

Abstract
Inherited neurodegenerative diseases, such as Huntington disease and subset of Alzheimer disease, Parkinson disease, and amyotrophic lateral sclerosis, are caused by the mutant genes that have gained undefined properties that harm cells in the nervous system, causing neurodegeneration and clinical phenotypes. Lowering the mutant gene expression is predicted to slow the disease progression and produce clinical benefit. Administration of small interfering RNA (siRNA) can silence specific genes. However, long term delivery of siRNA to silence the mutant genes, a requirement for treatment of these chronic central nervous system (CNS) diseases, remains a critical unsolved issue. Here we designed and tested a chemically stabilized siRNA against human Cu,Zn-superoxide dismutase (SOD1) in a mouse model for amyotrophic lateral sclerosis. We show that the modified siRNA has enhanced stability and retains siRNA activity. Administration of this siRNA at the disease onset by long term infusion into the CNS resulted in widespread distribution of this siRNA, knocked down the mutant SOD1 expression, slowed the disease progression, and extended the survival. These results bring RNA interference therapy one step closer to its clinical application for treatment of chronic, devastating, and fatal CNS disorders.
Treatment of age-dependent neurodegenerative diseases, including Huntington disease, Alzheimer disease, Parkinson disease, and ALS,5 is a serious challenge for 21st century medicine. Although all cases of Huntington disease are caused by mutations in the huntingtin (htt) gene, other diseases have both sporadic and familial cases. Regardless, highly efficacious treatment is lacking for all of these diseases. A promising therapeutic strategy for these diseases is to inhibit the expression of genes that are required in the pathogenic pathways. This concept is straightforward in cases where genetic mutations lead to a gain of toxicity for the gene product, which harm cells in the CNS. By inhibiting the expression of the toxic gene product, one may predict a reduction in the toxicity and a slower cell death, and consequently, a slower disease progression. The effectiveness of this therapeutic approach has been demonstrated by experiments in animal models using virus-delivered and transgenic RNAi (reviewed in Ref. 1).
RNAi is a conserved cellular mechanism in eukaryotes (2). Double-stranded RNA triggers RNAi, which destroys the target RNA that shares sequence homology with the double-stranded RNA. In mammalian cells, RNAi against specific mRNA can be induced using siRNAs of ∼21 nucleotides in length, which avoid nonspecific interferon response that can be triggered by long double-stranded RNAs (3, 4). The siRNA initiates RNAi by interacting with the Dicer and Argonaut 2 (AGO2) complex, which destroys one strand (the passenger strand) and incorporates the other strand (the guide strand) into the RNA-induced silencing complex. Composed of a single-stranded RNA, AGO2 and several other protein components, the RNA-induced silencing complex recognizes the target mRNA sequence by Watson-Crick base-pairing with the single-stranded RNA and cuts the target mRNA sequence, leading to gene silencing (33). Although high doses of siRNA are associated with off-target effects and nonspecific toxicity (5–8), these effects can be managed by careful design and chemical modifications of the siRNA and by controlling the doses of siRNA (9–11). Given the specificity and potency of RNAi for gene silencing, its potential for therapeutic gene silencing (TGS) has been increasingly recognized (12).
One obstacle in application of TGS for CNS disorders is delivery. Previous studies have shown that short hairpin RNA delivered by gene therapy can trigger RNAi against mutant sod1 and other disease genes and slow the disease progression in animal models (13). However, clinical application of this approach is hampered by its potential toxicity and its difficulty in stopping the therapy if adverse effects develop. In this regard, direct delivery of siRNA is advantageous because the dose can be controlled and therapy can be stopped if signs of toxicity emerge during treatment. siRNAs cross the blood-brain barrier poorly when administered peripherally (14). Although a recent study showed that siRNA conjugated to a peptide from rabies virus can enter the CNS (15), it remains unknown whether this delivery method can be used for repeated or continuous long term administration to deliver sustained TGS, which is required for treatment of chronic neurological disorders. An alternative method is to deliver siRNA directly into the CNS, which circumvents the blood-brain barrier. Several studies have shown that this delivery method is effective for short term, local treatment in acute disease models (16, 17). However, the feasibility of long term TGS against the disease-causing mutant gene for treatment of chronic CNS diseases remains unexplored.
A major challenge in delivering siRNA is to overcome the instability of siRNA (14, 18, 19), which limits the administration of siRNA to the short term and lowers the effective siRNA concentration in vivo. It has been demonstrated that chemical modifications of siRNA can improve siRNA stability and enhance target gene silencing in vivo (20–22). To apply chemically modified siRNA in sustained TGS against chronic neurological disorders, the siRNA has to meet several basic requirements. First, the siRNA should be sufficiently stable so that it can be carried by the patients for a prolonged period of time without significant degradation; second, the siRNA should be capable of diffusing throughout the CNS so that diseases that affect wide areas of the CNS can be treated; third, the siRNA should be able to cross the cell membrane and reach inside the cells; fourth, the siRNA should be able to mediate sustained gene silencing of the disease genes in vivo; fifth, the siRNA should be tolerated by the body and not trigger toxic side effects; and sixth, the siRNA should be beneficial at therapeutic doses.
In this study, we investigated these requirements using a chemically modified siRNA-targeting human sod1 gene in a transgenic mouse model of ALS. This model expresses human mutant SOD1G93A, which causes ALS by a gained toxicity to motor neurons (23, 24). By intrathecal infusion of this siRNA, we demonstrate that this siRNA is stable for a prolonged period of time in vivo, diffuses to all CNS regions, crosses the cell membrane into the cells, and silences the expression of the mutant sod1 gene. Furthermore, when infused at disease onset at the therapeutic dose for 4 weeks, this siRNA slows disease progression without detectable adverse effects. Our result demonstrates that long term CNS administration of chemically modified siRNA can treat chronic CNS disorders efficaciously.
EXPERIMENTAL PROCEDURES
siRNAs and Their Chemical Modifications—All siRNAs used in these studies were chemically synthesized using silyl ethers to protect 5′-hydroxyls and acid-labile orthoesters to protect 2′-hydroxyls with 2′-ACE (2′-bis(acetoxyethoxy)-methyl ether) (Dharmacon, Lafayette, CO). After deprotection and purification, siRNA strands were annealed as described (18). The unmodified SOD1 siRNA (U1) sequences (targeting open reading frame positions 288–308) are: sense 5′-CGAUGUGUCUAUUGAAGAUUC-3′, antisense 5′-AUCUUCAAUAGACACAUCGGC-3′. The chemically modified siRNA (R1) sequences are: sense 5′-DY547-CSGSASUFGUFGUCUAUUGAAGSASUFSUFSC-3′, antisense 5′-PAUFCFUFUCAAUAGACACFASUFSCFSGSGSC. The chemically modified R1 mismatch siRNA (Mm) sequences are: sense 5′-Dy547-CSGSAAGUFUUGGAUUCAUCAUFSUFSC-3′, antisense 5′-PASUFGAUGAAUCCAAACUUFCFGSGSC. The superscript letters F and S represent 2′-O-F and HS-backbone modifications, respectively. P is a phosphate group, and DY547 is an analog of CY3 dye molecule. RNA labeling with various functional groups, purification, and characterization were accomplished according to established methods in our laboratory (25, 26).
Plasmid Constructs—For luciferase target, pGL2-SOD1(239–314) were created by inserting the sequence of 239–314 nucleotides from the human SOD1 coding sequence into the PflMl site (2236) of the pGL2 control vector (Promega Corp., Madison WI) at the 3′-untranslated region of the firefly luciferase gene. The construct was verified by sequencing. The pRL-TK vector (Promega) that expresses Renilla luciferase was used as internal transfection control.
Cell Culture and Transfection—HEK293 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 100 unit/ml penicillin, and 100 μg/ml streptomycin. One day before transfection, cells were detached by trituration and plated at 70–90% confluency in a 96-well plate in medium without antibiotics. For the short term silencing assay, various amounts of siRNAs plus 0.2 μg of pGL2-SOD1 and 0.01 μg of pRL-TK were transfected in each well using Lipofectamine 2000™ (Invitrogen) according to the manufacturer's instructions. Twenty-four hours after the transfection, the cells were assayed for luciferase activity using the Dual-Luciferase reporter assay system (Promega). For a long term silencing assay, cells were transfected with 20 nm siRNAs. Four hours after transfection, the cells were transferred to a 6-well plate and allowed to grow. On the fifth day, cells were detached and plated at 70–90% confluency in a 96-well plate with antibiotic-free medium. On the sixth day, the cells were transfected with the reporter plasmids. On the seventh day, the cells were assayed for the luciferase activity. For fluorescent imaging, cells were plated in a 24-well plate at 50% confluency. On the following day, cells were transfected with 100 nm R1 siRNA and Cy3 dye. At various times after transfection, photos were taken using the same exposure settings. Cells were passed as needed to maintain 80–90% confluency on the day when the photo was taken.
Northern Blot and Western Blot—Mice under deep anesthesia were transcardially perfused with ice-cold PBS to wash out blood. Spinal cord, brain, and other tissues were quickly harvested, snap-frozen in liquid N2, and stored at -80 °C. For total RNA extraction, frozen tissues were homogenized in cold TRIzol reagent (Invitrogen) following the manufacturer's protocol. After the RNA extraction, the organic phase was cleared of DNA by ethanol precipitation. The supernatant was stored at -20 °C. For protein extraction, the supernatant was thawed and dialyzed for 25 h following the protocol described by Hummon et al. (27). Proteins in 200 μl of buffer containing 4 m urea and 0.5% SDS were incubated at 95 °C for 5 min and vortexed. After centrifugation at 16,000 × g for 1 min, proteins were diluted and quantified using the BCA™ protein assay (Pierce). For Northern blot detecting SOD1 mRNA, 3 μg of total RNA was electrophoresed on a 1% agarose denaturing gel in MOPS buffer, transferred to a nylon membrane (Roche Applied Science), and probed with a digoxigenin-labeled probe synthesized using a PCR digoxigenin probe synthesis kit (Roche Applied Science). After probing for SOD1, the blot was stripped and reprobed against β-actin coding region. For Northern blot detecting siRNA, 10 μg of total RNA/sample was separated by electrophoresis using a 14% polyacrylamide gel and then blotted on Hybond-XL membrane (Amersham Biosciences). The antisense strand of siRNA duplex was probed by DNA probe with the sequence of the sense strand (5′-GCCGATGTGTCTATTGAAGAT-3′). Hybridization was performed in 0.5 m phosphate buffer, pH 7.2, containing 1 mm EDTA and 7% SDS, at 42 °C overnight. For a Western blot to detect SOD1 protein, 15 μg of total protein was separated on a 15% SDS-polyacrylamide gel (Bio-Rad) and wet-transferred to a Protran® (Whatman GmbH) nitrocellulose transfer membrane. The membrane was probed with sheep anti-SOD1 (Biodesign) primary antibody and rabbit anti-sheep IgG (Calbiochem) secondary antibody. After detecting SOD1, the blot was striped and reprobed with anti-actin primary antibody JLA20 (Developmental Studies Hybridoma Bank) and goat anti-Mouse IgM (Bethyl) secondary antibody. The protein bands were visualized using SuperSignal West Pico kit (Pierce). Primary antibodies for 2′,5′-oligoadenylate synthetase 1 (OAS1) and signal transducer and activator of transcription 1 (STAT1) were purchased from Abgent and Cell Signaling Technology, respectively.
Immunofluorescent Staining and Microscopy—Mice were transcardially perfused with ice-cold PBS followed by the cold fixation buffer containing 4% paraformaldehyde and 0.1% of glutaraldehyde in PBS. Tissues were post-fixed by soaking in the same fixative for 48 h or longer. Brain and spinal cord from fixed mice were cut to 50-μm sections using a Vibratome. The sections were mounted with Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI, Vector Laboratories) and observed under conventional fluorescent microscope or confocal microscope.
Transgenic Mice and siRNA Administration—Transgenic mice SOD1G93A (28) were obtained from Jackson Laboratory and maintained on FVB/NJ background in the animal facility of University of Massachusetts Medical School (UMMS). All mouse experiments have been approved by the Institutional Animal Care and Use Committee and conducted abiding the UMMS policies and procedures regulating use of animals in research and the provisions of the Public Health Service/National Institutes of Health Guide for the Care and Use of Laboratory Animals. Infusion of siRNA into mice lumbar subarachnoid space was carried out using a method modified from Wu et al. (29). A thin catheter was made by stretching PE10 tube to the inner diameter ∼0.12 mm. The stretched section was cut to 1.9 mm, and two beads (1 mm apart) were made between the thin and the thick sections by heating and pressing the tube. The thick section of the tube was 2 cm long and was attached to a PE50 tube 1.5 cm in length. The Alzet osmotic pump (DURECT Corp.) was filled with modified siRNA or saline and primed for 24–48 h before implantation according to the manufacturer's instructions. To implant the catheter and the pump, the mouse was anesthetized by injection of Avertin (1.2% 2,2,2-tribromoethanol in 2% tert-amyl alcohol and PBS) intraperitoneally at 0.23 ml/10 g of body weight (30). The catheter was implanted between the L5 and L6 vertebra and connected to a primed Alzet osmotic pump with the PE50 tube. The catheter was stitched to the surface muscle, and the Alzet osmotic pumps were placed under the skin on the back of the mouse. The pump delivered 0.25 μl/h for 4 weeks or 0.5 μl/h for 1 week. For determining knockdown, the mice were sacrificed at the end of infusion, and their tissues were isolated. For testing therapy, only male mice were used for the convenience of surgery and to remove sex differences. The entire infusion set were surgically removed at the end of infusion. At the time when the pump was taken out or the animal was sacrificed, the position of the pump and the catheter was checked to ensure the correct positioning. The pump was also checked for correct infusion volume. All surgically operated mice were housed individually and checked for weight twice weekly. The end stage of G93A mice was defined as the day when complete paralysis of two limbs occurs, which was judged by an independent observer who did not know the treatment that the mouse received.
For testing interferon response, mice were infused with R1 siRNA as described above except the poly(IC)-treated group, which was infused with 16 μg of poly(IC) in PBS for 3 days (31) and was used as a positive control. These infused animals were sacrificed at 72 h after the start of infusion, and levels of interferon response markers such as OAS1, STAT1, and interferon-induced protein with tetratricopeptide repeat (IFIT) were measured using real-time PCR and Western blot.
Statistics—Disease onset, early stage, end stage, onset to early stage duration, and onset to end stage duration were obtained from individual animals. The averages and standard deviations were calculated for each treatment group. Statistical comparisons are done using Wilcoxon rank sum test.
RESULTS
We aimed at developing a therapeutic siRNA-targeting mutant SOD1 based on the rules for chemically modifying siRNA sequences to improve stability and cellular uptake without compromising their gene-silencing efficiency (Refs. 18 and 32 and reviewed in Ref. 33). Our previous studies have shown that modification of RNA at the ribose moiety enhanced RNA stability and prolonged siRNA half-life in vivo (18, 29, 30). We reasoned that siRNA sequences containing 2-fluoro modifications at the 2′-hydroxyl position (2′-fluoro) of nucleotides would enhance RNA stability in vivo, while retaining the A-helical geometry of the RNA duplex, which is required for efficient RNAi. In addition, we introduced phosphoro-thioate modifications in the siRNA to improve their cellular uptake. RNA labeling with various functional groups, purification, and characterization were accomplished according to established methods in our laboratory (25, 26). We introduced 2′-fluoro and phosphoro-thioate in the proximity of the terminal region of siRNA duplex to create a stabilized siRNA against human Sod1 gene, designated as R1.
To test whether R1 siRNA retains the RNAi efficacy, we cotransfected the unmodified and modified siRNAs into cultured cells with a luciferase reporter that carries the SOD1 target sequence in its 3′-untranslated region. The modified siRNA displayed the same silencing activity against this reporter as the unmodified siRNA (Fig. 1A). To determine whether the modified siRNA can be retained in cells for a prolonged period of time, we monitored Cy3 fluorescence in the transfected cells at different times after transfection. As a control, we also transfected unconjugated Cy3. The unconjugated Cy3 disappeared from the cells shortly after the transfection medium was replaced by the normal medium, indicating that it diffuses out of the cells quickly (Fig. 1B). In contrast, we observed Cy3 fluorescence in cells transfected with Cy3 conjugated to R1 siRNA for up to 6 days after transfection (Fig. 1B). To test whether the R1 siRNA retained in the cells had RNAi activity, we transfected cells with the luciferase reporter 6 days after the initial transfection of R1 siRNA. We observed silencing of the luciferase reporter (Fig. 1C), indicating that the R1 siRNA was still active 6 days after transfection.
FIGURE 1.

R1 siRNA knocks down sod1 gene expression and is stable in HEK293 cells. A, modified siRNA R1 and unmodified siRNA U1 were cotransfected with firefly luciferase reporter bearing the SOD1 target sequence in its 3′-untranslated region. The luciferase activity was measured at 24 h after transfection. R1 siRNA is as active in its silencing activity as its unmodified counterpart U1 siRNA. C represents control cells that were transfected with luciferase only. Mm, chemically modified mismatched control siRNA (see “Experimental Procedures”). B, R1 siRNA remains in cells for a long time. R1 siRNA and Cy3 were transfected into HEK293 cells, and Cy3 fluorescence was monitored before and after the transfection (TF) medium was replaced with the normal culture medium. Images on day 1 were taken at 20 min after the transfection medium (TF medium) was replaced with normal medium. All images were taken using the same exposure parameters except the ones for Cy3 day 1 with normal medium, R1 day 6, and R1 day 9, which were enhanced to ensure the observation of low level signals. C, R1 siRNA retained in cells at day 6 after transfection has silencing activity. R1 siRNA was transfected into HEK293 cells. Six days later, its target luciferase reporter was transfected into the same cells. The luciferase activity was measured on day 7.
For a siRNA to be effective in treating neurodegenerative diseases by direct CNS delivery, it has to meet several requirements as stipulated above. First, the siRNA should be sufficiently stable so that it can be delivered for a prolonged period. This is necessary because neurodegenerative diseases are chronic and progress over many years. To test whether R1 siRNA is stable at the body temperature, we implanted an osmotic pump that delivered R1 siRNA into the mouse spinal cord for 28 days. We then extracted the residual R1 siRNA from the infusion pump and examined the integrity of R1 siRNA by gel electrophoresis. We observed a major band with Cy3 fluorescence at the molecular weight of ∼23 nucleotides (Fig. 2A, left panel, upper arrow). SYBR Gold staining revealed an additional major band with lower molecular weight at ∼21 nucleotides (Fig. 2A, right panel, lower arrow), which corresponds to the other strand of R1 without Cy3. This result indicates that R1 is stable at the body temperature for at least one month.
FIGURE 2.

In vivo stability and distribution of R1 siRNA. A, R1 siRNA is stable in mice for 28 days. An Alzet osmotic pump filled with R1 siRNA was implanted in a mouse for infusion of the siRNA into the mouse spinal cord. After 28 days, the mouse was sacrificed, and the residual R1 siRNA was recovered from the pump and resolved on a polyacrylamide gel. The gel was photographed with illumination at 480 nm, and Cy3 fluorescence was monitored at 575 nm. The gel was then stained with SYBR and visualized using the same excitation wave length. Lane 2 was loaded twice as much extracted R1 as lane 1. C represents a control siRNA with different sequence from R1. B, R1 siRNA is distributed to all CNS area after 28 day intrathecal infusion at 4 μg/day. Total RNA was prepared from the fore brain (FB), brain stem (BS), cerebellum (CB), upper spinal cord (USC), and lower spinal cord (LSC). Total RNA from a PBS-infused mouse was used as a negative control. R1 siRNA was detected by Northern blot using a DNA probe with the sequence of the sense strand of the siRNA as probe. C, Cy3 fluorescence was detected in all cells in ventral horn of the spinal cord. D, confocal image of ventral horn of the spinal cord showing Cy3 fluorescence in motor neurons. DAPI, 4′,6-diamidino-2-phenylindole.
Second, the siRNA should be able to reach distant areas from the site of administration so that it can distribute widely throughout the CNS. This is necessary because ALS affects cells in wide areas of the CNS. To determine how widely the R1 siRNA is distributed in the CNS, we carried out Northern blot on RNA extracted from various CNS regions and detected the intact R1 siRNA in all CNS regions (Fig. 2B). This indicates that the R1 siRNA can distribute widely in the CNS by intrathecal infusion.
Third, the siRNA should be able to cross the cell membrane and accumulate intracellularly. To determine this, we examined Cy3 fluorescence on spinal cord sections. Under the light microscope, Cy3 fluorescence was distributed throughout the spinal cord and was not excluded from cells. Some cells in the ventral horn appeared particularly bright, suggesting that they accumulate more R1 siRNA than surrounding cells (Fig. 2C). A close examination on these cells using confocal microscopy confirmed that some of these bright cells are motor neurons (Fig. 2D). This indicates that R1 siRNA can accumulate inside cells, including motor neurons.
Fourth, the siRNA should be active in silencing its target in vivo. To determine whether this is the case, we infused R1 siRNA into mouse spinal cord for 1 week at the rates of 100–400 μg/day. We observed dramatic knockdown of SOD1 mRNA (Fig. 3A), confirming that R1 siRNA is active in vivo. However, infusion at these high doses was not feasible for long term administration because the animals develop signs of toxicity, including inactivity, poor grooming, and weight loss, when the infusion period exceeded 1 week. This agrees with a previous observation that high doses of siRNA expressed from viral vectors interferes with endogenous microRNA function and causes toxicity (8). To find out the therapeutic dose range of the R1 siRNA, we infused R1 siRNA at lower doses, 4, 8, and 16 μg/day for 28 days. These doses were generally tolerated and were effective in knocking down SOD1 levels in a dose-dependent manner (Fig. 3, B–D).
FIGURE 3.

R1 siRNA knocks down sod1 gene expression in vivo. A, 1 week after intrathecal infusion of R1 siRNA at high doses, total RNA was extracted from the spinal cord, and 3 μg was used for detection of human SOD1 mRNA by Northern blot. β-Actin was detected as loading control. B, Northern blots detecting human SOD1 mRNA and Western blots detecting human SOD1 protein 28 days after intrathecal infusion of low doses of R1 siRNA. C, quantification of human SOD1 mRNA and protein levels from three animals infused with low doses of R1 siRNA.
Fifth, the siRNA should not trigger an intolerable adverse effects at the therapeutically effective dose range. Although at doses of 4–16 μg/day, mice did not exhibit overt intolerable effects, a more stringent measure would be to measure molecular events such as interferon response. To test this, we infused mice with two different doses of R1 siRNA for 28 days, extracted mRNA and protein from mouse spinal cords, and examined markers for interferon response. Infusion of 16 μg/day of R1 siRNA caused significant interferon response, as shown by the increase of OAS1, STAT1, and IFIT mRNAs (Fig. 4A). In contrast, infusion of 4 μg/day did not trigger significant changes in the mRNA levels of these markers (Fig. 4A). This suggests that 4 μg/day can be safely used for therapy. To further confirm the safety at this dose, we examined the protein levels of OAS1 and STAT1 in the spinal cord and detected no changes (Fig. 4B).
FIGURE 4.

Long term CNS infusion of low dose R1 siRNA does not induce interferon response. Mice (three in each group) were infused with R1 siRNA at 4 or 16 μg/day for 28 days. At the end of infusion, their spinal cords were collected, and mRNA and protein levels of OAS1, STAT1, and IFIT were measured using quantitative reverse transcription-PCR and Western blots, respectively. The negative controls are uninfused mice. The positive controls are mice infused with poly(IC) (PIC) at 16 μg/day for 3 days. A, 1/ΔCT values of quantitative reverse transcription-PCR. B, Western blot for OAS1 and STAT1.
The preceding experiments demonstrate that R1 siRNA is stable, capable of distributing to all CNS areas and reach inside cells, is effective in knocking down SOD1, and is safe when administered at low doses for a prolonged time. Thus, we chose the dose of 4 μg/day for 28 days for the therapeutic trial in these mice. To simulate the clinical setting closely, we began this treatment at the disease onset. As controls, a cohort of mice was treated with PBS. We assigned mice randomly to the R1 siRNA- or PBS-treated groups. Because we began the treatment at the disease onset, we did not expect that the treatment would alter the disease onset. In fact, on average, the R1 siRNA group had the disease onset slightly earlier than the PBS group (Fig. 5A and Table 1), although this difference was not statistically significant. Even so, the R1 siRNA group reached early and end stage disease stages later than the PBS group (Fig. 5, B and C, and Table 1). Thus, R1 siRNA treatment significantly slowed the disease progression, extending the time span between the onset and the early stage disease from 14 days in the PBS group to 23 days and the time span between the onset and the end stage disease from 20 to 33 days, an ∼65% extension in both readouts (Table 1).
FIGURE 5.
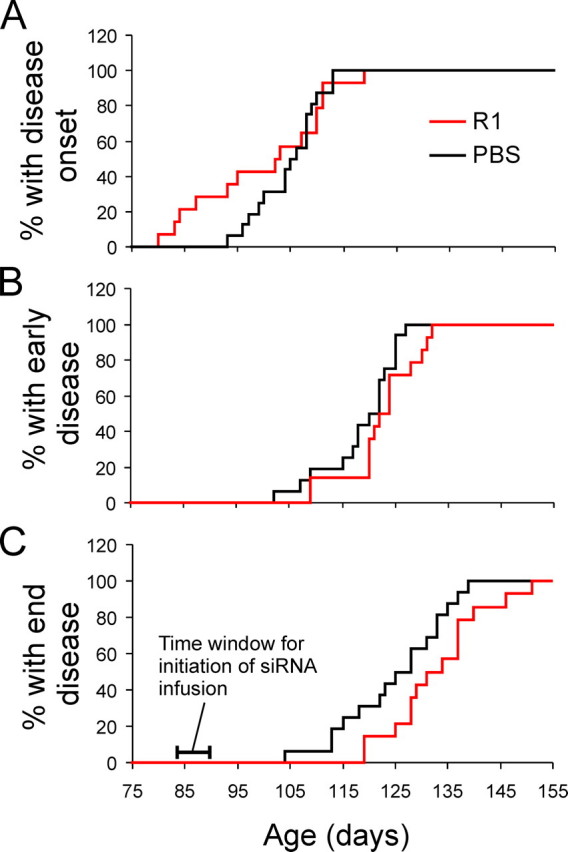
Long term intrathecal infusion of R1 siRNA starting at disease onset slows disease progression in mice expression mutant SOD1G93A. A, probability of disease onset, which is defined as the day of the peak body weight. B, probability of onset of the early disease stage, defined as the first day when the body weight became lower than 10% of the peak body weight. C, probability of the end disease stage, defined as the day when two of the four limbs are completely paralyzed. Mice were sacrificed at end stage for humane reasons.
TABLE 1.
Effects of R1 treatment on disease progression ErS: early stage; EdS: end stage; n = 14 for the R1-infused group; n = 16 for the PBS-infused group. The numbers are average days ± standard deviation. Onset-to-ErS and onset-to-EdS periods were calculated from individual animals in each group and averaged. Statistical comparisons between R1 and PBS groups were done using the Wilcoxon rank sum test, and the p values are the following: onset, 0.200; ErS, 0.085; EdS, 0.022; Onset to ErS, 0.024; Onset to EdS, 0.004.
| Treatment | Onset | ErS | EdS | Onset to ErS | Onset to EdS |
|---|---|---|---|---|---|
| R1 | 100 ± 13 | 122 ± 7 | 133 ± 9 | 23 ± 11 | 33 ± 12 |
| PBS | 105 ± 6 | 119 ± 7 | 125 ± 10 | 14 ± 6 | 20 ± 8 |
DISCUSSION
Using a transgenic mouse model for ALS, we have investigated critical parameters for developing RNAi therapy using siRNA for chronic CNS disorders. We show that chemical modification of a siRNA against mutant SOD1 can achieve long term drug stability in vivo and enables it to distribute to the distant regions of the CNS from the site of drug administration. Furthermore, we show that by adjusting the therapeutic dose of the siRNA, we can achieve a significant therapeutic benefit while controlling the toxicity. Our results demonstrate that sustained long term CNS infusion of a chemically modified siRNA is effective and safe in treating the mouse model for ALS, thereby illustrating that this approach can be used for treatment of chronic neurodegenerative diseases. Because surgical implantation of a catheter and pump is practical in humans (34), our study evokes a practical way for delivering RNAi therapy for fatal CNS disorders that are incurable at present.
Although the direct CNS administration of siRNA has disadvantages such as the requirement of a minor surgery to implant the catheter and the pump and the long term wearing of these devices for the patient, it also has some advantages. For example, restricted CNS administration can limit the target gene knockdown within the CNS, thus avoiding the potential side effects in the periphery. This is important in silencing SOD1 because lack of SOD1 activity has been shown to increase the incidence of liver cancer in aged mice (35). In addition, the infused siRNA can access all the cells including both neurons and glia. This is likely to enhance the therapeutic benefit when compared with motor neuron-specific silencing because mutant SOD1 expression in both neurons and glia in the CNS contributes to the disease progression (36, 37).
The overall therapeutic efficacy in this study is relatively modest, which is likely a result of several factors. First, we used a relatively low dose of R1 siRNA. At 4 μg/day infusion rate, a modest ∼15% knockdown of SOD1 protein was achieved (Fig. 3). Nevertheless, the modest therapeutic effect consequent to this knockdown is consistent with our previous results using genetic approaches to knockdown SOD1 (38), i.e. a significant therapeutic effect can be observed from a modest knockdown of mutant SOD1 expression.
Second, we began administering R1 siRNA at the disease onset, which is relatively late in disease progression in this disease model. Nevertheless, the fact that significant therapeutic benefit resulted from this treatment indicates for the first time that knockdown of mutant SOD1 expression even when the disease has begun can slow down the disease progression. This is important for TGS in humans because the therapy is mostly likely initiated after the diagnosis of the disease.
Third, we infused for 28 days, which is rather limited and does not cover the entire disease period. Finally, TGS is extremely challenging in this transgenic ALS model because these mice carry more than 20 copies of human mutant SOD1 transgene (38) and express ∼40 times the amount of mutant SOD1 mRNA and ∼17 times the amount of the mutant SOD1 protein over the endogenous SOD1 level, and their disease is dramatically accelerated when compared with human ALS (39). It is known that with the same dose of siRNA that silence mutant SOD1, the degree of mutant SOD1 knockdown and the extent of therapeutic benefit are significantly higher in mice expressing a low level of mutant SOD1 (8 times the endogenous SOD1 level) than in those expressing high level (17 times the endogenous SOD1 level) (38). Based on these data, we predict that a similar dose of R1 siRNA or doses lower than what we have used here could achieve a substantially greater degree of knockdown and therapeutic efficacy if the mutant is expressed at its natural levels by a single endogenous allele.
One recent study examined the therapeutic efficacy of silencing mutant SOD1 expression using an antisense oligonucleotide (40). Both their studies and our studies exploit the same therapeutic principle by lowering the level of toxic protein. Nevertheless, our study differs from theirs in several aspects. Although the antisense oligonucleotide was administered 30 days prior to the disease onset, we administered the R1 siRNA at the disease onset. Therefore, our results provide the basis for trying RNAi therapy in human cases at the disease onset. The therapeutic dose used in our study is lower than the dose of the antisense oligonucleotides used in their study, and the therapeutic efficacy is similar between both studies (40). Although differences in the disease models and methods of drug administration preclude a direct efficacy comparison between the two studies, it is generally known that siRNA is at least one order of magnitude more efficient in silencing genes than antisense oligonucleotides (41–43). It is worth noting that although the antisense oligonucleotide sequences against human SOD1 were exhaustively screened (40), we have not conducted such a screen for the best siRNA. Therefore, we cannot rule out the possibility that more potent silencing siRNAs against human SOD1 will be found and used for treatment of ALS caused by SOD1 mutants in the future.
The issue of dose-dependent toxicity for RNAi therapy can be a serious impediment in developing RNAi therapy. A recent study showed that administration of high doses of short hairpin RNA using viral vectors to mouse liver can cause serious toxicity and even death (8). Although it is not particularly surprising that an exceedingly high dose of a drug can cause toxicity, it is critical to have the ability to control the toxicity while offering therapeutic benefit. Our data show that high doses of R1 siRNA can cause toxicity. Nevertheless, we are able to control this toxicity by adjusting the therapeutic dose of the siRNA, thus highlighting a key advantage of delivering TGS using chemically modified siRNA.
In conclusion, we have demonstrated that chemically modified siRNA can be delivered directly to the CNS for TGS to treat chronic CNS disorders and attain the goal of significant therapeutic benefit and controllable toxicity. TGS using chemically modified siRNA for fatal neurological diseases should be further developed and tried clinically.
Acknowledgments
The JLA20 anti-actin monoclonal antibody developed by Dr. Jim Jung-Ching Lin was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD, National Institutes of Health, and maintained by The University of Iowa, Department of Biological Sciences, Iowa City, IA. We are grateful to Thomas Adolfsson (Luma Metall, SWEDEN) for a generous gift of tungsten wire; Kathryn Chase for teaching us the mouse surgery techniques; Sili Zhou for genotyping and assessing end disease stage; Dr. Hong Cao for assisting with plate reading; and Dr. Alonzo H. Ross and Phillip D. Zamore for sharing equipment.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1NS048145 and R21NS053770 from the NINDS (to Z. X.) and a National Institutes of Health grant (to T. M. R.). This work was also supported by grants from The Robert Packard Center for ALS Research at Johns Hopkins, CytRx Corp., and The ALS Association. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
This article was selected as a Paper of the Week.
Footnotes
The abbreviations used are: ALS, amyotrophic lateral sclerosis; siRNA, small interfering RNA; RNAi, RNA interference; SOD1, Cu,Zn-superoxide dismutase; CNS, central nervous system; TGS, therapeutic gene silencing; PBS, phosphate-buffered saline; OAS1, 2′, 5′-oligoadenylate synthetase 1; STAT1, signal transducer and activator of transcription 1; IFIT, interferon-induced protein with tetratricopeptide repeat; MOPS, 4-morpholinepropanesulfonic acid.
References
- 1.Gonzalez-Alegre, P., and Paulson, H. L. (2007) Nat. Clin. Pract. Neurol. 3 394-404 [DOI] [PubMed] [Google Scholar]
- 2.Mello, C. C., and Conte, D. (2004) Nature 431 338-342 [DOI] [PubMed] [Google Scholar]
- 3.Elbashir, S. M., Harborth, J., Lendeckel, W., Yalcin, A., Weber, K., and Tuschl, T. (2001) Nature 411 494-498 [DOI] [PubMed] [Google Scholar]
- 4.Caplen, N. J., Parrish, S., Imani, F., Fire, A., and Morgan, R. A. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 9742-9747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jackson, A. L., and Linsley, P. S. (2004) Trends Genet. 20 521-524 [DOI] [PubMed] [Google Scholar]
- 6.Judge, A. D., Sood, V., Shaw, J. R., Fang, D., McClintock, K., and MacLachlan, I. (2005) Nat. Biotech. 23 457-462 [DOI] [PubMed] [Google Scholar]
- 7.Hornung, V., Guenthner-Biller, M., Bourquin, C., Ablasser, A., Schlee, M., Uematsu, S., Noronha, A., Manoharan, M., Akira, S., de Fougerolles, A., Endres, S., and Hartmann, G. (2005) Nat. Med. 11 263-270 [DOI] [PubMed] [Google Scholar]
- 8.Grimm, D., Streetz, K. L., Jopling, C. L., Storm, T. A., Pandey, K., Davis, C. R., Marion, P., Salazar, F., and Kay, M. A. (2006) Nature 441 537-541 [DOI] [PubMed] [Google Scholar]
- 9.Semizarov, D., Frost, L., Sarthy, A., Kroeger, P., Halbert, D. N., and Fesik, S. W. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 6347-6352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jackson, A. L., Burchard, J., Leake, D., Reynolds, A., Schelter, J., Guo, J., Johnson, J. M., Lim, L., Karpilow, J., Nichols, K., Marshall, W., Khvorova, A., and Linsley, P. S. (2006) RNA (Cold Spring Harbor) 12 1197-1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.John, M., Constien, R., Akinc, A., Goldberg, M., Moon, Y.-A., Spranger, M., Hadwiger, P., Soutschek, J., Vornlocher, H.-P., Manoharan, M., Stoffel, M., Langer, R., Anderson, D. G., Horton, J. D., Koteliansky, V., and Bumcrot, D. (2007) Nature 449 745-747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gewirtz, A. M. (2007) J. Clin. Investig. 117 3612-3614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boudreau, R. L., Davidson, B. L., and Gerald, P. S. (2006) Curr. Top. Dev. Biol. 75 73-92 [DOI] [PubMed] [Google Scholar]
- 14.Braasch, D. A., Paroo, Z., Constantinescu, A., Ren, G., Oz, O. K., Mason, R. P., and Corey, D. R. (2004) Bioorg. Med. Chem. Lett. 14 1139-1143 [DOI] [PubMed] [Google Scholar]
- 15.Kumar, P., Wu, H., McBride, J. L., Jung, K.-E., Hee Kim, M., Davidson, B. L., Kyung Lee, S., Shankar, P., and Manjunath, N. (2007) Nature 448 39-43 [DOI] [PubMed] [Google Scholar]
- 16.Dorn, G., Patel, S., Wotherspoon, G., Hemmings-Mieszczak, M., Barclay, J., Natt, F. J., Martin, P., Bevan, S., Fox, A., Ganju, P., Wishart, W., and Hall, J. (2004) Nucleic Acids Res. 32 e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DiFiglia, M., Sena-Esteves, M., Chase, K., Sapp, E., Pfister, E., Sass, M., Yoder, J., Reeves, P., Pandey, R. K., Rajeev, K. G., Manoharan, M., Sah, D. W. Y., Zamore, P. D., and Aronin, N. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 17204-17209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chiu, Y. L., and Rana, T. M. (2003) RNA (Cold Spring Harbor) 9 1034-1048 [Google Scholar]
- 19.Bumcrot, D., Manoharan, M., Koteliansky, V., and Sah, D. W. Y. (2006) Nat. Chem. Biol. 2 711-719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morrissey, D. V., Lockridge, J. A., Shaw, L., Blanchard, K., Jensen, K., Breen, W., Hartsough, K., Machemer, L., Radka, S., Jadhav, V., Vaish, N., Zinnen, S., Vargeese, C., Bowman, K., Shaffer, C. S., Jeffs, L. B., Judge, A., MacLachlan, I., and Polisky, B. (2005) Nat Biotech. 23 1002-1007 [DOI] [PubMed] [Google Scholar]
- 21.Soutschek, J., Akinc, A., Bramlage, B., Charisse, K., Constien, R., Donoghue, M., Elbashir, S., Geick, A., Hadwiger, P., Harborth, J., John, M., Kesavan, V., Lavine, G., Pandey, R. K., Racie, T., Rajeev, K. G., Rohl, I., Toudjarska, I., Wang, G., Wuschko, S., Bumcrot, D., Koteliansky, V., Limmer, S., Manoharan, M., and Vornlocher, H.-P. (2004) Nature 432 173-178 [DOI] [PubMed] [Google Scholar]
- 22.Baigude, H., McCarroll, J., Yang, C. S., Swain, P. M., and Rana, T. M. (2007) ACS Chem Biol. 2 237-241 [DOI] [PubMed] [Google Scholar]
- 23.Gurney, M. E., Pu, H., Chiu, A. Y., Dal Canto, M. C., Polchow, C. Y., Alexander, D. D., Caliendo, J., Hentati, A., Kwon, Y. W., Deng, H.-X., Chen, W., Zhai, P., Sufit, R. L., and Siddique, T. (1994) Science 264 1772-1775 [DOI] [PubMed] [Google Scholar]
- 24.Xu, Z. (2000) Amyotroph. Lateral Scler. 1 225-234 [DOI] [PubMed] [Google Scholar]
- 25.Shah, K., Wu, H., and Rana, T. M. (1994) Bioconjugate Chem. 5 508-512 [DOI] [PubMed] [Google Scholar]
- 26.Hwang, S., Tamilarasu, N., Kibler, K., Cao, H., Ali, A., Ping, Y.-H., Jeang, K.-T., and Rana, T. M. (2003) J. Biol. Chem. 278 39092-39103 [DOI] [PubMed] [Google Scholar]
- 27.Hummon, A. B., Lim, S. R., Difilippantonio, M. J., and Ried, T. (2007) BioTechniques 42 467-472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gurney, M. E. (1994) N. Engl. J. Med. 331 1721-1722 [DOI] [PubMed] [Google Scholar]
- 29.Wu, W. P., Xu, X. J., and Hao, J. X. (2004) J. Neurosci. Methods 133 65-69 [DOI] [PubMed] [Google Scholar]
- 30.Papaioannou, V. E., and Fox, J. G. (1993) Lab. Anim. Sci. 43 189-192 [PubMed] [Google Scholar]
- 31.Flenniken, A. M., Galabru, J., Rutherford, M. N., Hovanessian, A. G., and Williams, B. R. (1988) J. Virol. 62 3077-3083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chiu, Y. L., and Rana, T. M. (2002) Mol. Cell 10 549-561 [DOI] [PubMed] [Google Scholar]
- 33.Rana, T. M. (2007) Nat. Rev. Mol. Cell Biol. 8 23-36 [DOI] [PubMed] [Google Scholar]
- 34.Rise, M. T. (2000) Arch. Med. Res. 31 237-247 [DOI] [PubMed] [Google Scholar]
- 35.Elchuri, S., Oberley, T. D., Qi, W., Eisenstein, R. S., Jackson Roberts, L., Van Remmen, H., Epstein, C. J., and Huang, T. T. (2005) Oncogene 24 367-380 [DOI] [PubMed] [Google Scholar]
- 36.Clement, A. M., Nguyen, M. D., Roberts, E. A., Garcia, M. L., Boillee, S., Rule, M., McMahon, A. P., Doucette, W., Siwek, D., Ferrante, R. J., Brown, R. H., Jr., Julien, J. P., Goldstein, L. S. B., and Cleveland, D. W. (2003) Science 302 113-117 [DOI] [PubMed] [Google Scholar]
- 37.Boillee, S., Yamanaka, K., Lobsiger, C. S., Copeland, N. G., Jenkins, N. A., Kassiotis, G., Kollias, G., and Cleveland, D. W. (2006) Science 312 1389-1392 [DOI] [PubMed] [Google Scholar]
- 38.Xia, X., Zhou, H., Huang, Y., and Xu, Z. (2006) Neurobiol. Dis. 23 578-586 [DOI] [PubMed] [Google Scholar]
- 39.Jonsson, P. A., Graffmo, K. S., Andersen, P. M., Brannstrom, T., Lindberg, M., Oliveberg, M., and Marklund, S. L. (2006) Brain 129 451-464 [DOI] [PubMed] [Google Scholar]
- 40.Smith, R. A., Miller, T. M., Yamanaka, K., Monia, B. P., Condon, T. P., Hung, G., Lobsiger, C. S., Ward, C. M., McAlonis-Downes, M., Wei, H., Wancewicz, E. V., Bennett, C. F., and Cleveland, D. W. (2006) J. Clin. Investig. 116 2290-2296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miyagishi, M., Hayashi, M., and Taira, K. (2003) Antisense Nucleic Acid Drug Dev. 13 1-7 [DOI] [PubMed] [Google Scholar]
- 42.Kretschmer-Kazemi Far, R., and Sczakiel, G. (2003) Nucleic Acids Res. 31 4417-4424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bertrand, J.-R., Pottier, M., Vekris, A., Opolon, P., Maksimenko, A., and Malvy, C. (2002) Biochem. Biophys. Res. Commun. 296 1000-1004 [DOI] [PubMed] [Google Scholar]