

Abstract
The question of whether distinct self-propagating structures could be formed within the same amino acid sequence in the absence of external cofactors or templates has important implications for a number of issues, including the origin of prion strains and the engineering of smart, self-assembling peptide-based biomaterials. In the current study, we showed that chemically identical prion protein can give rise to conformationally distinct, self-propagating amyloid structures in the absence of cellular cofactors, post-translational modification, or PrPSc-specified templates. Even more surprising, two self-replicating states were produced under identical solvent conditions, but under different shaking modes. Individual prion conformations were inherited by daughter fibrils in seeding experiments conducted under alternative shaking modes, illustrating the high fidelity of fibrillation reactions. Our study showed that the ability to acquire conformationally different self-propagating structures is an intrinsic ability of protein fibrillation and strongly supports the hypothesis that conformational variation in self-propagating protein states underlies prion strain diversity.
The existence of multiple prion strains is considered to be one of the most puzzling features of prion biology. According to the protein-only hypothesis of prion propagation, the normal cellular isoform of the prion protein, PrPC, can be converted into a range of self-propagating disease-related structures, referred to as strains of PrPSc (1). Although certain physical properties are common for all PrPSc strains, each strain also possesses unique strain-specific conformational features. Within the protein-only hypothesis, it is assumed that variation in PrPSc structure can give rise to a broad range of strain-specific disease phenotypes, including substantial variation in incubation time to disease, in pathology, and in behavioral symptoms.
In the past several years considerable evidence has accumulated indicating that prion strains do, indeed, exhibit notable differences in the amount and type of β-sheet structure, conformational stability, proteolytic resistance, and surface-exposed epitopes (2–7). Although the results of these aforementioned studies were consistent with the protein-only hypothesis, the primary origin of strain-specific conformational variations remains unclear. What is the possible source of strain-specific structural variation in PrPSc? A “unified theory” of prion propagation postulates that the strain-specific features of the prion infectious agent may be encoded by a coprion, or small nucleic acid (8). Alternatively, the ability to adopt multiple self-propagating structures may be an intrinsic property of the fibrillation reaction itself and can be achieved in the absence of a coprion. In the past, two distinct amyloid states were produced from chemically identical Aβ peptides (9); however, this property has never been demonstrated for fibrillation of larger polypeptides or global proteins such as PrP.
In the current study, we showed that two fibrillar structures could be generated from the same pool of highly purified full-length recombinant PrP under identical solvent conditions but different shaking modes. The unique conformational properties of the two amyloid “strains” were stably replicated by the daughter fibrils in seeding experiments conducted under alternative shaking conditions. Our study provides a proof of principal that the PrP molecule has an intrinsic propensity to form conformationally distinct self-propagating structures in the absence of cellular cofactors, the influence of glycosylation, internal mutations, or a PrPSc-specified template.
EXPERIMENTAL PROCEDURES
Expression and Purification of PrP—Hamster full-length recombinant PrP encompassing residues 23–231 was expressed and purified as described earlier (10, 11) with the following modifications. For the last step of purification conducted on a 25-mm × 25-cm C4 HPLC column, the percentage of HPLC buffer B (0.1% trifluoroacetic acid in acetonitrile) was increased from 0 to 25% during the first 15 min (at flow rate 5 ml/min). Then, a gentle gradient was applied (from 25 to 35% of buffer B in the next 65 min) to ensure efficient separation of PrP adducts. From there, the percentage of buffer B was increased from 35 to 100% in 15 min; the column was washed with 100% of buffer B for 15 min, and the gradient dropped down to 0% within 10 min. The correctly folded, α-helical form of PrP was eluted as a major peak between 52.5–54.5 min. Minor amounts of truncated PrP polypeptides (molecular mass ∼21–22 kDa, less than 1–2% of total PrP) were eluted in the tail of the major peak (elution time >54.5 min). The fractions corresponding to the peak's tail were not used for the fibrillation reaction. A shoulder to the major peak containing PrP with oxidized methionines and eluted at <52.5 min was also discharged. Fractions eluted at 52.5–54.5 min were combined, lyophilized, and stored at -20 °C for no longer than 2 weeks prior to use. The purity of the final PrP was estimated by SDS-PAGE and electrospray mass spectroscopy to be >99%; the α-helical conformation was confirmed by CD (Fig. 1).
FIGURE 1.

a, the purity of PrP was estimated by electrophoresis on precast 12% SDS-PAGE followed by staining with Coomassie Blue. b, CD spectrum of PrP (0.25 mg/ml) confirmed native α-helical conformation. c, electrospray ionization mass spectrometry spectrum of PrP collected on a Waters ZMD single quadrupole mass spectrometer operated in positive ion mode. The spectrum shows a single series of peaks corresponding to multiply charged ions confirming high purity of PrP. The labels on the peaks (A21, A22, etc.) indicate the mass/protonation (charge) ratio of the ion species. Deconvolution of the electrospray ionization mass spectrometry spectrum results in a single peak with a maximum of 23102.35 Da (inset), which is in good agreement with the predicted molecular mass of 23102.42 Da.
Formation of S- and R-fibrils—To form amyloid fibrils in non-seeded or seeded reactions, stock solutions of PrP were prepared immediately before use by resuspending lyophilized PrP powder in 5 mm HEPES, pH 7.0. Stock solutions of PrP were diluted with MES,2 pH 6.0, and GdnHCl to final concentrations of 50 mm and 2 m, respectively, and to a final protein concentration of 0.25 mg/ml. The protein concentration was determined by measuring the absorbance at 280 nm. The fibrillation reactions were carried out in 1.5-ml conical plastic tubes (Fisher) at a total reaction volume of 0.6 ml at 37 °C with continuous shaking at 600 rpm using a Delfia plate shaker (Wallac) or continuous rotation at 24 rpm using a Clay Adams Nutator (model 1105). The same stock solution of PrP was used for the reactions carried out under both shaking and rotation modes.
To be used as seeds, the R- or S-fibrils formed in previous experiments were stored in 2 m GdnHCl at 4 °C for no longer than 2 weeks. Prior to seeding, the fibrils were sonicated for 10 s in a volume of 20 μl using a Brasonic-2510 bath sonicator (Branson Ultrasonic). For seeded reactions, the fibrils were added to fresh reaction mixtures after mixing of all reagents.
SDS-PAGE in Denaturing and Non-denaturing Conditions— To estimate the yield of amyloid formation, aliquots were taken at the end points of the polymerization reactions and treated with two sample buffers, denaturing (final concentrations 60 mm Tris, 2% SDS, and 1.25% β-mercaptoethanol, 2.25 m urea, heating for 15 min at 90 °C) and non-denaturing (no SDS, β-mercaptoethanol or urea, no heating). 12% SDS-PAGE (precast NuPAGE gels; Invitrogen) was used for analysis of samples treated with both denaturing and non-denaturing sample buffers.
Immunostaining and Fluorescence Microscopy—PrP fibrils (2 μg/ml) were deposited onto Permanox 8-well Lab-Teks chamber slides and stained with antibody as previously described (12) with minor modifications. Formaldehyde fixation was omitted, and the staining was performed in the following order: 1) anti-PrP human Ab R1 (1:500, recognizes epitope 225–231); 2) mouse Ab AG4 (1:1000, recognizes N-terminal epitope 37–50); 3) the mixture of secondary antibodies, goat anti-human and goat anti-mouse labeled with Alexa-488 and Alexa-546, respectively (Invitrogen/Molecular Probes, 1:1000 for both antibodies). Fluorescence microscopy was carried out on an inverted microscope (Nikon Eclipse TE2000-U) using 1.3 aperture Plan Fluor ×100 numerical aperture objective. Collected images were processed with WCIF ImageJ software (National Institutes of Health) as described previously (12). Atomic force microscopy (AFM), negative staining electron microscopy (EM), and Fourier transform infrared spectroscopy (FTIR) were performed as described in our former publications (13, 14).
CD Spectroscopy—CD spectra were recorded in 1 mm ammonium acetate buffer, pH 5.5, in a 0.1-cm cuvette with a Jasco-810 CD spectrometer (Jasco, Easton, MD) at a protein concentration of 0.25 mg/ml and scanning rate of 20 nm/min; the background spectra were subtracted.
Electrospray Ionization Mass Spectrometry—Lyophilized PrP samples were first dissolved in water and then diluted to 0.1 μg/μl in 1:1 water:methanol containing 1% acetic acid. The solution was injected directly at 10 μl/min into a Waters ZMD single quadrupole mass spectrometer operated in positive ion mode. The instrument acquired full scan mass spectra (m/z 500–2000) every second for 2 min.
RESULTS
PrP Forms Structurally Different Fibrils under Different Shaking Modes—In the past, we have pursued multiple attempts to generate different amyloid strains within the same PrP primary sequence by (i) manipulating solvent conditions such as pH, temperature, concentrations of denaturants, salt, co-solvents, or agitation or (ii) in reactions cross-seeded with PrP variants (data not shown). However, regardless of the experimental approach, fibrils formed under different conditions displayed relatively similar physical properties. Following these attempts, we modified the protocol for PrP purification and improved the purity of PrP. When highly purified PrP (the purity was estimated to be >99%, Fig. 1) was used for fibrillation reactions, we noticed that the shape of fibrils was substantially affected by the mode of agitation used for the fibrillation reaction. As judged by EM and AFM, the fibrils formed under slow rotation at 24 rpm displayed a straight or rigid shape (this type of fibril will be referred to as R-fibrils, where R stands for Rigid or Rotation), whereas the fibrils produced under fast shaking at 600 rpm showed a curvy S-like shape (this type will be referred to as S-fibrils, where S stands for S-like fibrillar shape or for Shaking) (Fig. 2, a–c). These experiments were repeated multiple times in the past 2 years using PrP from different purifications. Noteworthy, in each experiment the S- and R-fibrils were formed from the same freshly prepared PrP stock solution, indicating that the amyloidogenic pathways depended on the agitation conditions, but not on pre-existing nuclei. To confirm that PrP stock did not contain preformed S- or R-specific nuclei, in additional experiments the stock solution was filtered through 0.22-μm filters or incubated in 6 m GdnHCl for 24 h prior to fibrillation. Filtration or incubation of stock solution in GdnHCl did not change the result of the fibrillation reactions, confirming that the two amyloidogenic pathways depend solely on the agitation conditions.
FIGURE 2.
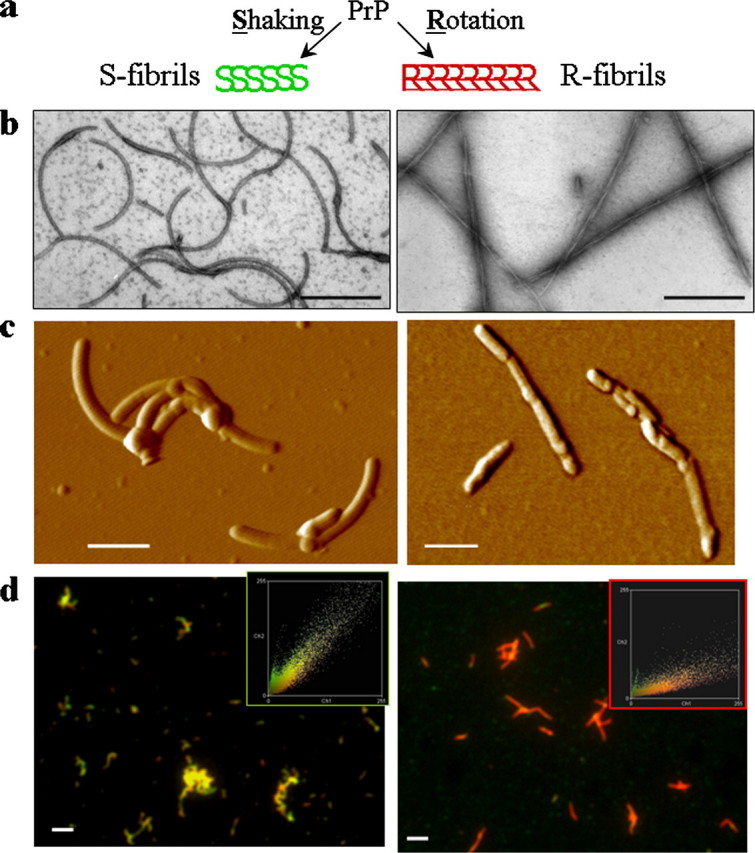
a, S- and R-fibrils were produced from the same stock of PrP under identical solvent conditions but different agitation modes (as described under “Experimental Procedures”). Negative staining EM images (b), AFM images (c), and double staining immunofluorescence microscopy images (d) of S-fibrils (left panels) and R-fibrils (right panels) are shown. The microscopy images were transformed into two-dimensional fluorescence intensity scattering plots (insets) as previously described (12). Red fluorescence intensities are plotted on the horizontal axis, and the green intensities are plotted on the vertical axis. Scale bars, 0.2 μmin b and c and 2 μm in d. The apparent discrepancy in thickness of fibrils in AFM versus EM images is due to the fact that the negative staining EM imaging reports the pattern of positive charges on the fibrillar surface and underestimates the real thickness, whereas the AFM imaging overestimates the thickness due to the convolution effect.
Both fibrillation reactions showed typical nucleation-elongation kinetics (data not shown), and both types of fibrils displayed a high Thioflavine T reading as judged by thioflavine fluorescence assay and thioflavine fluorescence microscopy (Fig. 3, a and b), suggesting that mature amyloid structures were formed under both shaking modes. Analysis of the fibrillation yield by non-denaturing PAGE revealed that at the final stage of polymerization the soluble PrP was consumed equally well in the reactions carried out in both agitation modes (Fig. 3c).
FIGURE 3.

a, fluorescence microscopy imaging of S-fibrils (left panel) and R-fibrils (right panel) stained with Thioflavin T. Thioflavin T staining and microscopy imaging were performed as described earlier (10). b, thioflavin T (ThT) fluorescence spectra of S-fibrils (dotted line), R-fibrils (dashed line), and α-PrP (solid line) recorded at a protein concentration equivalent to 0.3 μm and thioflavin concentration of 10 μm. The higher fluorescence intensity observed for S-fibrils was, presumably, due to larger surface area in S-fibrils than in R-fibrils and/or differences in conformation between S- and R-fibrils. c, PAGE analysis of fibrillation yield. S- and R-fibrils were treated with denaturing sample buffer (SDS + urea, lanes 4 and 5) or non-denaturing sample buffer (no SDS or urea, lanes 1 and 2) and analyzed in SDS-PAGE. When treated with non-denaturing sample buffer, only non-fibrillar PrP enters PAGE. For comparison, lane 3 was loaded with an equivalent amount of monomeric α-PrP.
Detail analysis of fibril morphology by EM revealed polymorphism within both fibrillar classes (R and S). Although all fibrils of the R-type were twisted, the number of constitutive filaments within individual fibrils and the twisting modes varied (Fig. 4b). For example, for a subclass of R-fibrils apparently consisting of two twisting filaments, the periodicity of twisting varied from 80 to 160 nm (Fig. 4b, panels 1, 2). None of the S-fibrils exhibited periodical twisting. The S-fibrils, however, showed variations with respect to their width (Fig. 4a) and height (supplemental Fig. S1). It is likely that the polymorphism within fibrils of R- or S-types is attributed to the variable number of constitutive protofilaments and variable modes of lateral association of protofilaments within individual fibrils (13, 15). Regardless of the polymorphism observed within each individual class, S- and R-fibrils displayed remarkable differences with respect to their substructures, as described below.
FIGURE 4.

Electron microscopy images of S-fibrils (a) and R-fibrils (b). Scale bars, 0.1 μm.
To assess the extent to which R- and S-fibrils are structurally different, we employed FTIR and a double immunostaining fluorescence microscopy assay (12). As judged from FTIR, the R-fibrils showed strong peaks at 1628 and 1613 cm-1 (both are characteristics of intermolecular β-sheet structures) and a peak at 1662 cm-1 that can be largely assigned to loop components with possible contributions from β-turns and α-helices (Fig. 5b). The β-sheet structure in S-fibrils was characterized by a single peak at 1626 cm-1 and a minor shoulder at 1613 cm-1 (Fig. 5a). The S-fibrils showed only a very minor peak at 1664 cm-1.
FIGURE 5.
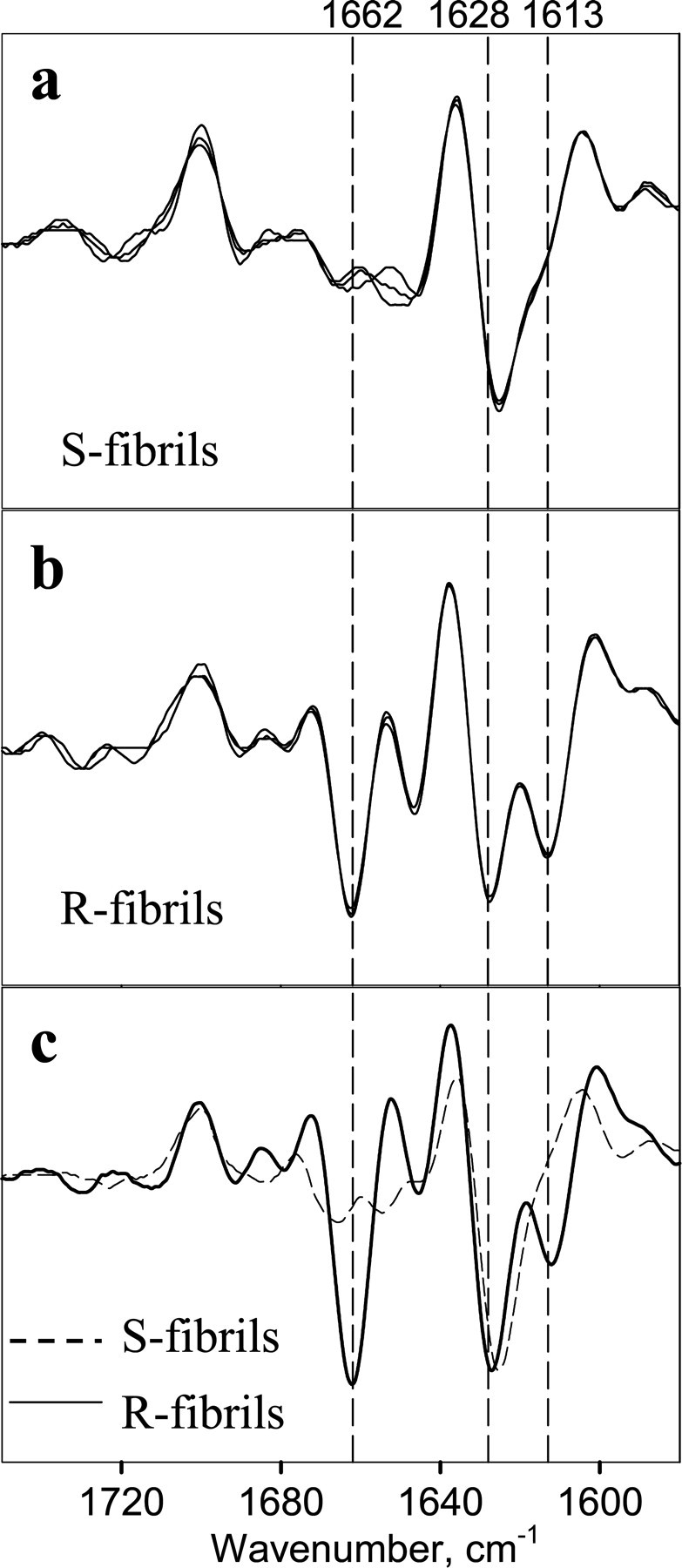
Second derivatives of the FTIR spectra obtained for S-fibrils (a), R-fibrils (b), and daughter fibrils (c) produced in the following seeded reactions: the reaction was seeded with R-fibrils (3%) and carried out under shaking (solid line) or the reaction was seeded with S-fibrils (3%) and carried out under rotation (dashed line). a and b show three independent preparations of S- and R-fibrils, respectively.
The immunostaining microscopy assay probes conformation within individual fibrils as well as across a whole fibrillar population. The assay consisted of immunostaining with a pair of PrP-specific antibodies (Ab), R1 (specific to the C-terminal epitope 225–231) and AG4 (recognizes the N-terminal region). AG4 was used as a reference Ab, because the N-terminal region was solvent-exposed in both types of fibrils regardless of their substructure. The secondary Ab to Ab AG4 was labeled with ALEXA-546 (red) and the secondary Ab to Ab R1 with ALEXA-488 (green); therefore, the color of fibrils in double staining reflected the extent to which the epitope to R1 was solvent-accessible or buried. Consistent with our earlier observations (12), the R1 epitope was found to be hidden in the R-fibrils (Fig. 2d). The previous studies showed that pretreatment of R-fibrils with 4–6 m of GdnHCl was required for the R1 epitope to become immunoreactive (12, 14). The same epitope, however, was found to be immunoreactive in the S-fibrils under native conditions (Fig. 2d).
Taken together, the data acquired by EM, AFM, FTIR, and immunofluorescence assays suggest that the S- and R-fibrils are conformationally different and represent two alternative self-propagating amyloid structures. These structures were formed from the same stock solutions of highly purified recombinant PrP under identical solvent conditions and protein concentrations, but using different agitation modes.
Do S- and R-fibrils Maintain Their Original Conformation under Alternative Agitation Conditions?— Next we were interested in whether the S- and R-fibrils preserve their individual structures when propagated under alternative agitation conditions. To address this question, we performed cross-seeding experiments in which preformed S- and R-fibrils were used as seeds to initiate the fibrillation reactions conducted under rotation or shaking, respectively. The conformation of daughter fibrils was assessed at the late stages of fibrillation using electron microscopy, the double staining fluorescence microscopy assay, and FTIR. We found that regardless of the agitation conditions, the daughter fibrils formed in the reactions seeded with 3% S-fibrils displayed S-like fibrillar shape (supplemental Fig. S2a) and were immunoreactive to R1 (Fig. 6a). Similarly, the daughter fibrils formed in the polymerization assays seeded with 3% R-fibrils inherited the fibrillar morphology typical for the R-fibrils (supplemental Fig. S2b) and showed negative immunoreactivity to R1 regardless of the agitation mode (Fig. 6b). FTIR spectroscopy confirmed that R- or S-specific structural properties were transferred to the daughter fibrils in the seeded reactions carried out under unfavorable agitation conditions (Fig. 5c). These experiments illustrated that the daughter fibrils inherited the conformational features of the original seeds even under agitation conditions that favored alternative amyloid structures.
FIGURE 6.
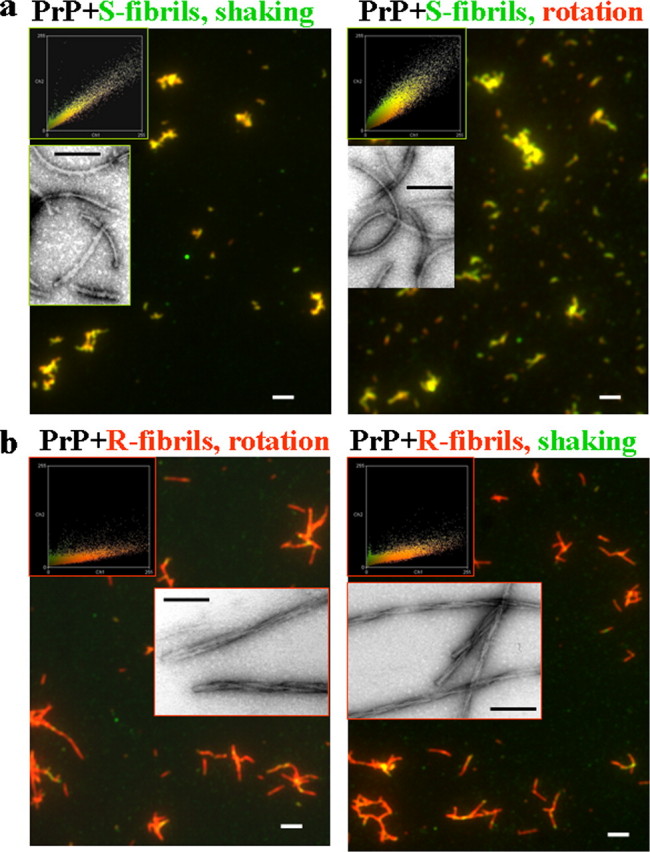
Double staining immunofluorescence microscopy images and EM images (insets) of daughter fibrils produced in seeded reactions. a, the fibrillation reactions were seeded with S-fibrils (3%) and carried out under shaking (left panel) or rotation (right panel). b, the fibrillation reactions were seeded with R-fibrils (3%) and carried out under rotation (left panel) or shaking (right panel). The microscopy images were transformed into two-dimensional fluorescence intensity scattering plots (insets) as previously described (12). Red fluorescence intensities are plotted on the horizontal axis, and the green intensities are plotted on the vertical axis. Scale bars, 2 μm (0.1 μmin insets).
DISCUSSION
The current studies illustrate that two conformationally distinct self-propagating structures, referred to as S- and R-fibrils, could be formed within the same PrP primary sequence in the absence of cellular cofactors, post-translational glycosylation, or a PrPSc-specified template. Although certain levels of polymorphism were observed within each fibrillar type, S- and R-fibrils were found to exhibit substantial differences with respect to immunoreactivity of the R1 epitope, their secondary structure, and fibril morphology. We would like to emphasize that a high quality of purified PrP was essential for producing the two amyloid strains.
Recent studies by Surewicz and co-workers (16, 17) demonstrated that conformationally distinct fibrils could be produced in vitro from PrP-derived peptides encompassing residues 23–144. In those studies, new amyloid strains were formed as a result of cross-seeding with fibrils prepared from PrP variants carrying species-specific amino acid substitutions. Therefore, the conformational differences in daughter fibrils arose from the substitution of amino acid residues in the parent fibrils. The experiments on fibrillation of PrP-derived peptides were inspired by the classical experiments on transmission of prions between mammalian species. In these experiments, new prion strains emerged when PrPSc derived from a host species was propagated in another, intermediate species and then returned to the original species. For instance, in classical studies by Kimberlin and co-workers (18, 19), a mouse strain gave rise to a new strain after being passaged in hamster and then returned to mice. The physical properties of the newly emerged strain appeared to be primarily determined by the structure of the original PrPSc strain that provided a template and the history of passages through different species (20).
Although passage of existing strains through different species was shown to result in emergence of new strains, it is not clear how new strains could originate in the absence of a PrPSc-specified template and whether new strains can arise in sporadic diseases. A “unified theory” of prion propagation postulates that although the transmissible spongiform encephalopathy infectious agent consists of the prion protein, the strain-specific properties are defined by a coprion, a nucleic acid, that can be recruited in the host cell and associated with PrPSc (8). Consistent with the coprion theory, nucleic acids are found capable of binding PrP and facilitating the cell-free conversion of recombinant α-PrP into β-sheet-rich isoforms (21) or conversion of PrPC into PrPSc (22). In recent studies, polyanions including poly(A) RNAs were shown to be essential for converting PrPC into infectious conformations in the absence of PrPSc (23). Although several lines of evidence strongly support the potential involvement of nucleic acids in prion replication, the critical role of nucleic acids in regulating strain-specific diversity has yet to be documented (24).
Although several possible mechanisms might account for strain diversity, the present studies illustrate that conformationally distinct self-propagating structures could be formed within the same PrP primary sequence in the absence of cellular cofactors such as nucleic acids. Our previous work revealed that recombinant mouse PrP encompassing residues 89–230, converted in vitro into the fibrillar form in the absence of a PrPSc template or nucleic acid (25), gave rise to a new prion strain referred to as mouse synthetic prion 1 (MoSP1) (26, 27). MoSP1 is distinguished from natural prion strains by its peculiar incubation time and neuropathological profile (26, 27). The unique biological properties of the MoSP1 were linked to the peculiar physical features of the fibrils generated in vitro from PrP89–230 and, specifically, to its high conformational stability (28–30). Just like the R-fibrils described here, the fibrils produced from mouse PrP89–230 had a hidden R1 epitope (residues 225–230). The mouse PrP89–230 fibrils cause transmissible prion disease in mice only after prolonged incubation time and, therefore, can be regarded as very inefficient in recruiting PrPC in vivo. Such low efficiency could be, at least in part, due to the conformational constraints created by the glycosylphosphatidylinositol anchor, which is attached to residue 230 in mouse PrPC and which cannot be easily accommodated within R-fibrils (14). This conformational constraint does not exist for S-fibrils, where the R1 epitope was found to be immunoreactive in a manner similar to natural PrPSc strains (31).
What is the possible mechanism for acquiring different amyloid conformations in the absence of seeding or cellular cofactors? In the past, two structurally different fibrillar forms were produced from Aβ1–40 peptide under fixed solvent conditions by different agitation modes, a work illustrating that this property is not unique to PrP but could be generic for amyloid fibrillation (9). Alternative nucleation pathways were suggested as a plausible mechanism for generating structurally different amyloid forms. Another study showed that α-synuclein forms two conformationally different types of fibrils as a mixture under single growth conditions (32). The differences in molecular structure of α-synuclein fibrils detected by solid state NMR were found to correlate with distinct fibril morphologies. In vitro, the nucleation pathway might depend on the interface between the solution and air or the solution and the wall of the reaction vial (33), whereas interactions of a polypeptide with the surface of biological membranes might regulate the nucleation step in a cell. Using insulin as a model amyloidogenic protein, Winter and co-workers (34) showed that subtle differences in the initial hydration state of a protein at the early stages of fibrillation impose profound consequences for its self-assembling pattern and the physical properties of mature amyloid structures. Our result is consistent with the hypothesis that different nucleation routes lead to well separated energetic minima that correspond to different amyloid strains. In contrast to amyloid fibrils, the structure of a native state does not seem to depend on specific routes of folding. Folding of globular proteins often proceeds via multiple kinetic routes that result in the same native conformation.
The results of cross-seeding experiments illustrate that the in vitro fibrillation reaction exhibits high fidelity and that S- and R-fibrils behave in a strain-like manner. Once mature S- or R-fibrils were formed, S- or R-specific properties were transferred in a template-dependent fashion to daughter fibrils despite unfavorable shaking modes (Figs. 5 and 6). Unless individual conformational properties are inherited in subsequent passages, morphologically different fibrils cannot be referred to as amyloid strains. It is important to acknowledge that the in vitro fibrillation reactions do not always display high fidelity of replication. Strong and weak strains of yeast prion protein Sup35 were found to exhibit substantial differences in their conversion efficiency in a cell. However, when replication of strong and weak [PSI+] strains ([PSI+] strains are the prion states of the Sup35 protein) was conducted in vitro, the differences in replication efficiency were lost in a second round of replication (35). Similarly, not all types of amyloid fibrils produced in vitro from PrP display equally high fidelity in cross-seeding reactions.3 Therefore, the templating activity of amyloid fibrils does not always accompany their catalytic activity, i.e. their ability to convert monomeric substrate into a fibrillar state. The molecular origin of high fidelity in self-replicating processes needs to be addressed in future studies. It remains to be established whether the high fidelity of fibrillation reactions depends strictly on peculiar structural elements present in some, but not in all, amyloid states. Answering this question will help to dissect the molecular mechanisms underlying the high fidelity of replication of PrPSc strains.
Supplementary Material
Acknowledgments
We thank Chen-I Lee for performing AFM imaging and Pamela Wright for editing the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant NSO45585 (to I. V. B.). This work was also supported by the Program in Prion Diseases at the Medical Biotechnology Center, UMBI. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement”in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
Footnotes
The abbreviations used are: MES, 4-morpholineethanesulfonic acid; PrPC, normal cellular isoform of the prion protein; PrPSc, abnormal, disease-associated isoform of the prion protein; PrP, recombinant PrP; EM, electron microscopy; AFM, atomic force fluorescence microscopy; Ab, antibody; GdnHCl, guanidine hydrochloride; FTIR, Fourier transform infrared spectroscopy.
L. Breydo and I. V. Baskakov, unpublished observation.
References
- 1.Prusiner, S. B. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 13363-13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Caughey, B., Raymond, G. J., and Bessen, R. A. (1998) J. Biol. Chem. 273 32230-32235 [DOI] [PubMed] [Google Scholar]
- 3.Safar, J., Wille, H., Itri, V., Groth, D., Serban, H., Torchia, M., Cohen, F. E., and Prusiner, S. B. (1998) Nat. Med. 4 1157-1165 [DOI] [PubMed] [Google Scholar]
- 4.Thomzig, A., Spassov, S., Friedrich, M., Naumann, D., and Beekes, M. (2004) J. Biol. Chem. 279 33847-33854 [DOI] [PubMed] [Google Scholar]
- 5.Peretz, D., Scott, M., Groth, D., Williamson, A., Burton, D., Cohen, F. E., and Prusiner, S. B. (2001) Protein Sci. 10 854-863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bessen, R. A., and Marsh, R. F. (1994) J. Virol. 68 7859-7868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Telling, G. C., Parchi, P., DeArmond, S. J., Cortelli, P., Montagna, P., Gabizon, R., Mastrianni, J., Lugaresi, E., Gambetti, P., and Prusiner, S. B. (1996) Science 274 2079-2082 [DOI] [PubMed] [Google Scholar]
- 8.Weissmann, C. (1991) Nature 352 679-683 [DOI] [PubMed] [Google Scholar]
- 9.Petkova, A. T., Leapman, R. D., Gua, Z., Yau, W.-M., Mattson, M. P., and Tycko, R. (2005) Science 307 262-265 [DOI] [PubMed] [Google Scholar]
- 10.Bocharova, O. V., Breydo, L., Parfenov, A. S., Salnikov, V. V., and Baskakov, I. V. (2005) J. Mol. Biol. 346 645-659 [DOI] [PubMed] [Google Scholar]
- 11.Breydo, L., Bocharova, O. V., Makarava, N., Salnikov, V. V., Anderson, M., and Baskakov, I. V. (2005) Biochemistry 44 15534-15543 [DOI] [PubMed] [Google Scholar]
- 12.Novitskaya, V., Makarava, N., Bellon, A., Bocharova, O. V., Bronstein, I. B., Williamson, R. A., and Baskakov, I. V. (2006) J. Biol. Chem. 281 15536-15545 [DOI] [PubMed] [Google Scholar]
- 13.Anderson, M., Bocharova, O. V., Makarava, N., Breydo, L., Salnikov, V. V., and Baskakov, I. V. (2006) J. Mol. Biol. 358 580-596 [DOI] [PubMed] [Google Scholar]
- 14.Breydo, L., Sun, Y., Makarava, N., Lee, C.-I., Novitskaia, V., Bocharova, O. V., Kao, J. P. Y., and Baskakov, I. V. (2007) Biochemistry 46 852-861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Makarava, N., Bocharova, O. V., Salnikov, V. V., Breydo, L., Anderson, M., and Baskakov, I. V. (2006) Protein Science 15 1334-1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vanik, D. L., Surewicz, K. A., and Surewicz, W. K. (2004) Mol. Cell 14 139-145 [DOI] [PubMed] [Google Scholar]
- 17.Jones, E. M., and Surewicz, W. K. (2005) Cell 121 63-72 [DOI] [PubMed] [Google Scholar]
- 18.Kimberlin, R. H., Cole, S., and Walker, C. A. (1987) J. Gen. Virol. 68 1875-1881 [DOI] [PubMed] [Google Scholar]
- 19.Kimberlin, R. H., Walker, C. A., and Fraser, H. (1989) J. Gen. Virol. 70 2017-2025 [DOI] [PubMed] [Google Scholar]
- 20.Peretz, D., Williamson, R. A., Legname, G., Matsunaga, Y., Vergara, J., Burton, D., DeArmond, S., Prusiner, S., and Scott, M. R. (2002) Neuron 34 921-932 [DOI] [PubMed] [Google Scholar]
- 21.Cordeiro, Y., Machado, F., Juliano, L., Juliano, M. A., Brentani, R. R., Foguel, D., and Silva, J. L. (2001) J. Biol. Chem. 276 49400-49409 [DOI] [PubMed] [Google Scholar]
- 22.Deleault, N. R., Lucassen, R. W., and Supattapone, S. (2003) Nature 425 717-720 [DOI] [PubMed] [Google Scholar]
- 23.Deleault, N. R., Harris, B. T., Rees, J. R., and Supattapone, S. (2007) Proc. Acad. Natl. Sci. U. S. A. 104 9741-9746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Safar, J. G., Kellings, K., Serban, A., Groth, D., Cleaver, J. E., Prusiner, S. B., and Riesner, D. (2005) J. Virol. 79 10796-10806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baskakov, I. V., Legname, G., Baldwin, M. A., Prusiner, S. B., and Cohen, F. E. (2002) J. Biol. Chem. 277 21140-21148 [DOI] [PubMed] [Google Scholar]
- 26.Legname, G., Baskakov, I. V., Nguyen, H.-O. B., Riesner, D., Cohen, F. E., DeArmond, S. J., and Prusiner, S. B. (2004) Science 305 673-676 [DOI] [PubMed] [Google Scholar]
- 27.Legname, G., Nguyen, H.-O. B., Baskakov, I. V., Cohen, F. E., DeArmond, S. J., and Prusiner, S. B. (2005) Proc. Natl. Aca. Sci. U. S. A. 102 2168-2173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bocharova, O. V., Breydo, L., Salnikov, V. V., Gill, A. C., and Baskakov, I. V. (2005) Protein Sci. 14 1222-1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baskakov, I. V., and Breydo, L. (2007) Biochim. Biophys. Acta 1772 692-703 [DOI] [PubMed] [Google Scholar]
- 30.Sun, Y., Breydo, L., Makarava, N., Yang, Q., Bocharova, O. V., and Baskakov, I. V. (2007) J. Biol. Chem. 282 9090-9097 [DOI] [PubMed] [Google Scholar]
- 31.Peretz, D., Williamson, R. A., Matsunaga, Y., Serban, H., Pinilla, C., Bastidas, R. B., Rozenshteyn, R., James, T. L., Houghten, R. A., Cohen, F. E., Prusiner, S. B., and Burton, D. R. (1997) J. Mol. Biol. 273 614-622 [DOI] [PubMed] [Google Scholar]
- 32.Heise, H., Hoyer, W., Becker, S., Andronesi, O. C., Reidel, D., and Baldus, M. (2005) Proc. Acad. Natl. Sci. U. S. A. 102 15871-15876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baskakov, I. V., Legname, G., Gryczynski, Z., and Prusiner, S. B. (2004) Protein Sci. 13 586-595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dzwolak, W., Grudzielanek, S., Smirnovas, S., Ravindra, R., Nicolini, C., Jansen, R., Loksztein, A., Porowski, S., and Winter, R. (2005) Biochemistry 44 8948-8958 [DOI] [PubMed] [Google Scholar]
- 35.Uptain, S. M., Sawicki, G., Caughey, B., and Lindquist, S. (2001) EMBO J. 20 6236-6245 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.