

Abstract
Pectin is a structural polysaccharide that is integral for the stability of plant cell walls. During soft rot infection, secreted virulence factors from pectinolytic bacteria such as Erwinia spp. degrade pectin, resulting in characteristic plant cell necrosis and tissue maceration. Catabolism of pectin and its breakdown products by pectinolytic bacteria occurs within distinct cellular environments. This process initiates outside the cell, continues within the periplasmic space, and culminates in the cytoplasm. Although pectin utilization is well understood at the genetic and biochemical levels, an inclusive structural description of pectinases and pectin binding proteins by both extracellular and periplasmic enzymes has been lacking, especially following the recent characterization of several periplasmic components and protein-oligogalacturonide complexes. Here we provide a comprehensive analysis of the protein folds and mechanisms of pectate lyases, polygalacturonases, and carbohydrate esterases and the binding specificities of two periplasmic pectic binding proteins from Enterobacteriaceae. This review provides a structural understanding of the molecular determinants of pectin utilization and the mechanisms driving catabolite selectivity and flow through the pathway.
INTRODUCTION
The plant cell wall is a formidable barrier to microbial infection. One integral component of this structure is pectin, a heterogeneous polysaccharide that is composed primarily of galacturonans. The biological function of pectin is to cross-link cellulose and hemicellulose fibers, providing rigidity to the cell wall. Degradation of this structure by pectinolytic microorganisms such Erwinia carotovora and Erwinia chrysanthemi during soft rot infection is a devastating process for the plant that leads to plant cell necrosis and tissue maceration. A large amount of biological study in this area has focused on the secreted virulence factors from these two phytopathogens, although other members of the Enterobacteriaceae, including several human gastrointestinal pathogens, now have been shown to contain abridged pectin degradation pathways (19, 56).
In general, pectin degradation is facilitated by a battery of pectinases, including pectate lyases (families 1, 2, 3, and 9), polygalacturonases and rhamnogalacturonases (glycoside hydrolase family 28), pectin methylesterases (carbohydrate esterase family 8) and pectin acetylesterases (carbohydrate esterase family 12) (30; http://www.cazy.org/index.html) (Table 1). Depolymerization and de-esterification of the polysaccharide is initiated by extracellular enzymes. The resulting oligogalacturonide chains are passively transported into the periplasmic space through anion-specific oligosaccharide porins of the KdgM family (8, 49), where the action of downstream pectinases further digests the substrates into di- and trigalacturonides. These oligogalacturonides are subsequently passaged into the cytoplasm through TogMNAB, a multisubunit CUT1 family ABC transporter that couples transport to ATP hydrolysis (1, 29, 32). Subsidiary transport systems, ExuT and KdgT, transport saturated and unsaturated monosaccharides, respectively (18, 31, 58), and TogT transports oligogalacturonides in a process that parallels the function of TogMNAB (32). Within the cell, oligogalacturonides are ultimately degraded into pyruvate and 3-phosphoglyceraldehyde, which enter the citric acid cycle and are converted into energy. Interestingly, the genes for virtually every protein involved in this process are regulated by the transcriptional repressor KdgR, an IclR family protein that is allosterically regulated by the effector metabolite 2-keto-3-deoxygluconate.
TABLE 1.
Pectinases and hexuronate transporters in pectinolytic Enterobacteriaceaea
| Organism | Pels | GHs | CEs | Transport and accumulation proteins |
|---|---|---|---|---|
| Erwinia carotovora subsp. | EcaPL1 | EcaGH28 | EcaCE8 | Outer membrane |
| atroseptica SCRI1043 | PelA (E), PelB (E), PelC (E), PelF (E), PelZ (E), Pnl (E) | PehA (E),b PehN (E), PehX (?) | PemA (E), PemB (P) | KdgM1, KdgM2, KdgM3, KdgM4 |
| EcaPL2 | EcaCE12 | Inner membrane | ||
| PelP (P), PelW (C) | PaeX (P), PaeY (E)PaeX (P), PaeY (E) | TogMNAB, TogT, ExuT | ||
| EcaPL3 | Polygalacturonate binding protein | |||
| PelI (E), HrpW (?) | SghX/CBM32 | |||
| EchPL9 | ||||
| PelL (E), PelX (P) | ||||
| Erwinia chrysanthemi | EchPL1 | EchGH28 | EchCE8 | Outer membrane |
| 3937/EC16 | PelA (E),b PelB (E), PelC (E),b PelD (E), PelE (E),b PelZ (E) | PehN (E), PehV (P), PehW (P), PehX (P) | PemA (E),b PemB (P) | KdgM, KdgN, KdgT |
| EchPL2 | EchCE10 | Inner membrane | ||
| PelW (C) | PaeX (P) | TogMNA(B),b TogT, ExuT | ||
| EchPL3 | EchCE12 | |||
| PelI (E) | PaeY (E) | |||
| EchPL9 | ||||
| PelL (E),b PelX (P) | ||||
| Escherichia coli CFTO73 | EcoPL9 | EcoCE8 | Outer membrane | |
| PelX (P) | YbhC/PemB (P) | KdgT | ||
| Inner membrane | ||||
| TogT, ExuT | ||||
| Yersinia enterocolitica subsp. | YePL2 | YeGH28 | YeCE8 | Outer membrane |
| enterocolitica 8081 | PelP (P),b PelW (C) | PehX (P)b | PemA (E)b | KdgM1, KdgM2 |
| Inner membrane | ||||
| TogMNAB, TogT, ExuT | ||||
| Polygalacturonate binding protein | ||||
| SghX/CBM32 |
Much of the work focusing on the activity and regulation of secreted pectinases has been explained by several extensive reviews and comparative genomics studies (30, 33, 56, 57, 74). For the purposes of this article, we have summarized what is presently known about the structural biology of extracellular and periplasmic pectin utilization within Enterobacteriaceae. Below, we discuss the protein folds of each pectinase family from Enterobacteriaceae and compare the distinct catalytic mechanisms for pectate lyases, glycoside hydrolases, and carbohydrate esterases. In addition, we also consider the structural and mechanistic aspects of ligand selectivity and transporter architecture for catabolite passage across both the outer and inner membranes.
Abbreviations
In this review, protein nomenclature is based upon both the common enzyme name and the CAZy classification system (www.cazy.org). The CAZy classification system lists each protein on the basis of organism source (genus and species name; e.g., Yersinia enterocolitica = Ye), carbohydrate active enzyme/protein types (glycoside hydrolase, polysaccharide lyase, carbohydrate esterase, and carbohydrate binding module), family number (carbohydrate active enzyme types are subdivided into sequence-based families), and isozyme designation (given alphabetically as designated by the common enzyme name). For example PelP from Y. enterocolitica is also known YePL2A (a family 2 polysaccharide lyase isozyme A). This nomenclature alleviates the difficulties associated with arbitrary classification systems and is particularly helpful when analyzing a consortium of enzymes from a structure-function perspective. For example, the pectate lyases from E. chrysanthemi (PelA to -E) are all from PL family 1, PelI is from family 3, PelL and PelX are from family 9, and PelW is from family 2. Although these distinct lyase families all catalyze the β-elimination of polygalacturonate, they have unrelated sequences (in each case) and folds (in the case of PelW). For the purposes of this review, both naming systems will be utilized in order to provide clarity for the broad readership within the field and for simplified cross referencing within the literature.
The following abbreviations are used in this review: CBM, carbohydrate binding module; CE, carbohydrate esterase; KDG, 2-keto-3-deoxygluconate; GH, glycoside hydrolase; Pae, pectin acetylesterase; Peh, pectate hydrolase; Pel, pectate lyase; Pem, pectin methylesterase; PL, polysaccharide lyase; and Pnl, pectin lyase.
PECTIN STRUCTURE
Pectin is a complex heterogeneous structural polysaccharide that is a major component of the primary cell walls of all terrestrial plants (14, 16-18). Pectin can be described as a glycan matrix rich in galacturonic acid that anchors and cross-links the load-bearing cellulosolic and hemicellulosolic fibers of the cell wall. There are three different classes of pectic species, which vary in the abundances and chemistries of their respective monosaccharide subunits: rhamnogalacturonan I, rhamnogalacturonan II, and homogalacturonan (14, 19, 20). Of these classes, homogalacturonan is the primary target of the enzymes considered within this review and therefore is referred to simply as “pectin” below.
At its simplest level of structure, pectin is composed of α-1,4-linked galacturonic acid subunits. This linkage creates an “accordion-like” conformation between neighboring residues due to its signature axial C-4 configuration. The overall fiber is extended and flexible and can adopt a 21 or 31 helix, depending on the degree of hydration and presence of cations (14, 21-23). Variation in the potential three-dimensional structure of pectic fibers was supported by the galacturonate pentasaccharide-Pel cocrystal structure, which revealed that the substrate was a mixture of both 21 and 32 helical conformations (59).
Heterogeneity in the chemical composition of pectin can result from xylose decorations at C-3; methoxyl and acetyl group esterifications at C-6 and C-2/C-3, respectively; and the presence of divalent cations such as calcium (14). These chemical modifications are associated with distinct structural and functional aspects of the polysaccharide. In particular, methylesterification of the C-6 uronate group facilitates “gellation” by neutralizing the negative charges of monosaccharide subunits and allowing more cohesive packing of pectic chains. Conversion of pectin to polygalacturonate during tissue remodeling and pectin degradation is caused by the enzymatic removal of methoxyl groups, which restores the inherent charges on the carboxylates. Several excellent reviews contain further discussion on the chemistry of pectin structure (33, 55, 74).
EXTRACELLULAR PECTIN DEGRADATION
Depolymerases encoded within the genomes of phytopathogens from Enterobacteriaceae are secreted into the periplasm through the SEC pathway (53) and into the environment through the outer membrane OUT system (16, 30, 56). Although the molecular determinants of selective extracellular secretion verses periplasmic localization are still unclear, two polypeptide components of the OUT system, OutC and OutD, have been identified to specifically interact with exoproteins and likely participate in this process (12, 46). Proteomic analysis of the E. chrysanthemi secretome detected 25 unique polypeptides involved in pectin degradation (40). These included eight endo-acting extracellular Pels (PelA to -E/EchPL1A to -E, PelZ/EchPL1Z, PelI/EchPL3I, and PelL/EchPL9A), an intracellular exo-acting Pel (PelX/EchPL9X), one extracellular polygalacturonase (PehN/EchGH28N), three periplasmic exopolygalacturonases (PehV to -X/EchGH28V to -X), one extracellular (PaeY/EchCE12Y) and one periplasmic (PaeX/EchCE12X) Pae, and one extracellular Pem (PemA/EchCE8A) (40) (Table 1). Although this is the first secretome reported for a pectinolytic bacterium from Enterobacteriaceae, comparative genome analysis of other species predicts that the repertoire of secreted enzymes within cellular locations varies considerably. Below we highlight what is currently known about the structures of extracellular pectinases from these microorganisms.
Depolymerization of Polygalacturonate by β-Elimination
Secreted endo-Pels are the primary virulence factors in soft rot infection. The genomes of phytopathogens from the genus Erwinia contain many genes encoding isozymes from families 1 to 3 and 9 (30; www.cazy.org). The presence of different enzymes with overlapping functions suggests that Pels may have preferential activities on microheterogeneous target substrates that vary in degree of methylation (charge state), degree of polymerization (size), and perhaps the nature of carbohydrate subunits (i.e., rhamnose versus galacturonate) (26).
There are currently two families (families 1 and 9) containing extracellular Pels with determined folds (Table 2). These enzymes operate by a common mechanism to cleave glycosidic linkages between two neighboring galacturonic acid monosaccharides. Pels in general utilize a two-step E1cb β-elimination reaction, producing a planar product with an unsaturated bond between C-4 and C-5 at the nonreducing end (Fig. 1A) (13, 59). In the first step, the C-5 hydrogen is abstracted by a catalytic arginine (Brønstead base). This process is coupled to H-5 acidification resulting from Ca2+ coordination by the C-5 uronate group. Not surprisingly, due to the specialized chemistry of this catalytic base, the optimal pH of these enzymes is alkaline and ranges between 7.5 and 10 (66). A titratable guanidino group (the pKa of arginine is 12.5) under these conditions is suggestive of localized pKa perturbations, such as proximal Ca2+ cofactors (25, 26, 37). Following H-5 abstraction, the transition state is stabilized by electron delocalization to the C-5 carboxylate “sink.” In the second step of the reaction, product resolution results from electron shuttling to O-4, triggering elimination of the leaving group.
TABLE 2.
Three-dimensional structures of pectinases from Enterobacteriaceae
| Pectinase group | Family | Organism | Name | CAZy | Locationa | PDB ID | Reference(s) |
|---|---|---|---|---|---|---|---|
| Pels | 1, β-helix | E. chrysanthemi EC16 | PelA | EchPL1A | E | 1JRG, 1JTA, 1OOC, 1PE9 | 21 |
| PelC | EchPL1C | E | 1AIR, 1O88, 1O8D-1O8M, 1PLU, 2EWE,b 2PEC | 21, 44, 59, 76-78 | |||
| PelE | EchPL1E | E | 1PCL | 43, 44 | |||
| 2, (α/α)7 barrel | Y. enterocolitca ATCC 9610D | PelP | YePL2A | P | 2V8I, 2V8J, 2V8Kb | 3 | |
| 3, β-helix | NDc | ||||||
| 9, β-helix | E. chrysanthemi 3937 | Pel9A/PelL | EchPL9 | E | 1RU4 | 37 | |
| GHs | 28, β-helix | E. carotovora SCC3193 | PehA | EcaGH28A | E | 1BHEb | 2, 51 |
| Y. enterocolitica ATCC 9610D | PehX | YeGH28 | P | 2UVE, 2UVFb | |||
| CEs | 8, β-helix | E. chrysanthemi 3937 | PemA | EchCE8A | E | 1QJV, 2NTB,b 2NTP,b | 23, 35 |
| 10 | ND | 2TTQ,b 2NT6,b 2NT9,b | |||||
| 12 | ND | 2NSP,b 2NSTb |
E, extracellular; P, periplasmic.
Enzyme-substrate/product complex.
ND, not determined.
FIG. 1.

The extracellular endo-Pels PelA/EchPL1A, PelC/EchPL1C, and PelL/EchPL9A. (A) A generalized reaction coordinate for calcium assisted β-elimination. (B) PelC/EchPL1C (PDB ID, 2EWE) is displayed in a “cartoon” format with a transparent solvent-accessible surface. The pentagalacturonate substrate is shown as sticks colored beige and the calcium ions as spheres colored mageneta. (C) The superimposed active sites of PelC/EchPL1C (green) and PelA/EchPL1A (yellow) (PDB ID, 1OOC) displayed in a wall-eyed format. The structurally conserved Brønstead base and calcium-coordinating aspartate residues are shown as sticks and labeled with PelC/EchPL1C numbering. (D) The superimposed active sites of PelC/EchPL1C (green) and PelL/EchPL9A (blue) based upon overall enzyme alignment displayed in stereo. The catalytic base, a lysine within PL family 9, and calcium ion (pink) are structurally conserved but shifted laterally toward the reducing end of the superimposed substrate.
The structure of the E. chrysanthemi family 1 Pel PelC/EchPL1C was the first structure of a petinolytic enzyme ever described (75-77) (Fig. 1B). The enzyme adopts a parallel β-helix topology with three distinct β-sheets formed from eight complete β-strand turns. The center of the enzyme is stabilized by a ladder of stacking residues, including a rich hydrogen bond network between repeating asparagines, and hydrophobic stacks between aliphatic and aromatic side chains. This architecture of intramolecular bonds generates a very stable protein fold, presumably enabling persistence of the virulence factor within the harsh extracellular environment during infection. When visualized from the side, the enzyme is asymmetrical with a noticeable protrusion formed by several loops (called the T3 loops) that are contributed from different β-strands. This region of the molecule contains the catalytic center of the enzyme, which is a region of noticeable structural heterogeneity compared to other β-helix enzymes. What is truly remarkable about the β-helix is that following its initial discovery in 1993 (77), the topology has proven to be a well-conserved scaffold for pectinases in general, as other Pels from sequence divergent families (PLs 1, 3, and 9), and enzymes harnessing distinct catalytic machinery (GH28s and CE8s) (see below) have been described (34, 36). The superimposition of two family 1 Pels (PelA/EchPL1A [PDB ID, 1O88] and PelC/EchPL1C [PDB ID, 2EWE]) has a calculated core root mean square deviation value of 1.73 Å for 260 aligned Cα. Beyond the similarities in overall fold, closer analysis of the active site reveals that there is a striking conservation of catalytic residue architecture (Fig. 1).
Much of what is known about Pel-catalyzed β-elimination is based upon the PelC/EchPL1C R218K catalytic mutant-pentagalacturonide complex, which was reported in 1999 by the lab of Frances Jurnak (59). The mutated enzyme has an attenuated activity, which enabled the detailed analysis of the catalytic machinery and subsite architecture (Fig. 1C). As described above, the two main components of β-elimination are the Brønstead base and divalent cation binding pocket. When R218 was reintroduced into the mutant structure, it was predicted to be disposed within hydrogen bonding distance of the Cα-H-5 targeted for abstraction from the substrate (59). This arginine is flanked by a Ca2+ coordination pocket consisting of E166, the uronate oxygens from subsites −1 and −2, the ring oxygen of −2, and three water ligands (59). In addition to the catalytic Ca2+, other Ca2+ binding sites that function to bridge the carboxylate groups of the substrate to aspartate and glutamate residues within the active site were also present (26, 59). These electrostatic contacts contribute to the extensive subsite architecture of the enzyme that is critical for substrate selectivity and complex formation. In addition, there are several tryptophan residues that form the surface of the active-site pocket and stack to the apolar face of galacturonic acid subunits, and there are numerous polar amino acids, including asparagines and arginines that form hydrogen bonds and electrostatic interactions with the substituent groups of the substrate.
When the active site of PelC/EchPL1C is compared to other family 1 Pel structures from E. chrysanthemi, there is a stringent conservation of catalytic amino acids (Fig. 1C). In the PelA/EchPL1A structure, both the catalytic base (R241) and calcium-coordinating residue (D184) are conserved. The lack of calcium complexes for this enzyme precludes any direct comparison of the metal coordination chemistries. Structural analysis of more distantly related enzymes, however, does reveal subtle structural differences between them. Superimposition of PelC/EchPL1C and the family 9 PelL/EchPL9A (PDB ID, 1OOC) has a root mean square deviation of 2.31 Å2 for 218 matched Cα. Analysis of the catalytic site architecture indicates that in the family 9 enzyme the catalytic base, a lysine in this enzyme (K273), is shifted two positions toward the reducing end of the sugar and the Ca2+ coordination site (D209, D237, and D233) is rotated around the substrate axis (Fig. 1D). However, based upon overall enzyme structural alignments, it is difficult to directly evaluate the similarities in catalytic site architecture for unrelated enzyme families. Superimposition of the catalytic bases revealed that the Ca2+ coordination pockets are in fact structurally conserved (37). This observation has proven to be reflective of Pels in general, as even diverse fold families have very similar active-site architectures (see below).
Depolymerization of Polygalacturonate by Hydrolysis
Cleavage of glycosidic linkages within pectic fragments by the addition of water is catalyzed exclusively by the family 28 polygalacturonases (GH28s) (Fig. 2A). In addition to homogalacturonan, GH28s are also involved in hydrolysis of heterogeneous pectin derivatives such as rhamnogalacturonan and xylogalacturonan. This enzyme family encapsulates one of the largest groups of sequence-related GHs described in nature, with members found in diverse species ranging from bacteria to insects (47).
FIG. 2.

The extracellular endopolygalacturonase PehA/EcaGH28A. (A) Generalized reaction mechanism for inverting family 28 GHs. (Based on data from reference 73.) (B) PehA/EcaGH28A (PDB ID, 1BHE) is displayed in a “cartoon” format with a transparent solvent-accessible surface. Catalytic residues are displayed as sticks. (C) Superimposed catalytic sites of the closely related endopolygalacturonase PelA/EcaGH28A (green) and periplasmic exopolygalacturonase PehX/YeGH28 (yellow) (PDB ID, 2UVF) displayed in wall-eyed format. The residues from both PehA/EcaGH28A (D202, D223, and D224) and PehX/YeGH28 (D381, D402, and D403) are labeled. The digalacturonate product from the exopolygalacturonase complex is shown in beige, and subsites −1 and −2 are labeled in red.
Polygalacturonases are considered, along with the endo-acting Pels, a key factor in plant tissue maceration during soft rot infection (51). Within Enterobacteriaceae, there are two classes of GH28s based upon activities, i.e., the endopolygalacturonases and the exopolygalacturonases, which generally localize to the extracellular environment and periplasm, respectively. Closer analysis, however, does expose species-specific localization and isozyme differences. For example, E. chrysanthemi contains one extracellular α-endo-poly-d-galacturonosidase and three periplasmic α-exo-poly-d-galacturonosidases that releases disaccharides (40, 64), and Y. enterocolitica contains only one periplasmic α-exo-poly-d-galacturonosidase with homologous activity (Table 1) (56). This distinction is in agreement with potential substrate accumulation within each environment, as endo-acting enzymes would degrade the extracellular highly polymerized forms of polygalacturonate present within the plant cell wall, and exo-acting enzymes would be most efficient at producing small oligogalacturonides for intracellular transport from pectic fragments that accumulate in the periplasm (see below). In this light, the pattern of isozyme expression between different species may reflect distinct modes of phytopathogenesis or perhaps symbiotic relationships with other pectinolytic bacteria, where less equipped pectinolytic bacteria depend upon upstream depolymerizations by more robust species (56).
The endopolygalacturonase from E. carotovora subsp. atroseptica, PehA/EcaGH28A, was the second class of pectinase described to adopt the right-handed parallel β-helix topology (51) (Fig. 2B). In addition to the similarity in its overall fold to β-helix Pels, the polygalacturonase displays a pronounced active-site cleft in an analogous position. The most prominent structural difference between the two enzyme classes is that the tertiary structure of GH28s is comprised of four β-sheets, which is one more than in the lyases.
The open-ended PehA/EcaGH28A active site has a well-designed topography for the recognition of polygalacturonate, an observation that is in agreement with its previously described endo mode of activity (64). Attack of internal galacturonide residues is enabled by the freedom of the substrate to extend out into solvent at either end. In addition to these steric allowances, the chemistry of the amino acid side chains is also tailored for substrate recognition. The electrostatic potential of the solvent-accessible surface within the active site of EchGH28A reveals two loops with basic patches composed primarily of lysines (51). These residues are suitable candidates for involvement in substrate recognition events, as the formation of salt bridges has been reported to be critical for catalysis by endo-Pels (3, 13, 59) and predicted by modeling of an octagalacturonate-polygalacturonase complex in Aspergillus aculeatus (14).
The majority of what is known about the mechanism of Enterobacteriaceae GH28s has come from complementary enzyme-substrate structural studies and catalytic amino acid mutagenesis in extracellular enzymes from divergent species: E. carotovora subsp. atroseptica (51), Stereum purpurem (65), and Aspergillus niger (73). PehA/EcaGH28A contains a catalytic cluster of three aspartate residues: D202, D223, and D224 (Fig. 2C). These amino acids are positioned within 5 Å of one another and approach the substrate in a “syn” conformation. Interestingly, D202 and D223 are conserved within the catalytic sites of all known GH28s, including rhamno- and xylogalacturonases (47). The hydrolysis reaction proceeds by a single-step inverting mechanism resulting in stereochemical inversion around the anomeric carbon of the leaving group (Fig. 2A). Based upon proximity to the scissile glycosidic oxygen and mutagenic studies, D223 is considered to be the general acid (65). Currently, it is not known which of the complementary aspartates operates as the general base by accepting a hydrogen atom and charging the nucleophilic water. Further experiments are required to detail the role of D203 and D224 along the reaction coordinate.
De-Esterification of Pectin by CEs
There is only one described Pnl within Enterobacteriaceae, the family 1 Pnl from E. carotovora, which suggests that depolymerization reactions occur primarily on de-esterified polygalacturonate substrates (19). Indeed, it has been shown that E. chrysanthemi cannot grow on heavily esterified pectin as a sole carbon source (60). In Erwinia spp., there is both an extracellular Pae (PaeY/CE12Y) and a methylesterase (PemA/CE8A), the latter of which is also present in Yersinia spp. (40, 56). The acetylesterase removes acetylations at C-2 and C-3 (62), and the methylesterase demethylates the methoxylated uronate groups, releasing methanol and restoring the inherent negative charge on the galacturonide product (9, 60) (Fig. 3A). There appears to be a hierarchy in preferential activities for these enzymes, as acetylesterase efficiency is increased when esterified substrates are pretreated with methylesterase (62, 63). The importance of de-esterification for the activity of Pels is clear in consideration of both the molecular determinants of substrate recognition and the β-elimination mechanism discussed above. However, there are examples of downstream periplasmic CEs in Erwinia spp. and Vibrio parahaemolyticus (PaeX/CE12X and PemB/CE8B) (40, 56, 60), which suggests some plasticity in the different stages of pectin degradation compared between species. In this regard, it is important to consider that organisms with periplasmic CEs also contain pectate depolymerases in the same cellular compartment, which may alter the required stringency of extracellular de-esterification events. As there currently is no available structure for an acetylesterase from Enterobacteriaceae, discussion in the following section will be restricted to the structural biology of Pems.
FIG. 3.

The extracellular Pem PemA/EchCE8. (A) Generalized mechanism for demethylation of pectin by Pems. The nucleophile (D199) attacks the carbonyl carbon, forming a tetrahedral intermediate that is stabilized by Q177. The general acid-base catalyst D178 protonates the ester-linked oxygen, and attack by a catalytic water releases methanol and polygalacturonate, recharging the active site. (B) PemA/EchCE8A (PDB ID, 1QJV) is displayed in a “cartoon” format with a transparent solvent-accessible surface. (C) The active site of PemA/EchCE8A displayed in wall-eyed stereo. The bound hexasaccharide substrate (compound II) from E. chrysanthemi (PDB ID, 2NST) is shown, and the catalytic residue D178 has been reintroduced for reference.
The majority of study on Pems has focused on plant enzymes that are operational during tissue development and fruit ripening (48, 52). These enzymes operate in a processive fashion, similarly to bacterial enzymes, to produce blocks of demethylated subunits along the polysaccharide, for which both single-chain and multichain mechanisms have been proposed (24, 60). This process contrasts with that for the fungal methylesterases, which tend to demethylate pectin in a distributive fashion by randomly selecting substrates (45, 70).
The crystal structure of PemA/EchCE8A from E. chrysanthemi was first published in 2001 (35). Although the amino acid sequence of PemA/EchCE8A is unrelated to that of any other known protein, the enzyme adopts the parallel β-helix fold described above for both Pels and polygalacturonases (Fig. 3B). Comparison of the tertiary structures of these different enzyme classes indicates that the esterase is more structurally similar to Pels, in that it contains the same numbers of complete coils (eight) and β-sheets (three). The most noticeable difference in the CE8 enzyme is that the T3 loops harnessing the putative catalytic site are shifted along the longitudinal axis of the protein toward the C terminus. In addition, there is an extensive C-terminal tail with α-helical character that packs antiparallel to the face of the β-helix.
The active-site architecture of PemA/EchCE8A is unique and lacks the serine and histidine residues of the Ser-His-Asp catalytic triad present in functionally unrelated esterases (35). A putative catalytic mechanism was originally predicted based upon structural analysis (35, 38). The floor of the catalytic site is coated with aromatic residues: Y158, Y181, F202, and W269. These amino acids likely function to dock the pectin substrate by selectively stacking with the apolar faces of individual residues. Of these, Y181, F202 and W269 may be critical, as they are highly conserved among eukaryotic Pems (35). The de-esterification reaction is believed to be facilitated by two aspartate residues (D178 and D199), which are positioned as suitable candidates for acid-base catalysis (Fig. 3C). At their closest point the oxygen atoms from each carboxylate group are within 4.2 Å of each other, which is noticeably shorter that the 5.5 Å typically observed in retaining GHs.
Recently, the mechanism of PemA/EchCE8A has been illuminated by the crystal structures of several catalytic mutants in complex with various substrates and a product (Fig. 3A and C) (23). Through this elegant structural analysis the nucleophilic aspartate was determined to be D199, which attacks the carbonyl carbon of the C-6 ester and generates a tetrahedral intermediate. The second proximal aspartate residue (D178) operates as the general acid-base catalyst and forms a strong hydrogen bond with the carbonyl oxygen of the methyl ester. The transition state is stabilized by interactions with Q177 and D178, which help to neutralize the oxyanion formed on the carbonyl oxygen. Protonation of the leaving group by D178 enables the release of methanol and the generation of a covalently bound anhydride intermediate. Subsequent hydrolysis of the anhydride by a D198 activated water molecule releases the aglycon group and regenerates the active site.
Outer Membrane Transport
Within Enterobacteriaceae, depolymerized and de-esterified pectic fragments are passaged across the outer membrane by facilitated diffusion into the periplasm through the water-filled porin, KdgM (56). E. chrysanthemi growth on pectin as a carbon source is dependent upon wild-type KdgM, and in the presence of pectic substrates this porin is one of the most abundant proteins in the outer membrane (8). Liposome reconstitution and electrophysiological experiments demonstrated that this protein behaves in a voltage-dependent fashion with selectivity for anionic solutes (8). However, the presence of periplasmic methylesterases in some species suggests that oligogalacturonides do not require a negative charge at every residue for conductance.
There are currently structures available for several porins, including the Escherichia coli proteins OmpF and PhoE (20). Both of these channels are homotrimers with 16 transmembrane antiparallel β-strands within each monomer. The periplasmic domain is composed of short loops protruding into solvent, whereas the extracellular component is connected by prominently longer loops, which are functional in gating the mouth of the channel. In particular, loop 3 is active in entrance constriction and solute selectivity (20). KdgM differs from these multimeric porins in both topology and higher-order structure, as it consists of a predicted 12 or 14 β-strands and functions as a monomer (49). Although this architecture would result in a transmembrane barrel with a smaller diameter than OmpF and PhoE, it is believed that the antiparallel strands within the channel walls must pack together with appropriate geometry to create a pore with sufficient size to allow oligosaccharide conductance (49). In addition to these macromolecular distinctions, KdgM is believed to gate its entrance with loop 6 (49). Based upon these observations, KdgM (and homologs) has been classified as a unique porin family. Recently, a second oligogalacturonide porin, KdgN, was discovered in E. chrysanthemi (17, 49). In contrast to the case for vast majority of pectin utilization genes, the KdgN gene is not regulated by KdgR, suggesting that it may be produced constitutively during saprogenesis (17). The presence of four paralogous KdgM porins (KdgM1 to -4) in E. carotovora and two in Y. enterocolitica further complicates the biological process of polygalacturonate outer membrane transport (56). Three-dimensional structure determination is required to establish the mechanisms of solute recognition and passage through these channels, which will help identify any functional distinctions between them.
PERIPLASMIC PECTIN DEGRADATION
Although there is strong evidence for the targeted secretion of pectinases into either the periplasm or the extracellular space, the mechanisms behind such selectivity are not understood. As described above, there are noticeable differences in enzyme class (PL, GH, or CE), mode of activity (endo or exo), and isozyme redundancy between species when the two environments are compared (56) (Table 1). What is clear from this comparison is that within the pectinolytic Enterobacteriaceae the greatest collection of extracellular secreted pectinases is found in Erwinia spp., whereas all identified species have some collection of periplasmic enzymes (Table 1). Indeed, beyond Erwinia spp., only Y. enterocolitica contains an extracellular pectinase, the CE PemA/YeCE8. In comparison, there are numerous examples of species producing periplasmic enzymes with overlapping functions, including E. coli, Salmonella enterica serovar Typhimurium, and Klebsiella pneumoniae. This observation suggests that the members of the Enterobacteriaceae that are not dedicated phytopathogens depend upon upstream pectin and polygalacturonate processing by symbiotic microflora. All of these species with abridged pectin degradation pathways can be gastrointestinal pathogens, and it is plausible that the resident microecology enables them to occupy a niche scavenging available pectic fragments.
Periplasmic pectin degradation can be grouped into three main stages: substrate accumulation and retention (SghX/CBM32), substrate depolymerization (PelP/PL2A, PelX/PL9, and PehV-X/exoGH28), and intracellular transport (TogMNAB and TogT). We have recently characterized the three-dimensional structures and mechanisms of substrate/ligand selectivity in each soluble protein involved in this process within Y. enterocolitica. As these are the only structures for periplasmic pectin utilization proteins currently available for Enterobacteriaceae, we will focus our discussion in the following section to these select examples.
Periplasmic Accumulation of Polygalacturonate Involves a Specialized Polygalacturonate Binding Protein
A family 32 CBM originally classified as SghX (56) is found in a number of Enterobacteriaceae genomes, including those of E. carotovora (EcaCBM32) and Y. enterocolitica (SghX/YeCBM32), and also in V. vulnificus (VvCBM32) from the Vibrionaceae. CBMs are defined as modular components of carbohydrate-active enzymes that bind target carbohydrates and recruit the appended catalytic module it to its substrate. Interestingly, family 32 CBMs display one the greatest examples of structural and functional diversity ever reported for this class of proteins (4). For example, the CBM32s involved in pectin utilization lack an appended catalytic module and operate in an independent fashion (5). This phenomenon is rarely documented within the literature for CBMs. The genomic organizations of SghX/YeCBM32 and its homologs suggest that these proteins have coevolved with a periplasmic lyase (PelP/PL2A), and not surprisingly, their functions appear to be causally linked (see below).
SghX/YeCBM32 binds oligogalacturonides (di- and trisaccharides) weakly and prefers highly polymerized forms of polygalacturonate, with a footprint of approximately 1 CBM to 10 galacturonate residues (5). Although, this observation may simply be explained by the presence of numerous subsites, the process may be more complex, with polygalacturonate binding involving a cooperative mechanism consisting of a high-affinity binding site and a low-affinity binding site. Although the two-site binding model is still poorly understood, it would explain the preference of the protein for highly polymerized polygalacturonate, as the proteins may require longer chains for the partnered CBM32s to tandemly bind the polysaccharide.
SghX/YeCBM32 displays a β-sandwich fold connected with a jelly roll topology (Fig. 4A). This is the most common fold for CBMs in general (11). The SghX/YeCBM32 β-sandwich consists of one sheet containing five antiparallel β-strands opposed by a second sheet with three antiparallel β-strands (5). There is a structural Ca2+ ion that does not appear to have any direct role in ligand recognition. Analysis of the electrostatic potentials of the SghX/YeCBM32 binding site revealed a prominent basic charge patch, which was proposed to form salt bridges with the polygalacturonate backbone (5). To test this hypothesis, site-directed mutagenesis was performed on K22, H24, R37, K65, and R69 and evaluated by affinity gel electrophoresis (Fig. 4C and D). Clearly, each of these amino acids contributes differently to polygalacturonate binding, as single mutations in K22, H24, and K65 had little effect, whereas mutation of R37 and double mutations had substantial effects. Importantly, this analysis is the first report of a CBM utilizing electrostatic interactions to bind its ligand.
FIG. 4.

The periplasmic polygalacturonic acid binding protein SghX/YeCBM32. (A) SghX/YeCBM32 (PDB ID, 2JDA) is displayed in a “cartoon” format with a transparent solvent-accessible surface. The structural calcium is shown as a sphere in magenta. (B) Binding site of SghX/YeCBM32 displaying the basic amino acids potentially involved in ligand recognition. (C) Native acrylamide gel electrophoresis of SghX/YeCBM32 mutants. Mutant protein was produced as described previously (5, 6), and ∼5 μg of purified SghX/YeCBM32 protein was electrophoresed through 10% acrylamide gels in the presence and absence of 0.1% polygalacturonate purified from citrus fruit at 100 V for 3.5 h. Lanes: 1, bovine serum albumin control; 2, wild type; 3, ΔK22A; 4, ΔH24A; 5, ΔK22A/H24A; 6, ΔR37A; 7, ΔK65A; 8, ΔK65A/R69A. (D) Polygalacturonate acrylamide (0.1%) gel electrophoresis of SghX/YeCBM32 mutants. Lanes are loaded in the same order as in panel C.
Although the role of SghX/YeCBM32 in pectin utilization has not been demonstrated in a biological system, we have proposed that it functions to bind and retain polygalacturonate within the periplasm as a substrate for downstream depolymerases. This model is based upon several factors. (i) SghX/YeCBM32 is predicted to be secreted into the periplasmic compartment. (ii) Pectic fragments enter the periplasm by facilitated diffusion through KdgM. In the absence of a retention mechanism, the substrate could diffuse back in the extracellular environment when the levels of carbohydrates are limiting. (iii) SghX/YeCBM32 has an affinity for polygalacturonate that is 2 to 3 orders of magnitude higher than that of small oligogalacturonides, which are the products of periplasmic pectinases. (iv) SghX/YeCBM32 does not have inherent catalytic activity and therefore likely operates in a combinatorial fashion with other proteins. Support of this model would benefit from a testable biological system and the elucidation of an SghX/YeCBM32-oligogalacturonide complex, which has proven elusive.
A Periplasmic Pel with a Rare Fold
Within the periplasm of pectinolytic Enterobacteriaceae there are three major classes of pectinases: endo-Pels (PelP), exo-Pels (PelX), and exo-GHs (PehV-X) (Table 1). As with the extracellular proteins in this pathway, the genetic repertoire of these enzymes varies between species. For example, only the genomes from E. carotovora and Yersinia spp. encode the family 2 lyase, PelP/PL2A (56). The activity of the Y. enterocolitica enzyme (PelP/YePL2A) was originally characterized by biochemical methods (5). Recently, we have provided structural evidence for its mode of activity and catalysis, including a β-elimination mechanism that utilizes a novel metal cofactor (3).
The overall structure of PelP/YePL2A is “vise-like” in shape (Fig. 5A). The core of the protein is an (α/α)7 barrel that has previously been reported only for family 47 α-1,2 mannosidases, GHs involved in glycoprotein folding quality control within the Golgi apparatuses of eukaryotes (39, 67, 69, 71). In PelP/YePL2A there are two arms, consisting primarily of β-strands, that harbor the catalytic machinery grafted onto the (α/α)7 platform. The active-site cleft running between these two arms spans the length of the enzyme (∼50 Å). This observation is consistent with its endo mode of activity that was defined by high-performance liquid chromatography with amperometric detection product profiling (3, 42). Kinetic analysis demonstrated that PelP/YePL2A is more active on polygalacturonate than on trigalacturonate and produces predominantly di- and trisaccharides following extensive digestion (3).
FIG. 5.

The periplasmic endo-Pel PelP/YePL2A. (A) PelP/YePL2A (PDB ID, 2V8J) is displayed in a “cartoon” format with a transparent solvent-accessible surface. The catalytic Mn2+ is shown as a light blue sphere. (B) Superimposition of the metal (green) and trigalacturonate (yellow) (PDB ID, 2V8K) complexes. Enzymes are rendered in a ribbon format and the substrate as sticks in beige. (C) Superimposition of the active sites from family 2 PelP/YePL2A and family 1 PelC/EchPL1C Pels displayed in wall-eyed format. The Mn2+ coordination pocket from the metal complex has been introduced for reference. The Mn2+ is shown in blue and the Ca2+ from PelC/EchPL1C in magenta.
In addition to its apo form, the structures of PelP/YePL2A in complex with a trigalacturonate substrate and transition metal were also determined. There are notable changes in the conformation of the catalytic arms within these two complexes, which reflects the inherent flexibility in these regions and may illuminate macromolecular details about its catalytic mechanism or processivity (Fig. 5B) (3). Structural analysis of the active sites within the two complexes enabled comparisons with previously described Pels (Fig. 5C). The intact trisaccharide is positioned with its reducing end in the +1 position and its nonreducing sugar in the −2 position. The functional Brønstead base (R171) is positioned within 2.9 Å of the H-5 targeted for abstraction with appropriate geometry. The enzyme is not active, however, as the complex did not crystallize in the presence of a cofactor. In addition to the catalytic base, a second arginine also interacts with the aglycon group. R272 is in hydrogen bond contact with O-2 and O-3, which contributes to substrate recognition and orientation. Surprisingly, analysis of the proximal subsite architecture failed to uncover many other interactions, which may be due to the enzyme not being in an active state.
Crystallization of PelP/YePL2A in a condition that lacked any chelating buffer generated a model with a bound cofactor. Based upon inductively coupled plasma mass spectrometry analysis and strong structural evidence, the metal cofactor was determined to be an Mn2+ or Ni2+ ion (3). This observation is consistent with metal supplementation assays of the closely related cytoplasmic paralog PelW/YePL2B (3) and ortholog PelW/EchPel2B (61). The metal-PelP/YePL2A complex documented the first Pel structure containing a metal cofactor other than calcium. Although the role of calcium in β-elimination is well established, any unique chemical contributions of the transition metal to the mechanism are unknown. The elucidation of a metal-oligogalacturonide-PelP/YePL2A complex would be very helpful in this regard.
Superimposition of the active sites from PelP/YePL2A and PelC/EchPL1C, two Pels with unrelated amino acid sequences and distinct folds, reveals that there is structural conservation of the Brønstead base, the metal coordination pocket, and a substrate-stabilizing interaction within the −1 subsite (Fig. 5C) (3). Previously, comparisons between a family 1 Pel and a family 10 Pel from Cellvibrio japonicus (CjPL10) also led to similar conclusions (13). This suggests that the β-elimination of polygalacturonate is dependent upon a strict catalytic framework (3, 13). Elucidation of active-site architectures from new Pel families in the future will likely benefit from this observation.
Structural Basis of Exopolygalacturonase Activity
There are only two species within Enterobacteriaceae that contain genes for periplasmic exopolygalacturonases: E. chrysanthemi (three isozymes, PehV to -X/EchGH28V to -X) and Y. enterocolitica (PehX/YeGH28). The higher ratio of exo-acting enzymes within the periplasm is consistent with the functional roles of these proteins (40, 56). Exopolygalacturonases exclusively produce digalacturonides by attacking the reducing ends of small pectic fragments, such as those that would be abundant within this cellular compartment (42, 64). Although there is an abundance of endopolygalacturonase structures available within the structural database for both bacteria (51) and fungi (14, 22, 50, 65, 72, 73), there is only one structure of an exopolygalacturonase currently known, that of PehX/YeGH28 (2).
The core of PehX/YeGH28 is structurally similar to the endopolygalacturonase from E. carotovora described above (Fig. 6A). It adopts a conventional right-handed parallel β-helix topology with 10 complete turns. Apart from this core scaffold, however, there are several distinct structural features within the exoenzyme (1). Most noticeably, there is an N-terminal FN3 domain comprised of ∼140 amino acids that is fused to the β-sheet on the opposite side of the protein to the active site. Using Dali structure alignments (27), this domain shows the most similarity to the human fibronectin binding protein (41). The function of FN3 domains in carbohydrate utilization remains a mystery. Currently there is virtually no functional evidence for their activity, despite the fact that they are commonly observed domains in carbohydrate-active enzymes (2). There are four loop insertions that cluster near the active site and function to close off one end of the active site. These loops are the structural determinants that transform the enzyme into an exopolygalacturonase (see below) (3). There are a unique β-sheet and α-helix encoded approximately halfway through the polypeptide that appear to stabilize the active-site loops and FN3 domain, respectively.
FIG. 6.
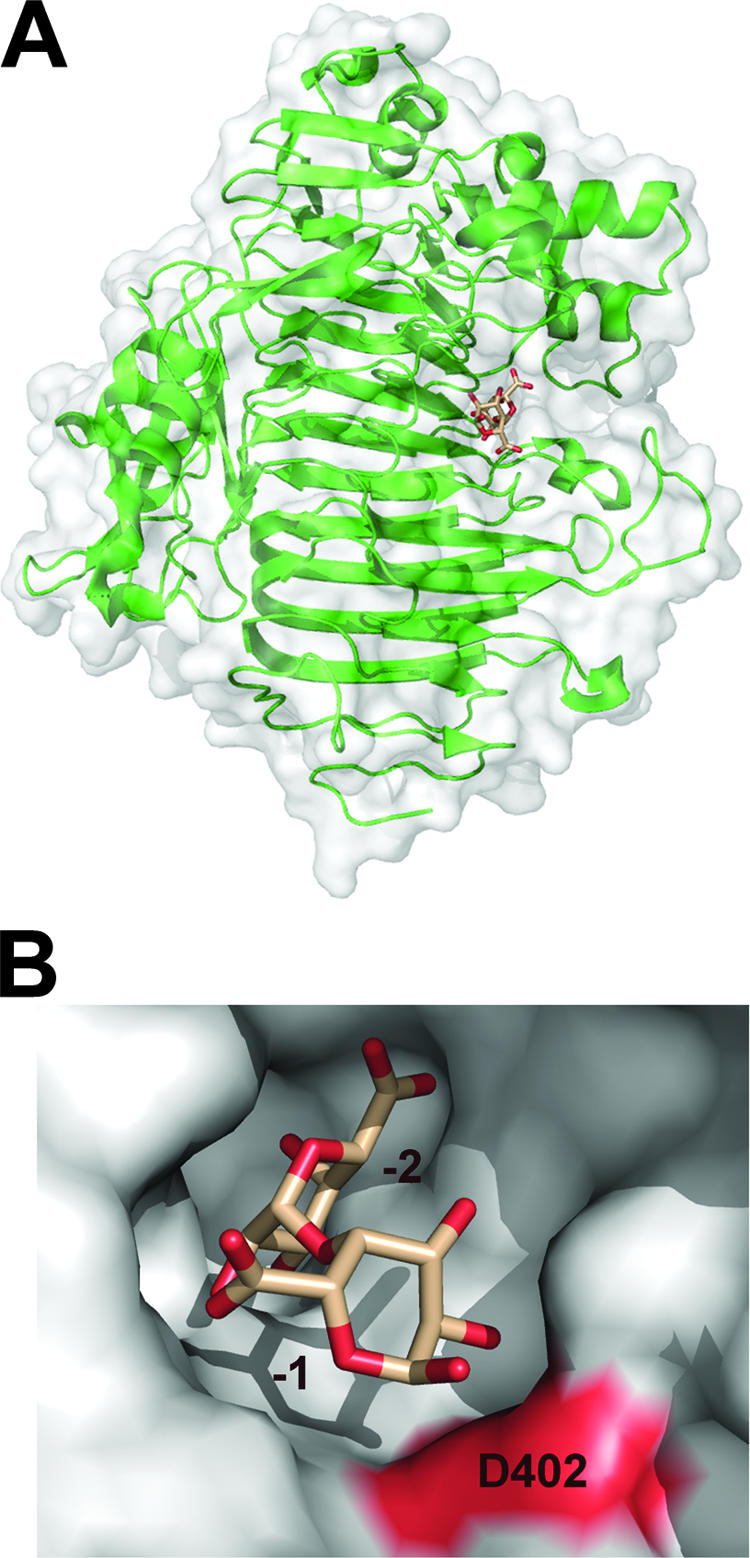
The periplasmic exopolygalacturonase PehX/YeGH28. (A) PehX/YeGH28 in complex with digalacturonate (PDB ID, 2UVF) is displayed in a “cartoon” format with a transparent solvent-accessible surface and the disaccharide in beige. (B) The active-site surface of the exopolygalacturonase is shown with its two accessible subsites (−1 and −2). The putative catalytic acid D402 is shown in red.
Analysis of the PehX/YeGH28 active site shows a stringent conservation of the three catalytic aspartate residues (D381, D402, and D403) characterized in PehA/EcaGH28A (D202, D223, and D224) (Fig. 2C). All three PehX/YeGH28 residues are within 5 Å of one another. The elucidation of PehX/YeGH28 in complex with a digalacturonide product enables the analysis of the protein-product interactions and subsite architecture. D402 is positioned 3.3 Å from the scissile glycosidic oxygen with appropriate geometry, supporting its potential role as the catalytic acid. The two putative catalytic bases are more distal to the cleavage site but in satisfactory position for charging the nucleophilic water. Interestingly, there were no other direct contacts between the monosaccharide and the enzyme in position −1. This observation may explain how the product is replaced within the active site by incoming substrate. Subsite −2 has stabilizing hydrogen bonds between R440 and N406 with the substituent O-2 and O-3, an electrostatic interaction between R240 and the uronate group, and a hydrogen bond between H355 and the uronate group (not shown). There were no reported molecular contacts between the protein and the C-4 hydroxyl, a stereochemical signature of galacto-configured sugars, which supports the observation that the enzyme is active on both saturated and 4,5-unsaturated oligogalacturonides (30).
The structure of PehX/YeGH28 beautifully illustrates the macromolecular determinants of the exclusive exo activity of the enzyme (Fig. 6B). As described above, when viewed down the active site the endopolygalacturonase homolog PehA/EchGH28A is a cleft that is open at both ends (Fig. 2B). Analogous to the activity of PelP/YePL2A described above, this configuration is designed for the attack of internal residues within a polygalacturonate chain. In contrast, the active-site cleft of PehX/YeGH28 resembles a pocket with one end sealed off by the inserted loop structures (Fig. 6B). The configurations of these loops within the enzyme restrict the substrate access to only two subsites (−1 and −2) and satisfactorily positions the scissile bond for hydrolysis. This topography explains structurally how the enzyme exclusively generates digalacturonide products during catalytic turnover.
Intracellular Transport Is an Active and Selective Process
The end result of extracellular and periplasmic pectin de-esterification and polygalacturonate depolymerization is the production of small (di- and tri-) oligogalacturonides (33, 54). The ultimate goal for these carbohydrates is to be passaged into the cytoplasm where intracellular metabolic pathways dedicated for the catabolism of both saturated and unsaturated oligogalacturonides converge to produce KDG (57). This molecule is the inducer molecule for the KdgR repressor and a substrate for the downstream generation of pyruvate and 3-phosphoglyceraldehyde. In this light, the inner membrane constitutes the final obstacle to the energy-harvesting stages of pectin utilization. Inner membrane transport is facilitated by four distinct transporters: ExuT, a symporter specific for monogalacturonate; KdgT a symporter specific for 5-keto-4-deoxyuronate, 2,5-di-keto-3-deoxygluconate, and KDG; and TogT and TogMNAB, which are specific for oligogalacturonides (1, 18, 29, 31, 32, 58). Of these four independent systems, TogMNAB is the most prominent transport mechanism during pectinolysis, as the majority of monosaccharides are generated within the cytoplasm by the disaccharide oligogalacturonate lyase, Ogl (30, 33).
TogB, the ligand selectivity determinant for the TogMNAB transporter, is the only example of a comprehensive structure-function analysis being available for hexuronate binding and transport (1). ABC transporters have an archetypical ultrastructure consisting of two transmembrane domains (TogM and TogN), two cytoplasmic ATPase domains (TogA2), and the periplasmic specificity determinant (TogB). This class of transporter couples the energy of ATP hydrolysis to the accumulation of oligogalacturonides within the cell. Depletion of periplasmic oligogalacturonide pools by intracellular transport is critical to prevent escape of the metabolites back through the KdgM porin, as SghX/YeCBM32 has low affinity for these smaller ligands.
There are many deposited structures of periplasmic binding domains within the database. Consistent with these other proteins, the TogB polypeptide from Y. enterocolitica contains two globular domains connected through a three-stranded hinge region (Fig. 7A). The C terminus of the protein is noticeably larger than the N-terminal portion of the protein and contains an extensive loop that navigates back into and out of the N-terminal region. In its unliganded state, TogB is an “open” conformation with its binding site exposed between the two domains (Fig. 7A). Following ligand binding, its two domains snap closed about the hinge region, in what is reminiscent of a “Venus flytrap”-like mechanism (Fig. 7B). This isomerization proceeds along a two-order coordinate, with transitions in a hinge and twist vector (1, 7, 54, 68). In its “closed” state, the ligand is completely inaccessible to bulk solvent (Fig. 7B).
FIG. 7.
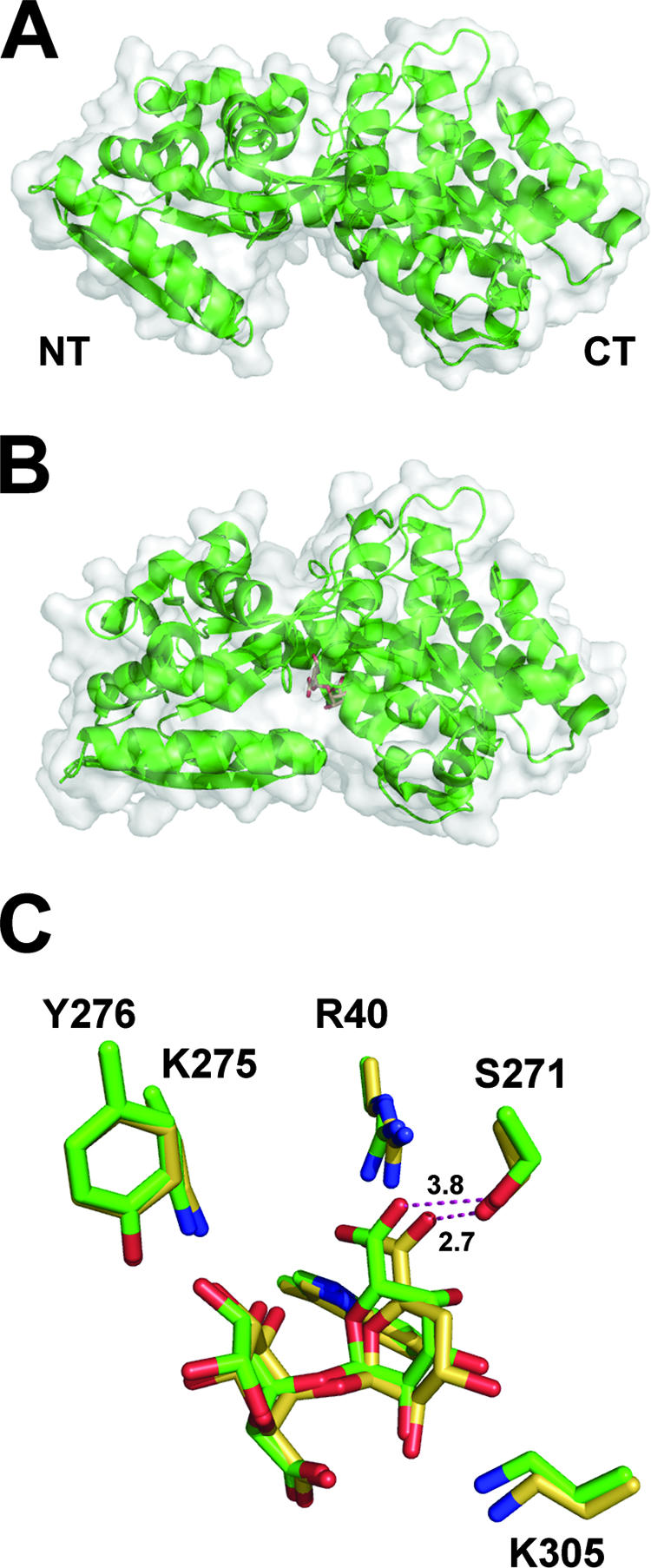
TogB, the periplasmic solute binding component of the oligogalacturonide transporter TogMNAB from Y. enterocolitica. (A) TogB (PDB ID, 2UVG) is displayed in a “cartoon” format with a transparent solvent-accessible surface. (B) TogB in complex with 4,5-unsaturated digalacturonate (PDB ID, 2UVI) displayed in a “cartoon” format with a transparent solvent-accessible surface in the same orientation as in panel A to demonstrate the large conformational change induced upon binding. (C) Superimposition on the ligands within the saturated (PDB ID, 2UVH) and 4,5-unsaturated digalacturonate complexes. The distances between the uronate groups and S271 are shown for the saturated ligand (3.8 Å) and the unsaturated ligand (2.7 Å).
Complementary biophysical techniques established a clear hierarchy in oligogalacturonide binding affinities: trigalacturonate < saturated digalacturonate < 4,5-unsaturated digalacturonate (1). Favorable enthalpic contributions were observed for complex formation with the unsaturated ligand compared to its saturated counterpart (∼5 kcal mol−1), which is approximately equivalent to the formation of one extra hydrogen bond. The observed ligand binding profile for TogB is in agreement with the most likely oligogalacturonide mixture present within the periplasmic compartment during active pectinolysis. Indeed, 4,5-unsaturated digalacturonate is generated by both endo-acting PelP/PL2A (3, 42) and PelX/EchPL9X and exo-acting PehX/exoGH28 polygalacturonases active upon substrates with an unsaturation at the nonreducing end (3, 42, 64), and there are no enzymes known that degrade digalacturonides within the periplasm. The weak affinity of TogB for trigalacturonate is also expected, as this carbohydrate is a substrate for further depolymerization reactions.
The analysis of TogB-oligogalacturonide complexes provides a structural explanation for the binding thermodynamics explained above. Within the binding site there are three subsites. The first two are well designed to accommodate digalacturonide binding (Fig. 7C). The third subsite tolerates trisaccharide occupancy by distorting two amino acids, Y276 and E187. A key interaction that drives hexuronate specificity is a salt bridge between R40 and the uronate of the nonreducing sugar in subsite 1. This interaction is complemented by a constellation of hydrogen bonds between the ligand and K305 and W35 in subsite 1 and K275 and Y276 in subsite 2. In addition, there are two stacking interactions between W269 and W67 and the planar faces of the ligand in subsites 1 and 2, respectively.
Closer analysis of the oligogalacturonide structures provides a structural explanation for the molecular determinant of binding selectivity between the two disaccharide species. The 4,5-unsaturation causes the pyranose ring to adopt a partially planar configuration. This structural transformation induces the formation of a novel hydrogen bond (2.7 Å) between S271 and the uronate group in subsite 1 (Fig. 7C). In contrast, the saturated disaccharide uronate oxygen is too far from S271 (3.8 Å) to interact. The energetic contributions of this exclusive interaction are in excellent agreement with the observed increase in binding enthalpy for the unsaturated ligand described above (∼5 kcal mol−1).
MODEL OF PECTIN DEGRADATION IN ENTEROBACTERIACEAE
This comprehensive structural review of pectinases and oligogalacturonide binding proteins from Enterobacteriaceae has provided a model for the conversion of extracellular pectin into intracellular digalacturonide catabolites. The process begins in the extracellular environment, spans the periplasm, and culminates in the cytoplasm of the bacterial cell (Fig. 8).
FIG. 8.

Structural biology of pectin degradation and transport within Enterobacteriaceae. The major stages of extracellular and periplasmic pectin utilization are shown. (1) Methylated polygalacturonate is de-esterified by PemA/EchCE8A (violet). Methoxyl groups are indicated by open circles. Extracellular depolymerization reactions occur predominantly by endo-acting enzymes. These reactions can occur by either a hydrolysis mechanism as shown for PehA/EcaGH28A (green) (2) or β-elimination by the Pels PelC/EchPL1C (red), PelA/EchPL1A (light purple), and PelL/EchPL9A (teal) (3). (4) The products of these reactions enter the periplasm by facilitated diffusion through the porin KdgM. (5) Retention of substrates within the periplasm is facilitated by SghX/YeCBM32 (yellow). Periplasmic depolymerizations are catalyzed by the endo-Pel PelP/YePL2A (blue) (6) or exopolygalacturonase PehX/YeGH28 (gray) (7). Oligogalacturonate products are bound by the TogB periplasmic binding protein (orange) and directed to the TogMNA components of the ABC transporter (8), where they are shuttled across the inner membrane in an ATP-coupled reaction (9).
Pectin utilization begins with endo-acting enzymes, such as the Pels PelA/EchPL1A and PelC/EchPL1C, being secreted into the extracellular environment. These pectinases attack polymerized pectin within the plant cell wall and liberate pectic fragments into solution. Accompanying these events, inhibitory esters such as uronate methoxyl groups are removed by secreted CEs, which include PemA (EchCE8A and YeCE8). De-esterification can occur on both sides of the outer membrane, as some pectinolytic species contain periplasmic CEs. The structural biology of pectin acetyl de-esterification within the Enterobacteriaceae remains to be explored. Processed pectic fragments enter the periplasm by facilitated diffusion through anion-specific porins of the KdgM family. Retention and accumulation of these carbohydrates within the periplasm are likely enabled by the polygalacturonate binding protein SghX/CBM32. Periplasmic depolymerization is catalyzed by resident Pels and family 28 GHs, the majority of which utilize an exo mode of activity. End products from these reactions include saturated and 4,5-unsaturated digalacturonate and trigalacturonate. Subsequent intracellular transport of these catabolites is facilitated in a hierarchical fashion by the ATP-dependent transporter TogMNAB.
The roles of two different classes of galacturonate-configured carbohydrate binding proteins, i.e., SghX/YeCBM32, a polygalacturonate binding protein, and TogB, a digalacturonate binding protein, suggest that catabolite flow occurs in what might best be described as a “pulling” mechanism. First, the retention of polygalacturonate within the periplasm by SghX/YeCBM32 draws substrates from the immediate environment and presents them to resident depolymerases. This process helps to keep the equilibrium shifted toward polygalacturonate flow through the KdgM porin and periplasmic accumulation. Similarly, the activity of TogB, in cooperation with other inner membrane transporters, TogT, KdgT, and ExuT, depletes the periplasmic product pools of upstream depolymerization reactions. This process keeps the equilibrium shifted toward substrate accumulation and prevents product inhibition of the periplasmic depolymerases.
FUTURE PERSPECTIVES
Although there is now ample structural information relating to both extracellular and periplasmic pectin utilization and transport, there are several major outstanding structural questions within this field. (i) What are the molecular determinants of hexuronate transmembrane passage? The three-dimensional structures and conductance mechanisms for the outer membrane porin KdgM (and its homologs) and the inner membrane transporters TogMN, TogT, and ExuT remain to be established. These transport systems are a potential repository of structural information in general for anion-specific transport and in some cases represent completely new families of transporters. (ii) Can pectin de-esterification, polygalacturonate hydrolysis, and β-elimination be inhibited? The architecture of many active sites and the basic mechanisms of catalysis are now understood, as are the enzymes that catalyze these distinct reactions. Despite this information, there has been no reported progress toward their inhibition by small molecules. Beyond the contributions that this would have for basic science, this development of mimetics would have enormous impact on the control of soft rot infection and food crop spoilage (3). The structural biology of intracellular pectin utilization is an unexplored field. The generation of products such as KDG has implications for the entire pathway, as this molecule controls the expression of virtually every gene involved in pectin utilization.
Although major milestones toward understanding the process of pectin degradation at the structural level have been established during the last 15 years, the future within this field likely holds many more exciting answers to difficult questions. The ability to perturb the regulation of gene expression and the activities of enzymes involved in pathogenic pectin utilization shows a promising course toward controlling pectinolytic activities of phytopathogens from the Enterobacteriaceae.
Acknowledgments
We declare that we have no competing financial interest or conflict of interest.
REFERENCES
- 1.Abbott, D. W., and A. B. Boraston. 2007. Specific recognition of saturated and 4,5-unsaturated hexuronate sugars by a periplasmic binding protein involved in pectin catabolism. J. Mol. Biol. 369759-770. [DOI] [PubMed] [Google Scholar]
- 2.Abbott, D. W., and A. B. Boraston. 2007. The structural basis for exopolygalacturonase activity in a family 28 glycoside hydrolase. J. Mol. Biol. 3681215-1222. [DOI] [PubMed] [Google Scholar]
- 3.Abbott, D. W., and A. B. Boraston. 2007. A family 2 pectate lyase displays a rare fold and transition metal-assisted beta-elimination. J. Biol. Chem. 28235328-35336. [DOI] [PubMed] [Google Scholar]
- 4.Abbott, D. W., J. M. Eirin-Lopez, and A. B. Boraston. 2008. Insight into ligand diversity and novel biological roles for family 32 carbohydrate-binding modules. Mol. Biol. Evol. 25155-167. [DOI] [PubMed] [Google Scholar]
- 5.Abbott, D. W., S. Hrynuik, and A. B. Boraston. 2007. Identification and characterization of a novel periplasmic polygalacturonic acid binding protein from Yersinia enterocolitica. J. Mol. Biol. 3671023-1033. [DOI] [PubMed] [Google Scholar]
- 6.Barik, S. 1996. Site-directed mutagenesis in vitro by megaprimer PCR. Methods Mol. Biol. 57203-215. [DOI] [PubMed] [Google Scholar]
- 7.Bjorkman, A. J., and S. L. Mowbray. 1998. Multiple open forms of ribose-binding protein trace the path of its conformational change. J. Mol. Biol. 279651-664. [DOI] [PubMed] [Google Scholar]
- 8.Blot, N., C. Berrier, N. Hugouvieux-Cotte-Pattat, A. Ghazi, and G. Condemine. 2002. The oligogalacturonate-specific porin KdgM of Erwinia chrysanthemi belongs to a new porin family. J. Biol. Chem. 2777936-7944. [DOI] [PubMed] [Google Scholar]
- 9.Boccara, M., and V. Chatain. 1989. Regulation and role in pathogenicity of Erwinia chrysanthemi 3937 pectin methylesterase. J. Bacteriol. 1714085-4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reference deleted.
- 11.Boraston, A. B., D. N. Bolam, H. J. Gilbert, and G. J. Davies. 2004. Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem. J. 382769-781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bouley, J., G. Condemine, and V. E. Shevchik. 2001. The PDZ domain of OutC and the N-terminal region of OutD determine the secretion specificity of the type II out pathway of Erwinia chrysanthemi. J. Mol. Biol. 308205-219. [DOI] [PubMed] [Google Scholar]
- 13.Charnock, S. J., I. E. Brown, J. P. Turkenburg, G. W. Black, and G. J. Davies. 2002. Convergent evolution sheds light on the anti-beta-elimination mechanism common to family 1 and 10 polysaccharide lyases. Proc. Natl. Acad. Sci. USA 9912067-12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cho, S. W., S. Lee, and W. Shin. 2001. The X-ray structure of Aspergillus aculeatus polygalacturonase and a modeled structure of the polygalacturonase-octagalacturonate complex. J. Mol. Biol. 311863-878. [DOI] [PubMed] [Google Scholar]
- 15.Reference deleted.
- 16.Condemine, G., C. Dorel, N. Hugouvieux-Cotte-Pattat, and J. Robert-Baudouy. 1992. Some of the out genes involved in the secretion of pectate lyases in Erwinia chrysanthemi are regulated by kdgR. Mol. Microbiol. 63199-3211. [DOI] [PubMed] [Google Scholar]
- 17.Condemine, G., and A. Ghazi. 2007. Differential regulation of two oligogalacturonate outer membrane channels, KdgN and KdgM, of Dickeya dadantii (Erwinia chrysanthemi). J. Bacteriol. 1895955-5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Condemine, G., and J. Robert-Baudouy. 1987. 2-Keto-3-deoxygluconate transport system in Erwinia chrysanthemi. J. Bacteriol. 1691972-1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coutinho, P. M., and B. Henrissat. 1999. Carbohydrate-active enzymes: an integrated database approach., p. 3-12. In G. D. H. J. Gilbert, B. Henrissat, and B. Svensson (ed.), Recent advances in carbohydrate bioengineering. The Royal Society of Chemistry, Cambridge, United Kingdom.
- 20.Cowan, S. W., T. Schirmer, G. Rummel, M. Steiert, R. Ghosh, R. A. Pauptit, J. N. Jansonius, and J. P. Rosenbusch. 1992. Crystal structures explain functional properties of two E. coli porins. Nature 358727-733. [DOI] [PubMed] [Google Scholar]
- 21.Dehdashti, S. J., C. N. Doan, K. L. Chao, and M. D. Yoder. 2003. Effect of mutations in the T1.5 loop of pectate lyase A from Erwinia chrysanthemi EC16. Acta Crystallogr. D 591339-1342. [DOI] [PubMed] [Google Scholar]
- 22.Federici, L., C. Caprari, B. Mattei, C. Savino, A. Di Matteo, G. De Lorenzo, F. Cervone, and D. Tsernoglou. 2001. Structural requirements of endopolygalacturonase for the interaction with PGIP (polygalacturonase-inhibiting protein). Proc. Natl. Acad. Sci. USA 9813425-13430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fries, M., J. Ihrig, K. Brocklehurst, V. E. Shevchik, and R. W. Pickersgill. 2007. Molecular basis of the activity of the phytopathogen pectin methylesterase. EMBO J. 263879-3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grasdalen, H., A. K. Andersen, and B Larsen. 1996. NMR spectroscopy studies of the action pattern of tomato pectinesterase: generation of block structure in pectin by a multiple-attack mechanism. Carbohydr. Res. 289105-114. [Google Scholar]
- 25.Guillen Schlippe, Y. V., and L. Hedstrom. 2005. A twisted base? The role of arginine in enzyme-catalyzed proton abstractions. Arch. Biochem. Biophys. 433266-278. [DOI] [PubMed] [Google Scholar]
- 26.Herron, S. R., J. A. Benen, R. D. Scavetta, J. Visser, and F. Jurnak. 2000. Structure and function of pectic enzymes: virulence factors of plant pathogens. Proc. Natl. Acad. Sci. USA 978762-8769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holm, L., and C. Sander. 1993. Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233123-138. [DOI] [PubMed] [Google Scholar]
- 28.Reference deleted.
- 29.Hugouvieux-Cotte-Pattat, N., N. Blot, and S. Reverchon. 2001. Identification of TogMNAB, an ABC transporter which mediates the uptake of pectic oligomers in Erwinia chrysanthemi 3937. Mol. Microbiol. 411113-1123. [DOI] [PubMed] [Google Scholar]
- 30.Hugouvieux-Cotte-Pattat, N., G. Condemine, W. Nasser, and S. Reverchon. 1996. Regulation of pectinolysis in Erwinia chrysanthemi. Annu. Rev. Microbiol. 50213-257. [DOI] [PubMed] [Google Scholar]
- 31.Hugouvieux-Cotte-Pattat, N., Y. Quesneau, and J. Robert-Baudouy. 1983. Aldohexuronate transport system in Erwinia carotovora. J. Bacteriol. 154663-668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hugouvieux-Cotte-Pattat, N., and S. Reverchon. 2001. Two transporters, TogT and TogMNAB, are responsible for oligogalacturonide uptake in Erwinia chrysanthemi 3937. Mol. Microbiol. 411125-1132. [DOI] [PubMed] [Google Scholar]
- 33.Hugouvieux-Cotte-Pattat, N., and J. Robert-Baudouy. 1987. Hexuronate catabolism in Erwinia chrysanthemi. J. Bacteriol. 1691223-1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jenkins, J., O. Mayans, and R. Pickersgill. 1998. Structure and evolution of parallel beta-helix proteins. J. Struct. Biol. 122236-246. [DOI] [PubMed] [Google Scholar]
- 35.Jenkins, J., O. Mayans, D. Smith, K. Worboys, and R. W. Pickersgill. 2001. Three-dimensional structure of Erwinia chrysanthemi pectin methylesterase reveals a novel esterase active site. J. Mol. Biol. 305951-960. [DOI] [PubMed] [Google Scholar]
- 36.Jenkins, J., and R. Pickersgill. 2001. The architecture of parallel beta-helices and related folds. Prog. Biophys. Mol. Biol. 77111-175. [DOI] [PubMed] [Google Scholar]
- 37.Jenkins, J., V. E. Shevchik, N. Hugouvieux-Cotte-Pattat, and R. W. Pickersgill. 2004. The crystal structure of pectate lyase Pel9A from Erwinia chrysanthemi. J. Biol. Chem. 2799139-9145. [DOI] [PubMed] [Google Scholar]
- 38.Johansson, K., M. El-Ahmad, R. Friemann, H. Jornvall, O. Markovic, and H. Eklund. 2002. Crystal structure of plant pectin methylesterase. FEBS Lett. 514243-249. [DOI] [PubMed] [Google Scholar]
- 39.Karaveg, K., A. Siriwardena, W. Tempel, Z. J. Liu, J. Glushka, B. C. Wang, and K. W. Moremen. 2005. Mechanism of class 1 (glycosylhydrolase family 47) α-mannosidases involved in N-glycan processing and endoplasmic reticulum quality control. J. Biol. Chem. 28016197-16207. [DOI] [PubMed] [Google Scholar]
- 40.Kazemi-Pour, N., G. Condemine, and N. Hugouvieux-Cotte-Pattat. 2004. The secretome of the plant pathogenic bacterium Erwinia chrysanthemi. Proteomics 43177-3186. [DOI] [PubMed] [Google Scholar]
- 41.Leahy, D. J., I. Aukhil, and H. P. Erickson. 1996. 2.0 A crystal structure of a four-domain segment of human fibronectin encompassing the RGD loop and synergy region. Cell 84155-164. [DOI] [PubMed] [Google Scholar]
- 42.Liao, C. H., L. Revear, A. Hotchkiss, and B. Savary. 1999. Genetic and biochemical characterization of an exopolygalacturonase and a pectate lyase from Yersinia enterocolitica. Can. J. Microbiol. 45396-403. [DOI] [PubMed] [Google Scholar]
- 43.Lietzke, S. E., R. D. Scavetta, M. D. Yoder, and F. Jurnak. 1996. The refined three-dimensional structure of pectate lyase E from Erwinia chrysanthemi at 2.2 A resolution. Plant Physiol. 11173-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lietzke, S. E., M. D. Yoder, N. T. Keen, and F. Jurnak. 1994. The three-dimensional structure of pectate lyase E, a plant virulence factor from Erwinia chrysanthemi. Plant Physiol. 106849-862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Limberg, G., R. Korner, H. C. Buchholt, T. M. Christensen, P. Roepstorff, and J. D. Mikkelsen. 2000. Quantification of the amount of galacturonic acid residues in blocksequences in pectin homogalacturonan by enzymatic fingerprinting with exo- and endo-polygalacturonase II from Aspergillus niger. Carbohydr. Res. 327321-332. [DOI] [PubMed] [Google Scholar]
- 46.Lindeberg, M., G. P. Salmond, and A. Collmer. 1996. Complementation of deletion mutations in a cloned functional cluster of Erwinia chrysanthemi out genes with Erwinia carotovora out homologues reveals OutC and OutD as candidate gatekeepers of species-specific secretion of proteins via the type II pathway. Mol. Microbiol. 20175-190. [DOI] [PubMed] [Google Scholar]
- 47.Markovic, O., and S. Janecek. 2001. Pectin degrading glycoside hydrolases of family 28: sequence-structural features, specificities and evolution. Protein Eng. 14615-631. [DOI] [PubMed] [Google Scholar]
- 48.Micheli, F. 2001. Pectin methylesterases: cell wall enzymes with important roles in plant physiology. Trends Plant Sci. 6414-419. [DOI] [PubMed] [Google Scholar]
- 49.Pellinen, T., H. Ahlfors, N. Blot, and G. Condemine. 2003. Topology of the Erwinia chrysanthemi oligogalacturonate porin KdgM. Biochem. J. 372329-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Petersen, T. N., S. Kauppinen, and S. Larsen. 1997. The crystal structure of rhamnogalacturonase A from Aspergillus aculeatus: a right-handed parallel beta helix. Structure 5533-544. [DOI] [PubMed] [Google Scholar]
- 51.Pickersgill, R., D. Smith, K. Worboys, and J. Jenkins. 1998. Crystal structure of polygalacturonase from Erwinia carotovora ssp. carotovora. J. Biol. Chem. 27324660-24664. [DOI] [PubMed] [Google Scholar]
- 52.Prasanna, V., T. N. Prabha, and R. N. Tharanathan. 2007. Fruit ripening phenomena—an overview. Crit. Rev. Food Sci. Nutr. 471-19. [DOI] [PubMed] [Google Scholar]
- 53.Pugsley, A. P. 1993. The complete general secretory pathway in gram-negative bacteria. Microbiol. Rev. 5750-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ravindranathan, K. P., E. Gallicchio, and R. M. Levy. 2005. Conformational equilibria and free energy profiles for the allosteric transition of the ribose-binding protein. J. Mol. Biol. 353196-210. [DOI] [PubMed] [Google Scholar]
- 55.Ridley, B. L., M. A. O'Neill, and D. Mohnen. 2001. Pectins: structure, biosynthesis, and oligogalacturonide-related signaling. Phytochemistry 57929-967. [DOI] [PubMed] [Google Scholar]
- 56.Rodionov, D. A., M. S. Gelfand, and N. Hugouvieux-Cotte-Pattat. 2004. Comparative genomics of the KdgR regulon in Erwinia chrysanthemi 3937 and other gamma-proteobacteria. Microbiology 1503571-3590. [DOI] [PubMed] [Google Scholar]
- 57.Rodionov, D. A., A. A. Mironov, A. B. Rakhmaninova, and M. S. Gelfand. 2000. Transcriptional regulation of transport and utilization systems for hexuronides, hexuronates and hexonates in gamma purple bacteria. Mol. Microbiol. 38673-683. [DOI] [PubMed] [Google Scholar]
- 58.San Francisco, M. J., and R. W. Keenan. 1993. Uptake of galacturonic acid in Erwinia chrysanthemi EC16. J. Bacteriol. 1754263-4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scavetta, R. D., S. R. Herron, A. T. Hotchkiss, N. Kita, N. T. Keen, J. A. Benen, H. C. Kester, J. Visser, and F. Jurnak. 1999. Structure of a plant cell wall fragment complexed to pectate lyase C. Plant Cell 111081-1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shevchik, V. E., G. Condemine, N. Hugouvieux-Cotte-Pattat, and J. Robert-Baudouy. 1996. Characterization of pectin methylesterase B, an outer membrane lipoprotein of Erwinia chrysanthemi 3937. Mol. Microbiol. 19455-466. [DOI] [PubMed] [Google Scholar]
- 61.Shevchik, V. E., G. Condemine, J. Robert-Baudouy, and N. Hugouvieux-Cotte-Pattat. 1999. The exopolygalacturonate lyase PelW and the oligogalacturonate lyase Ogl, two cytoplasmic enzymes of pectin catabolism in Erwinia chrysanthemi 3937. J. Bacteriol. 1813912-3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shevchik, V. E., and N. Hugouvieux-Cotte-Pattat. 1997. Identification of a bacterial pectin acetyl esterase in Erwinia chrysanthemi 3937. Mol. Microbiol. 241285-1301. [DOI] [PubMed] [Google Scholar]
- 63.Shevchik, V. E., and N. Hugouvieux-Cotte-Pattat. 2003. PaeX, a second pectin acetylesterase of Erwinia chrysanthemi 3937. J. Bacteriol. 1853091-3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shevchik, V. E., H. C. Kester, J. A. Benen, J. Visser, J. Robert-Baudouy, and N. Hugouvieux-Cotte-Pattat. 1999. Characterization of the exopolygalacturonate lyase PelX of Erwinia chrysanthemi 3937. J. Bacteriol. 1811652-1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shimizu, T., T. Nakatsu, K. Miyairi, T. Okuno, and H. Kato. 2002. Active-site architecture of endopolygalacturonase I from Stereum purpureum revealed by crystal structures in native and ligand-bound forms at atomic resolution. Biochemistry 416651-6659. [DOI] [PubMed] [Google Scholar]
- 66.Tardy, F., W. Nasser, J. Robert-Baudouy, and N. Hugouvieux-Cotte-Pattat. 1997. Comparative analysis of the five major Erwinia chrysanthemi pectate lyases: enzyme characteristics and potential inhibitors. J. Bacteriol. 1792503-2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tempel, W., K. Karaveg, Z. J. Liu, J. Rose, B. C. Wang, and K. W. Moremen. 2004. Structure of mouse Golgi alpha-mannosidase IA reveals the molecular basis for substrate specificity among class 1 (family 47 glycosylhydrolase) alpha1,2-mannosidases. J. Biol. Chem. 27929774-29786. [DOI] [PubMed] [Google Scholar]
- 68.Trakhanov, S., N. K. Vyas, H. Luecke, D. M. Kristensen, J. Ma, and F. A. Quiocho. 2005. Ligand-free and -bound structures of the binding protein (LivJ) of the Escherichia coli ABC leucine/isoleucine/valine transport system: trajectory and dynamics of the interdomain rotation and ligand specificity. Biochemistry 446597-6608. [DOI] [PubMed] [Google Scholar]
- 69.Vallee, F., F. Lipari, P. Yip, B. Sleno, A. Herscovics, and P. L. Howell. 2000. Crystal structure of a class I alpha1,2-mannosidase involved in N-glycan processing and endoplasmic reticulum quality control. EMBO J. 19581-588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.van Alebeek, G. J., K. van Scherpenzeel, G. Beldman, H. A. Schols, and A. G. Voragen. 2003. Partially esterified oligogalacturonides are the preferred substrates for pectin methylesterase of Aspergillus niger. Biochem. J. 372211-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Van Petegem, F., H. Contreras, R. Contreras, and J. Van Beeumen. 2001. Trichoderma reesei alpha-1,2-mannosidase: structural basis for the cleavage of four consecutive mannose residues. J. Mol. Biol. 312157-165. [DOI] [PubMed] [Google Scholar]
- 72.van Pouderoyen, G., H. J. Snijder, J. A. Benen, and B. W. Dijkstra. 2003. Structural insights into the processivity of endopolygalacturonase I from Aspergillus niger. FEBS Lett. 554462-466. [DOI] [PubMed] [Google Scholar]
- 73.van Santen, Y., J. A. Benen, K. H. Schroter, K. H. Kalk, S. Armand, J. Visser, and B. W. Dijkstra. 1999. 1.68-A crystal structure of endopolygalacturonase II from Aspergillus niger and identification of active site residues by site-directed mutagenesis. J. Biol. Chem. 27430474-30480. [DOI] [PubMed] [Google Scholar]
- 74.Willats, W. G., L. McCartney, W. Mackie, and J. P. Knox. 2001. Pectin: cell biology and prospects for functional analysis. Plant Mol. Biol. 479-27. [PubMed] [Google Scholar]
- 75.Yoder, M. D., and F. Jurnak. 1995. Protein motifs. 3. The parallel beta helix and other coiled folds. FASEB J. 9335-342. [DOI] [PubMed] [Google Scholar]
- 76.Yoder, M. D., and F. Jurnak. 1995. The refined three-dimensional structure of pectate lyase C from Erwinia chrysanthemi at 2.2 angstrom resolution (implications for an enzymatic mechanism). Plant Physiol. 107349-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yoder, M. D., N. T. Keen, and F. Jurnak. 1993. New domain motif: the structure of pectate lyase C, a secreted plant virulence factor. Science 2601503-1507. [DOI] [PubMed] [Google Scholar]
- 78.Yoder, M. D., S. E. Lietzke, and F. Jurnak. 1993. Unusual structural features in the parallel beta-helix in pectate lyases. Structure 1241-251. [DOI] [PubMed] [Google Scholar]