar]
- 9.Rigotti, A., Trigatti, B. L., Penman, M., Rayburn, M., Herz, J., and Krieger, M. (1997) Proc. Natl. Acad. Sci. U. S. A. 9412610 –12615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sorci-Thomas, M. G., Curtiss, L., Parks, J. S., Thomas, M. J., Kearns, M. W., and Landrum, M. (1998) J. Biol. Chem. 27311776 –11782 [DOI] [PubMed] [Google Scholar]
- 11.Barter, P. J. (1997) Clin. Exp. Pharmacol. Physiol. 24286 –2879131299 [Google Scholar]
- 12.Barter, P. J., Nicholls, S., Rye, K. A., Anantharamaiah, G. M., Navab, M., and Fogelman, A. M. (2004) Circ. Res. 95764 –772 [DOI] [PubMed] [Google Scholar]
- 13.Mineo, C., Deguchi, H., Griffin, J. H., and Shaul, P. W. (2006) Circ. Res. 981352 –1364 [DOI] [PubMed] [Google Scholar]
- 14.Reschly, E. J., Sorci-Thomas, M. G., Davidson, W. S., Meredith, S. C., Reardon, C. A., and Getz, G. S. (2002) J. Biol. Chem. 2779645 –9654 [DOI] [PubMed] [Google Scholar]
- 15.Mowri, H. O., Patsch, W., Smith, L. C., Gotto, A. M., and Patsch, J. R. (1992) J. Lipid Res. 331269 –1279 [PubMed] [Google Scholar]
- 16.Kontush, A., Therond, P., Zerrad, A., Couturier, M., Negre-Salvay de Souza, J. A., Chantepie, S., and Chapman, J. M. (2007) Arterioscler Throm. Vasc. Biol. 271843 –1849 [DOI] [PubMed] [Google Scholar]
- 17.Asztalos, B. F. (2004) Curr. Opin. Cardiol. 19385 –391 [DOI] [PubMed] [Google Scholar]
- 18.Rubin, E. M., Ishida, B. Y., Clift, S. M., and Krauss, R. M. (1991) Proc. Natl. Acad. Sci. U. S. A. 88434 –438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reardon, C. A., Kan, H. Y., Cabana, V., Blachowicz, L., Lukens, J. R., Wu, Q., Liadaki, K., Getz, G. S., and Zannis, V. I. (2001) Biochemistry 4013670 –13680 [DOI] [PubMed] [Google Scholar]
- 20.Thurberg, B. L., Reardon, C. A., and Getz, G. S. (1996) J. Biol. Chem. 2716062 –6070 [DOI] [PubMed] [Google Scholar]
- 21.Frank, P. G., and Marcel, Y. L. (2000) J. Lipid Res. 41853 –872 [PubMed] [Google Scholar]
- 22.Ajees, A., Anantharamaiah, G. M., Mishra, V. K., Hussain, M. M., and Murthy, M. M. (2006) Proc. Natl. Acad. Sci. U. S. A. 1032126 –2131 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Davidson, W. S., Hazlett, T., Mantulin, W. W., and Jonas, A. (1996) Proc. Natl. Acad. Sci. U. S. A. 9313605 –13610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saito, H., Katz-Lund, S., and Phillips, M. C. (2004) Prog. Lipid Res. 43350 –380 [DOI] [PubMed] [Google Scholar]
- 25.Borhani, D. W., Rogers, D. P., Engler, J. A., and Brouillette, C. G. (1997) Proc. Natl. Acad. Sci. U. S. A. 9412291 –12296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davidson, W. S., and Thompson, T. B. (2007) J. Biol. Chem. 28222249 –22253 [DOI] [PubMed] [Google Scholar]
- 27.Tanaka, M., Dhanasekaran, P., Nguyen, D., Ohta, S., Lund-Katz, S., Phillips, M. C., and Saito, H. (2006) Biochemistry 4510351 –10358 [DOI] [PubMed] [Google Scholar]
- 28.Klon, A. E., Segrest, J. P., and Harvey, S. C. (2002) Biochemistry 4110895 –10905 [DOI] [PubMed] [Google Scholar]
- 29.Bhat, S., Sorci-Thomas, M. G., Alexander, E. T., Samuel, M. P., and Thomas, M. J. (2005) J. Biol. Chem. 28033015 –33025 [DOI] [PubMed] [Google Scholar]
- 30.Martin, D. D., Budamagunta, M. S., Ryan, R. O., Voss, J. C., and Oda, M. N. (2006) J. Biol. Chem. 28120418 –20426 [DOI] [PubMed] [Google Scholar]
- 31.Saito, H., Dhanasekaran, P., Nguyen, D., Deridder, E., Holvoet, P., Lund-Katz, S., and Phillips, M. C. (2004) J. Biol. Chem. 27920974 –20981 [DOI] [PubMed] [Google Scholar]
- 32.Lazar, K. L., Miller-Auer, H., Getz, G. S., Orgel, J. P., and Meredith, S. C. (2005) Biochemistry 4412681 –12689 [DOI] [PubMed] [Google Scholar]
- 33.Ren, X., Zhao, L., Sivashanmugam, A., Miao, Y., Korando, L., Yang, Z., Reardon, C. A., Getz, G. S., Brouillette, C. G., Jerome, W. G., and Wang, J. (2005) Biochemistry 4414907 –14919 [DOI] [PubMed] [Google Scholar]
- 34.Mishra, V. K., and Palgunachari, M. N. (1996) Biochemistry 3511210 –11220 [DOI] [PubMed] [Google Scholar]