

Abstract
The glycosyltransferase termed MshA catalyzes the transfer of N-acetylglucosamine from UDP-N-acetylglucosamine to 1-l-myo-inositol-1-phosphate in the first committed step of mycothiol biosynthesis. The structure of MshA from Corynebacterium glutamicum was determined both in the absence of substrates and in a complex with UDP and 1-l-myo-inositol-1-phosphate. MshA belongs to the GT-B structural family whose members have a two-domain structure with both domains exhibiting a Rossman-type fold. Binding of the donor sugar to the C-terminal domain produces a 97° rotational reorientation of the N-terminal domain relative to the C-terminal domain, clamping down on UDP and generating the binding site for 1-l-myo-inositol-1-phosphate. The structure highlights the residues important in binding of UDP-N-acetylglucosamine and 1-l-myo-inositol-1-phosphate. Molecular models of the ternary complex suggest a mechanism in which the β-phosphate of the substrate, UDP-N-acetylglucosamine, promotes the nucleophilic attack of the 3-hydroxyl group of 1-l-myo-inositol-1-phosphate while at the same time promoting the cleavage of the sugar nucleotide bond.
Members of the Actinomycetales family function in diverse and important roles to humans. On the positive side, Streptomyces species produce over two-thirds of the clinically useful antibiotics of natural origin (1) and Corynebacteria species are heavily utilized in industrial synthesis (1, 2), whereas on the negative side many Actinomycetales are pathogens, for example, Mycobacterium are the causative agents of the diseases tuberculosis (3) and leprosy, infecting millions worldwide each year (3, 4). An intimate knowledge of biochemistry common to all Actinomycetales allows their utilization as chemistry toolboxes while mitigating their pathogenic nature.
For most organisms, maintenance of the appropriate reducing environment in the cell is required for proper cellular function, and this is usually achieved through the synthesis and cellular balance of low molecular weight thiols such as glutathione (5). Although glutathione is the predominate thiol in Gram-negative bacteria and eukaryotes, in the Actinomycetales, the major thiol is instead mycothiol (MSH),2 1-d-myoinosityl-2-(n-acetyl-l-cysteinyl)amido-2-deoxy-α-d-glucopyranoside (6). The synthetic pathway for MSH is believed to be conserved in all Actinomycetales species and requires at least four enzymes (MshA through MshD, Fig. 1). The first enzyme, MshA, catalyzes the transfer of N-acetylglucosamine from UDP-N-acetylglucosamine (UDP-GlcNAc) to 1-l-myo-inositol 1-phosphate (1-l-Ins-1-P) to produce 3-phospho-1-d-myo-inosityl-2-acetamido-2-deoxy-α-d-glucopyranoside (GlcNAc-Ins-P) (7, 8). A yet to be discovered phosphatase is proposed to dephosphorylate GlcNAc-Ins-P to produce GlcNAc-Ins (7). GlcNAc-Ins is then deacetylated (MshB) and subsequently cysteinylated (MshC) at the amino group to produce Cys-GlcN-Ins, which then is acetylated (MshD) to produce AcCys-GlcN-Ins or MSH (9–13). In Mycobacterium smegmatis, mutants blocked in MSH biosynthesis exhibit enhanced sensitivity to cellular stress reagents, including hydrogen peroxide and rifampicin (14, 15), whereas in Mycobacterium tuberculosis (Erdman strain) mutants blocked in MSH production were not viable (16). Of the four genes, mshA and mshC were found to be critical to the production of MSH and therefore viability of the organism (16, 17). Interruption of mshB or mshD was either complemented by a promiscuous cellular activity or the product of interrupted synthesis acted as a poor analog of MSH (14, 18). Therefore MshA and MshC are important potential drug targets for treatment of tuberculosis and other Actinomycetales infections. The three-dimensional structures of both MshB (19) and MshD (20, 21) have been determined; however, there have been no reports of structures for MshA or MshC.
FIGURE 1.

Steps in the biosynthesis of mycothiol.
Glycosyltransferases (GTs), such as MshA, can be grouped based on sequence into what is currently a total of 90 distinct families (CAZy, Carbohydrate-Active Enzymes data base) (22, 23). GTs can also be grouped based on structure, with most GTs determined to date having one of two folds, termed GT-A and GT-B (24). Recently two new GT folds have been described for CAZY family GT51 (25) and GT66 (26). Finally GTs can be termed “retaining” or “inverting” based on the configuration of the sugar nucleotide-derived carbohydrate in the final product. There is no absolute correlation between the fold (GT-A or GT-B) and the stereochemical outcome of the reaction as examples of both inverting and retaining GTs have been found in both families; however, all members of a CAZy family are expected to have the same fold (23, 27). Based on sequence similarity MshA is grouped into CAZy family GT4, all of which have been found to be retaining glycosyltransferases and have the GT-B fold. GT4 is the second largest CAZy data base family (6563 CAZY entries at the time of this writing) and have a diverse set of donor and acceptor molecules. Only recently a limited number of GT4 family members have been structural characterized, including WaaG, which transfers glucose from UDP-glucose onto l-glycero-d-mannoheptose II; AviGT4, which is involved in avilamycin A biosynthesis; and PimA, a phosphatidylinositol mannosyltransferase (28, 29).
Here we present the structure of unliganded MshA from Cornybacterium glutamicum and its complex with UDP and 1-l-Ins-1-P. Enzymatic studies demonstrate that UDP-GlcNAc and 1-l-Ins-1-P are substrates for CgMshA, and that the product is GlcNAc-Ins-P, as was previously determined for MshA from crude lysates of Mycobacterium smegmatis (7). Initial velocity studies indicate a sequential mechanism with UDP-GlcNAc almost certainly binding first followed by 1-l-Ins-1-P. The structural and kinetic data are most consistent with a mechanism involving the β-phosphoryl group of the substrate, UDP-GlcNAc, acting as the general base and not a mechanism involving a covalent enzyme-N-acetylglucosamine intermediate. The crystal structure highlights residues involved in binding and catalysis and serves as a starting point for inhibitor design.
EXPERIMENTAL PROCEDURES
Cloning—The MshA gene (residues 1–418) was amplified from C. glutamicum genomic DNA (ATCC 13032) using PCR (primers 5-TTTTTCATATGCGCGTAGCTATGATTTCC-3′ and 5′-TTTTTCTCGAGTTAGCCGTGATGCGTTTCAC-3′) and blunt-end-cloned into pCR-BLUNT (Invitrogen). An internal Nde1 site was removed using PCR mutagenesis and primers containing the desired alteration (primers, 5′-CCGGATACCTATAGGCAcATGGCAGAGGAACTGGGC-3′ and 5′-GCCCAGTTCCTCTGCCATgTGCCTATAGGTATCCGG-3′). The CgMshA gene was subcloned into the Nde1 and HindIII sites of pET28a(+) utilizing a HindIII site of the pCR-BLUNT vector as the downstream restriction site. The CgMshA gene was also cloned into the Nde1 and XhoI sites of pET29a(+) utilizing PCR amplification and the CgMshA: pET28a(+) vector as a template (primers 5′-TTTTTCATATGCGCGTAGCTATGATTTCC-3′ and 5′-ATCCCGCTCTCGAGGCCGTGATGCGTTTCACC-3′).
Expression—Overexpression vectors containing the CgMshA gene were transformed into Rosetta2 cells (Invitrogen) and grown in 8-ml overnight cultures of MDG media (30). These cultures were then made 10% in glycerol and stored at -80 °C in 2-ml aliquots. A 100-ml starter culture of MDG media was inoculated with one 2-ml storage stock and grown at 37 °C to late-log phase. This was used to inoculate 4 liters of ZYP5052 autoinduction media in six 2-liter baffled flasks (30). The cultures were grown at 23 °C with 300 rpm shaking and were harvested at saturation (36–48 h, A600 > 12). All growth media contained 30 μg/ml chloramphenicol and 200 μg/ml kanamycin.
Purification—Frozen cell paste was resuspended in three times its volume of buffer A (100 mm TEA, pH 7.8, 200 mm (NH4)2SO4, 10% glycerol, 15 mm imidazole), and lysozyme was added to 1 mg/ml. After incubation at 4 °C for 30 min the cell slurry was disrupted by sonication and was spun at 25,000 × g for 30 min. The supernatant was applied to a nickel-Sepharose HP (GE-Healthcare) column equilibrated against buffer A. Bound proteins were eluted with a gradient of 15–300 mm imidazole over 10 column volumes. Fractions with greater than 20% of the peak activity were pooled, made 1 m in (NH4)2SO4, and applied to a Phenyl-Sepharose HP (GE-Healthcare) column equilibrated against buffer B (20 mm TEA, pH 7.8, 1 m (NH4)2SO4, 10% glycerol, 0.5 mm EDTA, 1 mm β-mercaptoethanol). Bound proteins were separated with a 1 to 0 m (NH4)2SO4 gradient over 10 column volumes with properly folded CgMshA eluting between 500 and 300 mm (NH4)2SO4. Pooled fractions were concentrated by ultrafiltration (Amicon) to a concentration of 20–40 mg/ml and stored at -80 °C.
Crystallization and Phasing—Crystallization of CgMshA was by vapor diffusion under silicon oil (Fisher) utilizing 96-well round bottom assay plates stored open to room humidity at 18 °C. Crystals of APO CgMshA with a hexagonal bipyramidal morphology were obtained from drops containing 2 μl of protein (CgMshA:pET28a(+), 15 mg/ml, 400 mm (NH4)2SO4, 10% glycerol, 0.5 mm EDTA, 1 mm β-mercaptoethanol) with 2 μl of precipitant (20% polyethylene glycol 4000, 100 mm Tris, pH 8.5, 200 mm LiSO4). Crystals were soaked in a stabilization solution of 40% polyethylene glycol 4000, 100 mm Tris, pH 8.5, 200 mm LiSO4, prior to vitrification by plunging in liquid nitrogen. Data were collected on an MSC R-AXIS-IV++ image plate detector using CuKα radiation from a Rigaku RU-H3R x-ray generator and processed using MOSFLM and SCALA (31, 32). The space group was determined to be P31 with approximate cell dimensions of a = b = 79.7 Å, and c = 148.4 Å. There is a molecular dimer per asymmetric unit with a solvent content of 59%. The structure of APO CgMshA was determined by SIRAS using a single mercury derivitized crystal, which was prepared by soaking a crystal for 2 h in the stabilization solution with the addition of saturated p-chloromercuribenzoate. Heavy atom positions and initial phases were determined using PHENIX (33). The solvent flattened SIRAS map was of sufficient quality for ARP/WARP (34) to autobuild a majority of the structure. Iterative rounds of modeling building within the molecular graphics program COOT (35) followed by refinement against the data using REFMAC (36) were used to build the remainder of the structure. The final rounds included TLS refinement.
A second crystal form of CgMshA was obtained in drops that contained 2 μl of protein (CgMshA:pET29a(+), 40 mg/ml, 400 mm (NH4)2SO4, 10% glycerol, 0.5 mm EDTA, 1 mm β-mercaptoethanol, 10 mm UDP-GlcNAc) combined with 2 μl of precipitant (0.8 m sodium citrate, pH 5.5). This crystal form was cryoprotected by soaking in 1.2 m sodium citrate, pH 5.5, 10% glycerol, and 200 mm (NH4)2SO4 prior to vitrification by plunging in liquid nitrogen. For the UDP·1-l-Ins-1-P dataset the (NH4)2SO4 was replaced with 50 mm 1-l-Ins-1-P, and the crystals were soaked for 1 h. Data were collected at Brookhaven National Laboratories at beamline X25 and processed using MOSFLM (32) and SCALA (31). The space group was determined to be I422 with approximate cell dimensions of a = b = 223.9 Å, and c = 125.0 Å. This crystal form has two molecules per asymmetric unit with a solvent content of 71.5%.
Phasing of the UDP dataset utilized the molecular replacement program AMORE (37) and the APO form of CgMshA as the model. Searches with the complete APO structure and or either of the N- or C-terminal domains did not produce a convincing molecular replacement solution. However, utilizing a dimerized N-terminal domain produced two solutions that were significantly above other solutions despite (or possibly because of) the fact that both of the molecular dimers in this crystal form are acted upon by crystallographic symmetry. Molecular replacement searches for the missing C-terminal domain after placement of the N-terminal domains were unsuccessful. The recently determined structure of PimA (PDB ID: 2GEJ) was used as a guide for the structure of a closed GT-B:GT-4 glycosyltransferase family member (29). Utilizing electron density maps phased with only the two placed N-terminal domains, and the PimA structure as a guide, there was enough secondary structure visible in a skeletonized map to manually place both C-terminal domains, which were then successfully refined into their correct positions with rigid body refinement. Molecular constraints for UDP and 1-l-Ins-1-P were calculated using the PRODRG server (38). Refinement of this crystal form utilized REFMAC with the final rounds incorporating TLS refinement (36).
DLS measurements were performed with a 4 mg/ml CgMshA solution in 250 mm (NH4)2SO4, 50 mm TEA, pH 7.8, at 25 °C on a DynaPro MS/X instrument (Protein Solutions). Data collection and deconvolution were performed using the DYMAMICS 6.2.05 software (Protein Solutions).
Purified CgMshA was subjected to gel-filtration chromatography at a flow rate of 0.5 ml/min on a Superose-12 column (10/300, Amersham Biosciences). The column was pre-equilibrated with 50 mm TEA, pH 7.8, 200 mm (NH4)2SO4, 10% glycerol. The following proteins were used as molecular mass standards: thyroglobulin, 669 kDa; bovine γ-globulin, 158 kDa; chicken albumin, 44 kDa; equine myoglobin, 17.0 kDa; and vitamin B12, 1.3 kDa.
Production of 1-l-Ins-1-P—The gene for inositol-1-phosphate synthase from the hyperthermophile Archaeoglobus fulgidus (Af_INO) was cloned as described by a previous group (39). Briefly, the Af_INO gene was PCR-amplified from genomic DNA and ligated into pET23a(+) vector utilizing Nde1 and HindIII restriction sites. The resultant plasmid was transformed into Escherichia coli strain BL21DE3/pLysS, and the cells were grown and protein was expressed by autoinduction in an identical fashion as CgMshA (see above), except that the antibiotic was ampicillin (100 μg/ml), and the cells were grown at 37 °C for 12–18 h. Cells were sonicated in three times the weight/volume of buffer A (50 mm Tris, pH 8.5), and spun at 25,000 × g to remove cell debris. The supernatant, contained in a steel vessel, was heated in an 80 °C water bath, with constant stirring for 5 min. The vessel was then placed on ice and cooled to 4 °C as quickly as possible by constant stirring. The precipitated proteins were removed by centrifugation (25,000 × g). The supernatant was applied to a 2.6 × 12 cm Poros-HQ anion exchange (POROS MEDIA) column, equilibrated with buffer A. Bound proteins were eluted with a 0 to 0.25 m (NH4)2SO4 gradient over ten column volumes. Fractions, which related to peak concentrations of Af_INO, by SDS gels, were pooled, made 1 m in (NH4)2SO4 and applied to a 2.6 × 12 cm phenyl-Sepharose (GE-Healthcare) hydrophobic exchange column equilibrated with buffer A plus 1 m (NH4)2SO4. Bound proteins were eluted with a 1.0–0 m (NH4)2SO4 gradient in buffer A over ten column volumes, pooled based on purity by SDS gels, and dialyzed overnight in buffer A. Purified Af_INO was concentrated to 10 mg/ml by Amicon ultrafiltration, snap frozen in liquid nitrogen, and stored at -80 °C.
1-l-Ins-1-P was synthesized using Af_INO and glucose 6-phosphate. The starting solution was 125 mm glucose 6-phosphate, 0.625 mm ZnCl2, 1.25 mm NAD+, and 3.6 mg of Af_INO in 50 mm Tris, pH 7.5 (NH4OH). The reaction was held at 85 °C with a heat block, and every 45 min the NAD was increased by 0.5 mm and another 2 mg of Af_INO was added. The progress of the reaction was monitored utilizing the CgMshA/pyruvate kinase/lactate dehydrogenase assay with substoichiometric 1-l-Ins-1-P (potential from reaction, 0.20 mm) to NADH (0.25 mm). The completed reaction (∼2–3 h) was diluted to 100 mm (taking into account additions during the reaction) and stored at -80 °C to be used directly in assays for MshA activity.
Measurement of Enzymatic Activity—The production of UDP was measured using a coupled assay system and monitoring the reaction at 340 nm. Standard conditions were 30 nm enzyme, 50 mm TEA, pH 7.8, 200 μm NADH, 500 μm phosphoenol pyruvate, 10 mm MgCl2, 20 units of pyruvate kinase (ammonium sulfate suspension), and 55 units of lactate dehydrogenase (ammonium sulfate suspension) in a 1-ml reaction at 25 °C. Except for the enzyme, all components were mixed in the cuvette and allowed to equilibrate for 2 min. Reactions were initiated by the addition of enzyme. The amount of coupling enzymes was sufficient to not limit the rate of reaction and was required to minimize a lag in the assays.
Data Analysis—To determine the basic kinetic parameters for each substrate, initial velocity plots at saturating concentrations of one substrate were fit to the Michaelis-Menten equation (Equation 1) where Vmax is the maximal velocity, A is the concentration of the varied substrate, and Ka is the Michaelis constant for substrate A. When both substrates were varied, intersecting initial velocity plots were fit to Equation 2.
 |
(Eq. 1) |
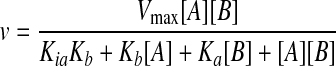 |
(Eq. 2) |
Data described with single variable equations were analyzed using KaleidaGraph (Synergy Software). Data described using equations with multiple independent variables were analyzed using GraFit (Erithacus Software).
Isolation of GlcNAc-inositol 3-Phosphate—CgMshA (80 μg) was added to a 1-ml solution containing 100 mm TEA, pH 7.8, 100 mm 1-l-Ins-1-P, and 100 mm UDP-GlcNAc. The production of UDP was monitored using a high-performance liquid chromatography-based assay and a UV detector (260 nm). The separation of compounds was accomplished using a 1-ml Mono Q ion-exchange column (GE-Healthcare) with the following programmed gradient: 0–5 min (0% B), 20 min (35% B), 25–30 min (100% B), 33 min (0%B) where Buffer A is 20 mm ammonium bicarbonate and Buffer B is 600 mm ammonium bicarbonate and a flow rate of 1 ml/min. Retention times for UDP-GlcNAc and UDP were 12 and 20 min, respectively. When the reaction had run to completion (∼5 h), the entire reaction was injected and fractionated using the above high-performance liquid chromatography method. Fractions (1 ml) were treated with alkaline phosphatase and then assayed for inorganic phosphate using a malachite green phosphate assay kit (Bioassay Systems). Fractions 9–12 were found to contain phosphate but did not correlate to a peak at 260 nm. These fractions were pooled and lyophilized. The lyophilized white powder was analyzed by 1H NMR, 31P NMR, and by mass spectrometry. Electrospray ionization-mass spectrometry yielded a value of m/z 462.36 (M-H)-1 (C14H25NO14P-1 calculated 462.10).
RESULTS AND DISCUSSION
Expression and Purification—Initial screens of full-length clones of MshA from Mycobacterium tuberculosis, Streptomyces coeliclor, Nocardia farcina, and C. glutamicum as N-terminally hexahistidine-tagged constructs all resulted in high overexpression, but only the MshA from C. glutamicum (CgMshA) resulted in large amounts of soluble protein. Typically, yields of soluble, active CgMshA using autoinduction media were 200 mg/liter. After nickel-nitrilotriacetic acid affinity chromatography, purification on Phenyl Sepharose hydrophobic exchange media was critical to separating active, monodisperse protein from inactive, polydisperse protein. Ammonium sulfate (>100 mm) was required to maintain activity, whereas protein kept in a minimal ionic strength buffer was subject to degradation and loss of activity. The initial construct of MshA utilized an N-terminal thrombin-cleavable 6× His tag to facilitate purification. Attempts at thrombin cleavage resulted in nonspecific degradation, so the N-terminal tag was not removed. The N-terminally tagged protein was utilized for the initial structure determination of unliganded MshA. A second C-terminally tagged MshA was used to determine the structure of MshA in complex with UDP and 1-l-Ins-1-P.
Enzymatic Characterization—From the initial velocity plots for 1-l-Ins-1-P and UDP-GlcNAc, both substrates exhibited Michaelis-Menten kinetics, and no curvature was seen in the Lineweaver-Burk linear replots (supplemental Fig. S1). The kinetic parameters for each substrate were determined from fits of the data to Equation 1. A value of 12.5 ± 0.2 s-1 was determined for the kcat, and the Michaelis constants for 1-l-Ins-1-P and UDP-GlcNAc were 240 ± 10 and 210 ± 20 μm, respectively. The Km values are consistent with those determined previously for MshA from Mycobacterium smegmatis crude lysates (7). To determine whether MshA proceeds through a sequential or ping-pong mechanism, an initial velocity plot was made by varying both substrates. The plot shows intersecting lines, and the data fit well to Equation 2, consistent with a sequential mechanism (supplemental Fig. S2). The pseudo-disaccharide phosphate monoester product of the reaction was purified using ion-exchange chromatography. 31P and 1H NMR spectroscopy of the product was consistent with GlcNAc-Ins-P (supplemental Figs. S3 and S4). A coupling constant of 3.3 Hz was determined for the anomeric proton confirming that the product retains the alpha sugar configuration (inset, supplemental Fig. S4).
GT-B Fold Monomer Structure—The structure of unliganded CgMshA (APO form) was determined by single isomorphous replacement with anomalous scattering utilizing a CuKα home source and a mercury derivative to 2.1-Å resolution (Table 1). The structure of CgMshA has two domains each exhibiting the β/α/β Rossmann-fold type typical of the GT-B fold superfamily (Fig. 2). The N-terminal domain has an 8-stranded β-sheet (β8, β7, β6, β5, β1, β2, β4, and β3) that is bounded by six α-helices (α1–α5 and α14). The C-terminal domain has a six-stranded β-sheet (β11, β10, β9, β12, β13, and β14), which is bounded by eight α-helices (α6–α13). The C-terminal domain β-sheet has all parallel β-strands, whereas the N-terminal β-sheet is a mixture of parallel (β1, β5, β6, β7, and β8) and antiparallel (β1, β2, β4, and β3) β-strands. The C-terminal domain ends at α13 with the protein chain crossing back to the N-terminal domain where the terminal α-helix (α14) interacts with the β-sheet of the N-terminal domain.
TABLE 1.
Data collection and refinement statistics
| Data collection | ||||
| Data set | APO | APO-PCMB | UDP | UDP/1-l-Ins-1-P |
| Space group | P31 | P31 | 1422 | 1422 |
| Resolution (Å) | 25-2.1 (2.21-2.1)a | 25-2.8 (2.95-2.8) | 25-2.8 (2.95-2.8) | 25-2.6 (2.74-2.6) |
| Completeness (%) | 98.7 (91.5) | 99.9 (100.0) | 96.0 (96.8) | 99.6 (100.0) |
| Redundancy | 2.9 (2.5) | 8.6 (8.2) | 5.4 (5.3) | 5.2 (4.9) |
| I/σI | 16.5 (2.9) | 28.5 (10.7) | 22.4 (3.9) | 23.0 (4.0) |
| Rmerge | 0.052 (0.238) | 0.064 (0.143) | 0.066 (0.32) | 0.056 (0.31) |
|
Mean FOM/phasing power
|
0.45/1.03
|
|||
| Refinement statistics | ||||
| Resolution (Å) | 25-2.1 (2.16-2.1) | 25-2.8 (2.8-2.73) | 25-2.6 (2.69-2.6) | |
| Rfactor (%) | 18.0 (22.0) | 18.4 (28.6) | 19.3 (30.2) | |
| Rfree (%)b | 22.1 (29.9) | 21.8 (34.2) | 21.4 (34.6) | |
| Residues fit | A(-1):15, A23:142, A147:205, A210:266, A273:418, B1:15, B23:139, B148:266, B274:418 | A1:138, A147:265, A274:409, B1:138, B146:265, B274:409 | A1:138, A147:265, A274:409, B1:138, B146:265, B274:409 | |
| Small molecules | 6 SO4 | 2UDP, 2 SO4, 2 Mg+2 | 2 UDP, 1 1-l-Ins-1-P, 2 Mg+2 | |
| Number of atoms | ||||
| Protein | 6148 | 6072 | 6051 | |
| Solvent/UDP/1-l-Ins-1-P | 405/-/- | 55/50/- | 70/50/16 | |
| Average B-factors (Å2) | ||||
| Protein | 36.4 | 28.6 | 44.6 | |
| Solvent/UDP/1-l-Ins-1-P | 36.7/-/- | 20.2/30.3/- | 38.7/38.8/83.9 | |
| Root mean square deviations | ||||
| Bonds (Å) | 0.015 | 0.013 | 0.014 | |
| Angles (°) | 1.494 | 1.733 | 1.606 |
Numbers in parentheses apply to the highest resolution shell
Calculated using 5% of the reflections omitted from refinement
FIGURE 2.
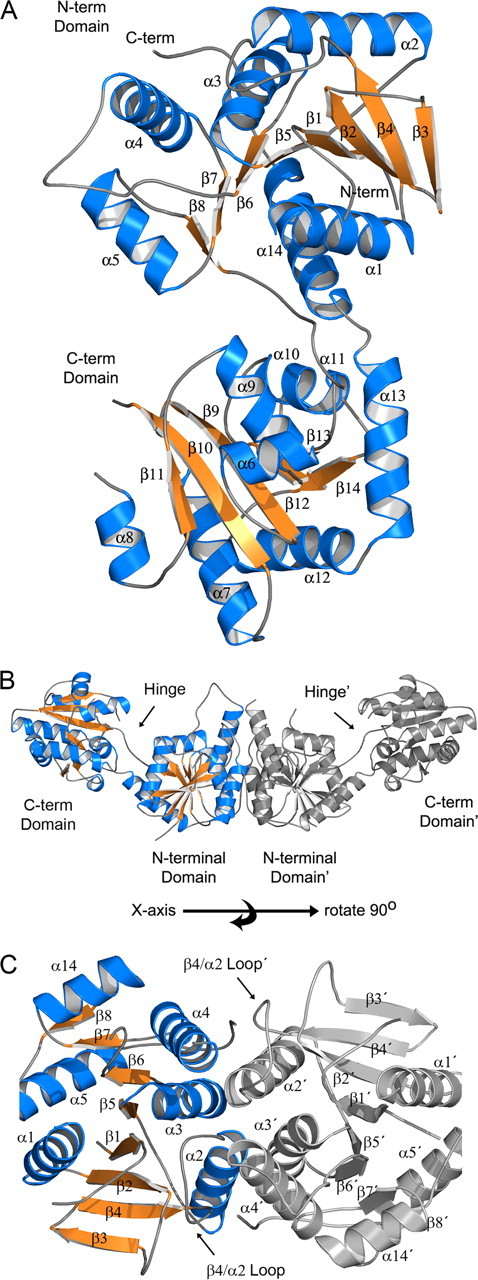
Structure of APO CgMshA. A, ribbon diagram of an APO-CgMshA monomer (open conformation). Helices are in blue, strands are in orange, and coils are in gray. B, molecular dimer of CgMshA. C, illustration of the secondary structure involved in formation of the molecular dimer. In B and C, the 2-fold-related monomer is colored gray with secondary structure labeled with prime (′) designations (i.e. α1′ and β12′).
Structure homolog searches using SSM, and the closed UDP·1-l-Ins-1-P complex yielded a number of other members of the GT-B family. The closest structural homologs were other members of the GT-4 family; the α1,3-glucosyltransferse WaaG (SSM Z-score of 11.5, r.m.s.d. of 2.10 Å, and 21% sequence identity) and the phospatidylinositol mannosyltransferase PimA (Z-score of 9.4, r.m.s.d. of 2.14 Å, 26% sequence identity) (28, 29). A slightly lower score was observed for the only other GT-4 family member with a known structure, an enzyme involved in avilamycin A biosynthesis, AviGT4 (Z-score of 7.4, r.m.s.d. of 2.85 Å, and 21% sequence identity) (28). Structure homolog searches using SSM and the open APO form of the enzyme yielded no structurally homologous structures with >70% structural overlay, indicating the open form of the enzyme is a newly observed conformation between the N- and C-terminal domains in GT-B family members.
MshA Oligomeric State—MshA is composed of 418 residues with a monomer molecular weight of 45,669. Analysis of CgMshA by gel filtration chromatography and dynamic light scattering yielded apparent molecular weights of 97,700 and 111,000 respectively, suggesting CgMshA is a dimer in solution. There is a conserved dimer interface in the two crystal forms. In the P31 crystal form (APO form) there is a molecular dimer in the asymmetric unit, whereas in the I422 crystal form (UDP·1-l-Ins-1-P ternary complex) there are two monomers per asymmetric that, when acted upon by the crystallographic symmetry, yield molecular dimers with identical interfaces observed in the P31 crystal form. Superposition of the two N-terminal domains that form the dimer from the two crystal forms yields an r.m.s.d. of 0.53 Å (405 common Cαs). The CgMshA APO-form dimer, when viewed perpendicular to the molecular 2-fold, has the shape of a slightly bent rod with dimensions of 40 × 40 × 135 Å (Fig. 2B). The dimer interface is entirely composed of residues from the N-terminal domain, wherein the dimer interface and the C-terminal domain interact on opposite sides of the N-terminal domain. The dimer interface is confined to α2, α3, α4, and β4/α2 and their 2-fold symmetrically related counterparts (Fig. 2C). A total of 1488 Å2 is buried per subunit within the dimer interface and includes both polar and non-polar interactions. A comparison of various Actinomycetes MshA orthologs suggests that the dimer interface is most likely conserved (supplemental Fig. S5). Some of the conserved interactions are a hydrophobic interaction between α2 and α3′ (Leu-93 and Leu-119′), and also α4 with the β4/α2 loop (Ile-156, Pro-72′, and Leu-76′), and a bifurcated polar interaction between the side chains of Gln-84 and Gln-160′. The active site is located between the N- and C-terminal domains and is >30 Å from the dimer interface at its closest points. However, three of the four residues that coordinate the phosphate of 1-l-Ins-1-P (Lys-78:β4/α2 loop, Tyr-110:α3, and Arg-154:α4) are located on the opposite face of structural elements used to construct the dimer interface (see below). Dimerization therefore might stabilize these structural elements and contribute secondarily to enzyme catalysis.
Donor Substrate Binding—Crystallization of CgMshA in the presence of UDP or UDP-GlcNAc resulted in a tetragonal crystal form (Table 1). The structure of the binary complex was determined by molecular replacement using the individual domains as search models (see “Experimental Procedures”). There are two subunits per asymmetric unit, with the CgMshA molecular dimer created by crystallographic symmetry. The two subunits are essentially equivalent with an r.m.s.d. of 0.31 Å over 392 residues. Similarly, the two subunits of the APO crystal form are essentially equivalent with an r.m.s.d. of 0.37 Å over 391 Cα residues. Comparisons of the APO and ternary complex will therefore be confined to the subunits with the best crystallographic statistics. Data collected on cocrystals with UDP or UDP-GlcNAc resulted in similar maps showing only UDP bound to the active site (Fig. 3A, data not shown). Crystals grown with UDP-GlcNAc and subsequently soaked in high concentrations of UDP-GlcNAc (50 mm), neither degraded the diffraction pattern nor resulted in any observable UDP-GlcNAc binding suggesting that the closed form of the protein, as held by crystal contacts, is not free to exchange sugar nucleotide. The lack of sugar nucleotides either arises from minor contaminating UDP in the UDP-GlcNAc preparation or slow hydrolysis of the UDP-GlcNAc bond over the lifetime of the crystals and the lack of free exchange of the bound glucoside with free UDP-GlcNAc.
FIGURE 3.
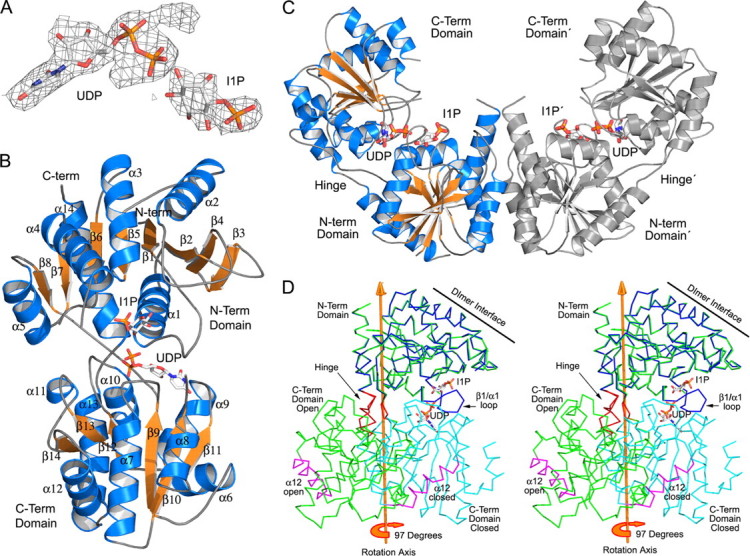
Structure of the CgMshA UDP·1-l-Ins-1-P complex. A, final 2Fo - Fc density for UDP and 1-l-Ins-1-P contoured at 1.0σ. B, ribbon diagram of a CgMshA monomer in complex with UDP and 1-l-Ins-1-P (closed conformation). UDP and 1-l-Ins-1-P are shown as sticks colored by atom type. C, molecular dimer of CgMshA after domain reorientation. Electron density for 1-l-Ins-1-P was sufficient only to fit in one subunit but was modeled in the dimer shown here for illustrative purposes. D, stereo illustration of DYNDOM rotation axis, relating the N- and C-terminal domain before and after binding of nucleoside. The APO structure is illustrated by the green trace, whereas the N- and C-terminal domains of the ternary complex are colored blue and cyan, respectively. α12 is colored maroon in both structures to help in orientation, whereas the hinge residues (196–197 and 386–392) are colored red.
Upon binding nucleotide there is a large conformational change bringing the binding sites for sugar nucleotide and 1-l-Ins-1-P into close proximity (Fig. 3, B and C, and supplemental Fig. S6). Rotation of the C-terminal domain yields a V-shape oligomer and decreases the longest dimension of the molecular dimer from 135 to 105 Å. Submission of the APO- and UDP-bound structures to the DYNDOM server (40) indicated a 97° rotation (0 Å translation and 38° closure) of the C-terminal domain (residues 197–390) relative to the N-terminal domain (residues 1–196 and 391–418). The axis of rotation is approximately parallel to α13 and α14 and intersects the hinge residues 196–197 and 386–392 (Fig. 3D). The large conformational change does not significantly change the overall structure of the individual domains with the N- and C-terminal domain exhibiting r.m.s.d. values of 0.32 Å (198 Cαs) and 0.50 Å (176 Cα), respectively. Two of the largest changes in the interactions between the two domains involve residues of the C-terminal domain structurally adjacent to the hinge residues (axis of rotation) and, separately, an N-terminal loop involved in interactions with nucleoside. In the APO structure there are no direct interactions between the N- and C-terminal domain outside of interactions of each domain with the linker region. One of these domain-to-linker interactions is the packing interactions of Phe-318 and Leu-320 with the linker region: Ala-393/Gln-397/Ser-194 and Gly-196/Ala-197, respectively. Upon nucleotide binding Phe-318 and Leu-320 rotate about the hinge axis moving by ∼14.5 Å and 11.0 Å, respectively (Cα to Cα). Phe-318 and Leu-320 form a new hydrophobic pocket with Pro-195, Trp-391, Gly-23, and Val-26. Their previous position is filled by the side chain of Phe-389, which was previously positioned out into solvent. Interestingly, Phe-318 and Leu-320 are on an important loop β12/α10, which is implicated in recognition of the sugar portion of UDP-GlcNAc. Interdomain flexibility has been observed or predicted for many members of the GT-B structural family. Examples include MurG (41), TDP-epi-vancosaminyltransferase (42), and glycogen synthase (43) with typical interdomain motions of 10–25°.
Electron density for UDP suggests that the pyrophosphate is in two conformations, most likely due to the lack of the anchoring interactions of GlcNAc (Fig. 3A). In addition, residual electron density adjacent to the α-phosphate may be the result of residual UDP-GlcNAc or an unknown small molecule binding at this position with low occupancy. The majority of the residues that interact with UDP are located in the C-terminal domain, with the uracil moiety forming polar interactions with the main-chain atoms of Arg-294 (α9/β11) and the side chain of Cys-262, the ribose with Glu-324 (α10), and the pyrophosphate with the main-chain amides of Leu-320, Val-321 (α10), and Arg-231 along with the side chains of Arg-231 and Arg-236 (β9/α7) (Fig. 4, A and B, and supplemental Fig. S7). Upon sugar nucleotide binding and domain rotation, residues from the N-terminal domain contribute interactions. Residues 16–22 are disordered in the APO state but become ordered in the binary complex. In the newly ordered state, Pro-16 and Gly-17 lay against the face of the uracil moiety, Asn-15 hydrogen bonds with the uracil (O2), and the backbone amide of Gly-23 is hydrogen bonded to the pyrophosphate moiety. A singular interdomain hydrogen bond is formed between the backbone carbonyl of Gly-17 and the backbone amide of Gly-264.
FIGURE 4.

Interactions in the ternary complex. Illustrations of the interaction of CgMshA with the UDP-GlcNAc model and 1-l-Ins-1-P are shown. A, Chemdraw diagram. Interactions shown with UDP (maroon) and 1-l-Ins-1-P (blue) are those observed in the ternary complex, whereas those with GlcNAc (red) are those found in the molecular model. B, stereo stick diagram.1-l-Ins-1-P carbons are colored yellow, and UDP-GlcNAc carbons are colored green. Important hydrogen bonding interactions are shown as gray dotted lines, and the interaction of the 3-OH of 1-l-Ins-1-P with the UDP-GlcNAc at the site of reaction chemistry is shown as a cyan dotted line. C, proposed SNi-substrate-assisted catalysis mechanism, featuring oxocarbenium-ion like transition state, with asymmetric phosphor bond breakage, and glycosidic bond formation.
There are three relevant structures of related glycosyltransferases with bound sugar nucleotides. The structures of WaaG and OtsA were determined with UDP-2-deoxy-2-fluoroglucose (UDP-2FGlc) (28, 44), and PimA was determined with GDP-mannose (GDP-man) (29). The binding mode of UDP-2FGlc to WaaG and OtsA were essentially identical, whereas the binding of GDP-man to PimA resulted in an alternate conformation for the pyrophosphate-sugar moiety, such that the faces of 2FGlc and mannose are perpendicular to each other, but binding in a similar “bent back” conformation relative to the pyrophosphate. Because UDP-2FGlc and UDP-GlcNAc share a common nucleoside base and sugar stereochemistry, the UDP-2FGlc structures were used to construct a model of UDP-GlcNAc binding to CgMshA (Fig. 4, A and B). As in all retaining glycosyltransferases the sugar moiety is bent back over the pyrophosphate moiety, exposing the anomeric sugar nucleotide carbon to nucleophilic attack. In this conformation the sugar 2-amino and 4-hydroxy groups would form hydrogen bonds with the β-phosphate and α-phosphate, respectively. In this binding mode, the GlcNAc molecule would have steric collisions with the β12/α10 loop, especially with Ser-317, which projects into the active site and interacts with the side chain of Glu-316 in the UDP binary complex. In the APO structure this loop is in a slightly different conformation and Ser-317 is positioned away from the UDP binding site. Therefore, in the UDP-GlcNAc model the β12/α10 loop was modeled based partially on its conformation in the APO state and utilizing main-chain conformations seen for this loop in OtsA and WaaG binary complexes with UDP-2FGlc (28, 44). In the molecular model the C3,C4-diol moiety of the sugar moiety plugs into the β12/α10 loop, forming hydrogen bonds between the 4-OH and the main-chain amides of Gly-319 and Phe-318, and between the 3-OH and the main-chain amides of Ser-317 and the side chain of Glu-316. In addition, the 6-OH forms hydrogen bonds with the side chains of His-133 and Asn-171 in a conserved interaction seen in the structure of OtsA with UDP-2FGlc (44). The acetyl group of UDP-GlcNAc would lie in a slot bordered by the β12/α10 loop and the 1-l-Ins-1-P binding site (see below), with potential interactions with the side chains of Asn-315, Thr-134, Phe-235, and Arg-231.
Acceptor Substrate Binding—Crystals of the binary complex could be soaked in high concentrations (50 mm) of 1-l-Ins-1-P without damaging the diffraction pattern. Examination of electron density maps after most of the structure was fit, suggested that only one of the subunits contained significant electron density for 1-l-Ins-1-P, so only that subunit will be discussed (Fig. 3A). There were no significant structural changes between the binary and ternary complexes (data not shown). The B-factors for 1-l-Ins-1-P average 84 Å3, nearly twice the surrounding residues (∼45 Å3), suggesting <100% occupancy, and perhaps a less than optimal binding site for 1-l-Ins-1-P. Despite this the structure of the ternary complex highlights several key features required for binding of the acceptor substrate. The side chains of Lys-78, Arg-154, Thr-134, and Tyr-110 all form hydrogen bonds with the phosphate moiety of 1-l-Ins-1-P with Tyr-110 in an allowed but disfavored phi-psi conformation (phi = 76, psi = 151) (Fig. 4, A and B, and supplemental Fig. S7). In the APO structure, and the UDP binary complex, this site is occupied by a sulfate ion, which may be one reason enzyme activity is stabilized by ammonium sulfate. The inositol moiety interacts with the β1/α1 loop and the N-terminal end of α1. There are polar interactions to the 3-OH (Met-24N), 4-OH (Asn-25), and 5-OH (His-9 and Asp-20O), whereas the side chains of Met-24 and Arg-231 form walls adjacent to the faces of the inositol. In the UDP·1-l-Ins-1-P structure the 3-OH is within hydrogen bonding distance (2.95 Å) to one of the oxygens of the β-phosphate and, therefore, is well positioned to participate in glycosidic bond formation.
Based on the structure of the UDP·1-l-Ins-1-P complex, three major binding determinants for 1-l-Ins-1-P are proposed. First the large number of contact points, both general electrostatic and hydrogen bonding interactions with the phosphate moiety are proposed to provide a large positive binding energy and limit the possible orientations of the inositol. Second, Arg-231 is important in delineating the substitution and conformation of the sugar. Arg-231 is in two conformations in the APO state, and becomes ordered in the binary complex through its interactions with the β-phosphate of UDP-GlcNAc. The side chain of Arg-231 lies against the face of the inositol and is ∼4.5 Å from the 1-l-Ins-1-P. Its position against the face requires that the 3, 4, 5, and 6 substituents of the ligand be in equatorial positions. Finally, the 2-OH of 1-l-Ins-1-P is the only substituent in an axial position and points into a small pocket created by the side chains of Met-24, Tyr-110, Thr-134, and UDP-GlcNAc. The axial nature of the 2-OH allows a closer approach of the 3-OH to the β-phosphate–GlcNAc bond.
Mechanistic Analysis—Determination of the APO and UDP·1-l-Ins-1-P ternary complex in conjunction with the modeling of the CgMshA·UDP-GlcNAc structure permits analysis of potential enzyme mechanisms. First, the open conformation and closed conformations are most likely distinct low energy states and not an artifact of crystallization because in each crystal form the two copies of the protein per asymmetric unit are in similar conformations, despite having dissimilar crystal contacts. In the APO structure the binding sites for UDP and 1-l-Ins-1-P are ∼30 Å from each other and, therefore, require a large domain reorientation to bring the reactants in close proximity. In addition, a portion of the β1/α1 loop is disordered in the APO structure, and therefore several of the binding determinates between CgMshA and the inositol moiety are either absent or oriented incorrectly. Binding of UDP-GlcNAc to the C-terminal domain presents an alternative surface to the N-terminal domain resulting in the stabilization of a closed form of the enzyme through interactions of the β1/α1 loop with the pyrophosphate and uracil moieties. Therefore, binding of UDP-GlcNAc and domain reorientation result in the structuring of the β1/α1 loop and the completion of the binding site for 1-l-Ins-1-P. These structures suggest that CgMshA proceeds by an ordered mechanism with UDP-GlcNAc binding first and 1-l-Ins-1-P binding second.
Chemical mechanisms proposed for glycosyltransferases share many of the features proposed for glycosidases. Glycosidases can be described as glycosyltransferases where the acceptor is a water molecule and, like glycosyltransferases, can be defined as inverting or retaining depending on the stereochemical outcome at the anomeric carbon. The involvement of an oxocarbenium ion transition state in the chemical mechanisms of glycosyltransferases and glycosidases is well supported. Significant normal α-deuterium secondary isotope effects have been reported for inverting and retaining glycosyltransferases (45–47) and glycosidases (48, 49) consistent with C1–O bond cleavage occurring prior to attack by the acceptor sugar or water. For inverting glycosidases and glycosyltransferases, the proposed single displacement mechanism is well established (50). There is also structural and kinetic evidence that retaining glycosidases follow a double displacement mechanism with participation of a covalent enzyme-substrate intermediate (49, 51).
Not surprisingly, prior to any structural evidence, retaining glycosyltransferases were proposed to utilize a double displacement mechanism. However, as the first structures were solved, it became apparent that there was not a readily identifiable nucleophile in the active site to support the formation of a covalent intermediate (44, 52, 53). In the absence of direct evidence of a viable covalent intermediate, an alternative mechanism, termed as an SNi “internal return,” has been proposed invoking an oxocarbenium ion-like transition state, with phospho-sugar bond breakage and glycosidic bond formation occurring in a concerted, but necessarily asynchronous manner, on the same face of the sugar (52, 53). This mechanism was first proposed nearly 30 years ago to describe anomalous results seen in the solvolysis of glucopyransoyl fluorides where a kinetically unimolecular reaction (SN1-like) resulted in retention of configuration of the sugar (SN2-like) (54).
Two recent reports, however, provide indirect support for the double displacement mechanism. Using a mutant form of the retaining glycosyltransferase LgtC, Lairson and coworkers (55) were able to isolate a covalently bound adduct. However, the adduct was unexpectedly formed on the neighboring residue, some 9.0 Å away from the anomeric carbon. A second report from Monegal and Planas describes the chemical rescue of a mutant form of a retaining α3-glycosyltransferase by sodium azide (56). The product of the chemical rescue is the inverted version of the sugar azide, which would be consistent with the first step in a double displacement mechanism. Although these findings provide evidence that retaining glycosyltransferases can accommodate parts of a double displacement mechanism, their support is inconclusive.
Similar to previous reports of retaining glycosyltransferases, the structure of CgMshA does not support a double displacement mechanism. In the CgMshA UDP-GlcNAc model, residues His-133:Thr-134 form a cap over the anomeric carbon on the β-face (closest distance, 5.5 Å; non-polar interactions). There are no side chains in proximity to assist in a double displacement mechanism (Fig. 4B). It should be noted, however, that as observed in the conversion of the APO- to the nucleoside-bound structure there are large conformational changes (domain-domain reorientation, β12/α10 loop), and it cannot be ruled out that somewhere along the reaction coordinate that a potential protein nucleophile would make a close approach to the β-face of UDP-GlcNAc.
The CgMshA UDP-GlcNAc model complex is consistent with an SNi mechanism where nucleophilic attack by the hydroxyl of 1-l-Ins-1-P and departure of UDP occur on the same face. In such a mechanism the transition state is highly dissociative with the donor having significant oxocarbenium ion character (Fig. 4C). In the UDP-GlcNAc model the 3-OH of 1-l-Ins-1-P approaches UDP-GlcNAc from the β-face and is 2.3 Å from the phosphorous oxygen and 2.5 Å away from the anomeric carbon. There are no protein groups near enough to the 3-OH to act as a general base consistent with the formation of a cyclic intermediate involving the 3-OH, the β-phosphate, and the anomeric carbon (Fig. 4, B and C). The conformation of UDP-GlcNAc with the carbohydrate located over the phosphates in a bent-back conformation appears to be a key factor in catalysis. This conformation may promote the significant oxocarbenium ion character in the donor through the formation of electrostatic interactions between the phosphates and the sugar and through the formation of hydrogen bonds between the 2-NH and 5-OH of UDP-GlcNAc to the β- and α-pyrophosphate oxygens, reducing the electron withdrawing effects of these polar substituents. In addition, in the bent-back conformation the β-side of the anomeric carbon can be presented to incoming substrates without the steric constraints that would have been imposed by the β-phosphoryl oxygens. The oxocarbenium ion-like nature of the transition state can assist in acid-base chemistry by raising the pKa of the β-phosphate such that it can act as an active site base abstracting a proton from the approaching 3-OH of 1-l-Ins-1-P. The dissociative nature of the transition state may be further stabilized by Arg-261, which forms an ion pair with the β-phosphate. A comparison of the active site architecture of CgMshA with other retaining glycosyltransferases suggests that the bent-back conformation of the donor molecule and the interaction of the β-phosphate with a positive charge (typically an arginine or metal ion) is a conserved feature of retaining glycosyltransferases (50, 57).
Mycothiol is the major low molecular weight thiol in Actinomycetes such as the human pathogen Mycobacterium tuberculosis, and therefore the enzymes involved in its biosynthesis are important drug targets. The structure determination of CgMshA, which catalyzes the first step in mycothiol biosynthesis, as APO and in complex with UDP and UDP·1-l-Ins-1-P highlights residues that are critical to the binding of substrates and catalysis and suggests an ordered binding mechanism. The structure of the CgMshA-UDP·1-l-Ins-1-P complex is noteworthy, because very few retaining glycosyltransferases have been visualized with bound acceptor, and it adds credence to a proposed SNi “internal return” mechanism instead of a covalent intermediate/double displacement mechanism. In addition these structures shed light on the structural flexibility of a large subset of glycosyltransferases. The large 97° interdomain motion upon binding sugar nucleotide depicted here for CgMshA highlights the flexibility of the interdomain “hinge” region of GT-B glycosyltransferases and widens the possible dynamic range other family members may undergo during substrate binding. Due to the lack of sequence conservation between or even within CAZy families, it is difficult to predict the degree of structural motions for individual family members, but the structural data so far indicates interdomain flexibility is an important feature of sugar nucleotide recognition of the GT-B family and needs to be taken into consideration in the design of inhibitors.
Supplementary Material
Acknowledgments
Data for this study were measured at beamline X25 of the National Synchrotron Light Source. Financial support comes principally from the Offices of Biological and Environmental Research and of Basic Energy Sciences of the U. S. Department of Energy and from the National Center for Research Resources of the National Institutes of Health.
The atomic coordinates and structure factors (codes 3C48, 3C4Q, and 3C4V) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported, in whole or in part, by National Institutes of Health Grant AI33696 (to J. S. B.). This work was also supported by a fellowship from the Charles H. Revson Foundation (to P. A. F.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S7.
Footnotes
The abbreviations used are: MSH, mycothiol or 1-d-myoinosityl-2-(n-acetyl-l-cysteinyl)amido-2-deoxy-α-d-glucopyranoside; CgMshA, C. glutamicum MshA; 1-l-Ins-1-P, 1-l-myo-inositol 1-phosphate; GlcNAc-Ins-P, 3-phospho-1-d-myo-inosityl-2-acetamido-2-dexoy-α-d-glucopyranoside; GT, glycosyltransferase; Af_INO, inositol-1-phosphate synthase from A. fulgidus; man, mannose; TEA, triethanolamine; CAZy, Carbohydrate-Active Enzymes data base.
References
- 1.Demain, A. L. (1999) Appl. Microbiol. Biotechnol. 52 455-463 [DOI] [PubMed] [Google Scholar]
- 2.Hermann, T. (2003) J. Biotechnol. 104 155-172 [DOI] [PubMed] [Google Scholar]
- 3.Dye, C., Scheele, S., Dolin, P., Pathania, V., and Raviglione, M. C. (1999) J. Am. Med. Assoc. 282 677-686 [DOI] [PubMed] [Google Scholar]
- 4.Britton, W. J., and Lockwood, D. N. (2004) Lancet 363 1209-1219 [DOI] [PubMed] [Google Scholar]
- 5.Hand, C. E., and Honek, J. F. (2005) J. Nat. Prod. 68 293-308 [DOI] [PubMed] [Google Scholar]
- 6.Newton, G. L., Arnold, K., Price, M. S., Sherrill, C., Delcardayre, S. B., Aharonowitz, Y., Cohen, G., Davies, J., Fahey, R. C., and Davis, C. (1996) J. Bacteriol. 178 1990-1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Newton, G. L., Ta, P., Bzymek, K. P., and Fahey, R. C. (2006) J. Biol. Chem. 281 33910-33920 [DOI] [PubMed] [Google Scholar]
- 8.Newton, G. L., Koledin, T., Gorovitz, B., Rawat, M., Fahey, R. C., and Av-Gay, Y. (2003) J. Bacteriol. 185 3476-3479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Newton, G. L., Av-Gay, Y., and Fahey, R. C. (2000) J. Bacteriol. 182 6958-6963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koledin, T., Newton, G. L., and Fahey, R. C. (2002) Arch. Microbiol. 178 331-337 [DOI] [PubMed] [Google Scholar]
- 11.Newton, G. L., and Fahey, R. C. (2002) Arch. Microbiol. 178 388-394 [DOI] [PubMed] [Google Scholar]
- 12.Anderberg, S. J., Newton, G. L., and Fahey, R. C. (1998) J. Biol. Chem. 273 30391-30397 [DOI] [PubMed] [Google Scholar]
- 13.Sareen, D., Steffek, M., Newton, G. L., and Fahey, R. C. (2002) Biochemistry 41 6885-6890 [DOI] [PubMed] [Google Scholar]
- 14.Newton, G. L., Ta, P., and Fahey, R. C. (2005) J. Bacteriol. 187 7309-7316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rawat, M., Newton, G. L., Ko, M., Martinez, G. J., Fahey, R. C., and Av-Gay, Y. (2002) Antimicrob. Agents Chemother. 46 3348-3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sareen, D., Newton, G. L., Fahey, R. C., and Buchmeier, N. A. (2003) J. Bacteriol. 185 6736-6740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buchmeier, N., and Fahey, R. C. (2006) FEMS Microbiol. Lett. 264 74-79 [DOI] [PubMed] [Google Scholar]
- 18.Buchmeier, N. A., Newton, G. L., and Fahey, R. C. (2006) J. Bacteriol. 188 6245-6252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maynes, J. T., Garen, C., Cherney, M. M., Newton, G., Arad, D., Av-Gay, Y., Fahey, R. C., and James, M. N. (2003) J. Biol. Chem. 278 47166-47170 [DOI] [PubMed] [Google Scholar]
- 20.Vetting, M. W., Yu, M., Rendle, P. M., and Blanchard, J. S. (2006) J. Biol. Chem. 281 2795-2802 [DOI] [PubMed] [Google Scholar]
- 21.Vetting, M. W., Roderick, S. L., Yu, M., and Blanchard, J. S. (2003) Prot. Sci. 12 1954-1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Campbell, J. A., Davies, G. J., Bulone, V. V., and Henrissat, B. (1998) Biochem. J. 329 719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coutinho, P. M., Deleury, E., Davies, G. J., and Henrissat, B. (2003) J. Mol. Biol. 328 307-317 [DOI] [PubMed] [Google Scholar]
- 24.Bourne, Y., and Henrissat, B. (2001) Curr. Opin. Struct. Biol. 11 593-600 [DOI] [PubMed] [Google Scholar]
- 25.Lovering, A. L., de Castro, L. H., Lim, D., and Strynadka, N. C. (2007) Science 315 1402-1405 [DOI] [PubMed] [Google Scholar]
- 26.Igura, M., Maita, N., Kamishikiryo, J., Yamada, M., Obita, T., Maenaka, K., and Kohda, D. (2008) EMBO J. 27 234-243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Breton, C., Snajdrova, L., Jeanneau, C., Koca, J., and Imberty, A. (2006) Glycobiology 16 29R-37R [DOI] [PubMed] [Google Scholar]
- 28.Martinez-Fleites, C., Proctor, M., Roberts, S., Bolam, D. N., Gilbert, H. J., and Davies, G. J. (2006) Chem. Biol. 13 1143-1152 [DOI] [PubMed] [Google Scholar]
- 29.Guerin, M. E., Kordulakova, J., Schaeffer, F., Svetlikova, Z., Buschiazzo, A., Giganti, D., Gicquel, B., Mikusova, K., Jackson, M., and Alzari, P. M. (2007) J. Biol. Chem. 282 20705-20714 [DOI] [PubMed] [Google Scholar]
- 30.Studier, F. W. (2005) Protein Expression Purif. 41 207-234 [DOI] [PubMed] [Google Scholar]
- 31.Evans, P. (2006) Acta Crystallogr. D Biol. Crystallogr. 62 72-82 [DOI] [PubMed] [Google Scholar]
- 32.Leslie, A. G. (2006) Acta Crystallogr. D Biol. Crystallogr. 62 48-57 [DOI] [PubMed] [Google Scholar]
- 33.Adams, P. D., Gopal, K., Grosse-Kunstleve, R. W., Hung, L. W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Pai, R. K., Read, R. J., Romo, T. D., Sacchettini, J. C., Sauter, N. K., Storoni, L. C., and Terwilliger, T. C. (2004) J. Synchrotron Rad. 11 53-55 [DOI] [PubMed] [Google Scholar]
- 34.Perrakis, A. (1997) Acta Crystallogr. Sect. D Biol. Crystallogr. 53 448-455 [DOI] [PubMed] [Google Scholar]
- 35.Emsley, P., and Cowtan, K. (2004) Acta Crystallogr. Sect. D Biol. Crystallogr. 60 2126-2132 [DOI] [PubMed] [Google Scholar]
- 36.Murshudov, G. N., Vagin, A. A., and Dodson, E. J. (1997) Acta Crystallogr. Sect. D Biol. Crystallogr. 53 240-255 [DOI] [PubMed] [Google Scholar]
- 37.Navaza, J. (1994) Acta Crystallogr. Sect. A 50 157-163 [Google Scholar]
- 38.Schuttelkopf, A. W., and van Aalten, D. M. (2004) Acta Crystallogr. Sect. D Biol. Crystallogr. 60 1355-1363 [DOI] [PubMed] [Google Scholar]
- 39.Chen, L., Zhou, C., Yang, H., and Roberts, M. F. (2000) Biochemistry 39 12415-12423 [DOI] [PubMed] [Google Scholar]
- 40.Hayward, S., and Lee, R. A. (2002) J. Mol. Graphics Model. 21 181-183 [DOI] [PubMed] [Google Scholar]
- 41.Hu, Y., Chen, L., Ha, S., Gross, B., Falcone, B., Walker, D., Mokhtarzadeh, M., and Walker, S. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 845-849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mulichak, A. M., Losey, H. C., Lu, W., Wawrzak, Z., Walsh, C. T., and Garavito, R. M. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 9238-9243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buschiazzo, A., Ugalde, J. E., Guerin, M. E., Shepard, W., Ugalde, R. A., and Alzari, P. M. (2004) EMBO J. 23 3196-3205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gibson, R. P., Tarling, C. A., Roberts, S., Withers, S. G., and Davies, G. J. (2004) J. Biol. Chem. 279 1950-1955 [DOI] [PubMed] [Google Scholar]
- 45.Kim, S. C., Singh, A. N., and Raushel, F. M. (1988) Arch. Biochem. Biophys. 267 54-59 [DOI] [PubMed] [Google Scholar]
- 46.Kim, S. C., Singh, A. N., and Raushel, F. M. (1988) J. Biol. Chem. 263 10151-10154 [PubMed] [Google Scholar]
- 47.Murray, B. W., Wittmann, V., Burkart, M. D., Hung, S. C., and Wong, C. H. (1997) Biochemistry 36 823-831 [DOI] [PubMed] [Google Scholar]
- 48.Tanaka, Y., Tao, W., Blanchard, J. S., and Hehre, E. J. (1994) J. Biol. Chem. 269 32306-32312 [PubMed] [Google Scholar]
- 49.Vocadlo, D. J., Wicki, J., Rupitz, K., and Withers, S. G. (2002) Biochemistry 41 9727-9735 [DOI] [PubMed] [Google Scholar]
- 50.Lairson, L. L., and Withers, S. G. (2004) Chem. Comm. (Camb.) 21 2243-2248 [DOI] [PubMed] [Google Scholar]
- 51.Vocadlo, D. J., Davies, G. J., Laine, R., and Withers, S. G. (2001) Nature 412 835-838 [DOI] [PubMed] [Google Scholar]
- 52.Gibson, R. P., Turkenburg, J. P., Charnock, S. J., Lloyd, R., and Davies, G. J. (2002) Chem. Biol. 9 1337-1346 [DOI] [PubMed] [Google Scholar]
- 53.Persson, K., Ly, H. D., Dieckelmann, M., Wakarchuk, W. W., Withers, S. G., and Strynadka, N. C. (2001) Nat. Struct. Biol. 8 166-175 [DOI] [PubMed] [Google Scholar]
- 54.Sinnot, M. L., and Jencks, W. P. (1980) J. Am. Chem. Soc. 102 2026-2032 [Google Scholar]
- 55.Lairson, L. L., Chiu, C. P., Ly, H. D., He, S., Wakarchuk, W. W., Strynadka, N. C., and Withers, S. G. (2004) J. Biol. Chem. 279 28339-28344 [DOI] [PubMed] [Google Scholar]
- 56.Monegal, A., and Planas, A. (2006) J. Am. Chem. Soc. 128 16030-16031 [DOI] [PubMed] [Google Scholar]
- 57.Goedl, C., and Nidetzky, B. (2008) FEBS J. 275 903-913 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.