

Abstract
In response to a variety of extracellular ligands, nuclear factor-κB (NF-κB) signaling regulates inflammation, cell proliferation, and apoptosis. It is likely that cells are not continuously exposed to stimulating ligands in vivo but rather experience transient pulses. To study the temporal regulation of NF-κB and its major regulator, inhibitor of NF-κBα (IκBα), in real time, we utilized a novel transcriptionally coupled IκBα-firefly luciferase fusion reporter and characterized the dynamics and responsiveness of IκBα processing upon a short 30-s pulse of tumor necrosis factor α (TNFα) or a continuous challenge of TNFα following a 30-s preconditioning pulse. Strikingly, a 30-s pulse of TNFα robustly activated inhibitor of NF-κB kinase (IKK), leading to IκBα degradation, NF-κB nuclear translocation, and strong transcriptional up-regulation of IκBα. Furthermore, we identified a transient refractory period (lasting up to 120 min) following preconditioning, during which the cells were not able to fully degrade IκBα upon a second TNFα challenge. Kinase assays of IKK activity revealed that regulation of IKK activity correlated in part with this transient refractory period. In contrast, experiments involving sequential exposure to TNFα and interleukin-1β indicated that receptor dynamics could not explain this phenomenon. Utilizing a well accepted computational model of NF-κB dynamics, we further identified an additional layer of regulation, downstream of IKK, that may govern the temporal capacity of cells to respond to a second proinflammatory insult. Overall, the data suggested that nuclear export of NF-κB·IκBα complexes represented another rate-limiting step that may impact this refractory period, thereby providing an additional regulatory mechanism.
Adequate resolution of an inflammatory reaction is as equally important as initiation. Persistent or fulminant responses can cause detrimental consequences both locally and systemically (1), and resolution of inflammation is important for both termination of an acute response as well as for prevention of destructive chronic responses. It is therefore not surprising that mechanisms aimed at rapid and specific initiation of proinflammatory reactions have co-evolved with mechanisms that provide timely termination of such processes. From a systems biology perspective, such “switchability” can be achieved by intracellular feedback loops that permit ligand-induced desensitization and resensitization of proinflammatory signaling cascades (2).
In this regard, recent studies have shown that nuclear factor-κB (NF-κB)4 signaling plays a critical role in both initiation and resolution of inflammation (2, 3). The transcription factor NF-κB is a key regulator of innate and adaptive immune responses as well as a mediator of cell survival and proliferation (4). Improper regulation of NF-κB contributes to induction and progression of a wide range of human disorders, including a variety of pathological inflammatory conditions, neurodegenerative diseases as well as many types of cancer (5, 6). In resting cells, inactive NF-κB is sequestered in the cytoplasm by binding to members of the inhibitor of NF-κB(IκB) family. Canonical activation of NF-κB depends on IκB kinase (IKK)-regulated proteasomal degradation of IκBα, an event that frees NF-κB for nuclear translocation within minutes (4, 7). Upon nuclear transport, NF-κB regulates the transcription of a few hundred genes (8-10) that can be divided into four major families (10, 11): 1) proinflammatory genes (e.g. cyclooxygenase 2, interleukin-1 (IL-1), tumor necrosis factor α (TNFα), inducible nitricoxide synthase, intercellular adhesion molecule-1, E-selectin, etc.), 2) proproliferative genes (e.g. cyclin D, and cellular myelocytomatosis), 3) antiapoptotic genes (B-cell leukemia/lymphoma 2, B-cell leukemia/lymphoma extra long, X-linked inhibitors of apoptosis protein, and cellular inhibitors of apoptosis protein), and 4) autoinhibitory genes (e.g. A20, cylindro-matosis, suppressor of cytokine signaling 1, and IκBα).
With respect to the last, other transcriptionally independent processes, aimed at autoinhibition of NF-κB activity, do exist. Such mechanisms down-regulate NF-κB signaling on a much shorter time frame (seconds to minutes). These include homologous receptor desensitization (12, 13), asymmetric heterologous receptor desensitization (13, 14), autocatalytic C-terminal IKK hyperphosphorylation (15), and protein phosphatase 2C-dependent dephosphorylation of IKK (16).
Considering the complex nature of the inflammatory milieu, one would expect that stationary tissue-residing cells are exposed to a myriad of temporally distinct NF-κB-stimulating cues. For instance, cells can be directly stimulated by pathogen-derived products (e.g. lipopolysaccharide through TLR4 (toll-like receptor 4) receptors (17)), exposed to numerous soluble proinflammatory stimuli produced by circulating effector cells (e.g. cytokines, chemokines, etc.), and/or experience inflammation-induced oxidative stress (18). These signals can occur simultaneously or sequentially to one another. For example, systemic administration of bacterial lipopolysaccharide to mice was shown to induce transient production of TNFα (serum levels peaking at ∼1.5 h and quickly returning to base line), but IL-1β production was delayed and prolonged (first detected at 2 h, but lasting >5-6 h) (19). Thus, cells co-expressing TLR4, IL-1, and TNFα receptors would sequentially interrogate signals arising from lipopolysaccharide, TNFα, and IL-1β, each of which could independently activate NF-κB.
Central to any signaling desensitization mechanism is a refractory period during which cells cannot fully respond to a second insult (autologous or heterologous desensitization). Therefore, consideration of the dynamic pattern of stimulus exposure described above begs the immediate question of whether cells can instantly initiate an NF-κB response to a second activating stimulus, and if not, when will such cells be able to remount a full response again? Specifically, are ligand-preconditioned cells capable of eliciting NF-κB activation to the same extent as naive cells?
Little is known about the capacity of cells to activate NF-κB in response to a second activating challenge, since the highly dynamic nature of this process presents many technical difficulties. These include low temporal resolution of conventional transcriptionally dependent NF-κB reporter gene assays, low throughput, inability to acquire longitudinal data, and the semiquantitative nature of traditional biochemical assays (e.g. electrophoretic mobility shift assay, immunoblotting, etc.). Such limitations render these assays incapable of accurate analysis of the early, ligand-induced dynamic changes in the capacity of cells to elicit a response to a second challenge.
To efficiently address this question, we generated an improved, transcriptionally coupled version of a previously published genetically encoded IκBα-firefly luciferase (IκBα-FLuc) fusion reporter (20) in conjunction with dynamic, live cell bioluminescence imaging of cultured cells. We chose to focus on HepG2 human hepatoma cells as a model system, because 1) NF-κB signaling has been extensively studied in these cells, 2) HepG2 cells have been shown to activate NF-κB in response to a variety of proinflammatory ligands (21), 3) these cells can be easily transfected with readily available reagents, and, most importantly, 4) the pivotal role that NF-κB signaling plays in hepatocytes to regulate inflammation, apoptosis, and carcinogenesis (22).
Using bioluminescence imaging of live cells in conjunction with a variety of biochemical assays, we demonstrate herein that a 30-s preconditioning exposure to TNFα is sufficient to robustly activate IKK, culminating in IκBα degradation, NF-κB nuclear translocation, and strong transcriptional up-regulation of IκBα. Furthermore, the capacity of preconditioned cells to degrade IκBα in response to a second TNFα challenge is transiently refractory, regaining full responsiveness ∼120 min later. Finally, both IKK regulation and possibly NF-κB nuclear export, but not receptor dynamics, govern this transient refractory period. This study highlights the interlocking layers of NF-κB regulation, ensuring efficient and timely propagation as well as termination of proinflammatory signals.
EXPERIMENTAL PROCEDURES
Materials—d-Luciferin (potassium salt) was from Biosynth (Naperville, IL). Human TNFα and IL-1β were from R&D Systems (Minneapolis, MN). Complete protease inhibitor mixture was from Roche Applied Science (Basel, Switzerland). [γ-32P]ATP was from PerkinElmer Life Sciences. Carbenicillin, isopropyl β-d-1-thiogalactopyranoside, ampicillin, kanamycin, glutathione S-transferase (GST), β-glycerolphosphate, NaCl, NaF, Na3VO4, KOH, MgCl2, EDTA, phenylmethylsulfonyl fluoride, Nonidet P-40, Tween 20, Triton X-100, ATP, dithiothreitol, paraformaldehyde, cycloheximide, and HEPES were from Sigma.
Plasmids—pκB5→FLuc (Stratagene, La Jolla, CA) contains five repeats of a κB motif upstream of a minimal TATA box controlling expression of firefly luciferase. pκB5→IκBα-FLuc was produced by cloning an EcoRI/HpaI (blunt) fragment from pCMV→IκBα-FLuc (20) into the EcoRI and EcoRV (blunt) sites of pκB5→FLuc. pκB5→FLuc, pCMV→IκBα-FLuc, and pκB5→IκBα-FLuc were propagated in TOP10 electrocompetent Escherichia coli (Invitrogen) and purified using Qiagen HiSpeed Maxi Kits (Qiagen, Valencia, CA). pGST-IκBαN (encoding for GST fused to the N-terminal fragment of human IκBα-(1-54)) was a kind gift from Prof. Alexander Hoffmann (University of California San Diego). pGST-IκBαN was propagated in BL21 codon+ E. coli cells (Stratagene).
Cells and Transfections—HepG2 human hepatoma cells were from the American Type Culture Collection (Manassas, VA). Cells were cultured in DMEM supplemented with heat-inactivated fetal bovine serum (10%) and l-glutamine (2 mm). Cell cultures were grown at 37 °C in a humidified atmosphere of 5% CO2. HepG2 cells (105) were transiently transfected (Fugene 6; Roche Applied Science) with pκB5→IκBα-FLuc (200 ng/well) and plated in black-coated 24-well plates (In Vitro Systems GmbH, Gottingen, Germany). Cells were then allowed to recover for 48 h prior to imaging.
Dynamic Bioluminescence Live Cell Imaging—Prior to imaging, cells were washed with prewarmed phosphate-buffered saline (PBS, pH 7.4) and placed into 900 μl of colorless HEPES-buffered DMEM, supplemented as above and with d-luciferin (150 μg/ml). Cells were allowed to equilibrate for 1 h (37 °C) before proceeding with ligand stimulation and imaging. Four different stimulation regimens were included in this study. 1) For continuous TNFα (C), TNFα (final concentration 20 ng/ml) or vehicle (colorless DMEM) was added (100 μl) to d-luciferin-containing DMEM, and imaging was performed before and at the indicated time points after the addition of TNFα.2) For TNFα pulse (30 s, P), cells were pulsed for 30 s with TNFα (20 ng/ml) or vehicle, washed with PBS, returned to d-luciferin-containing DMEM, and imaged before and at the indicated time points after the pulse of TNFα. 3) For TNFα preconditioning (30-s pulse) followed by continuous TNFα challenge (P + C), at t0 cells were pulsed for 30 s with TNFα (20 ng/ml) or vehicle, washed with PBS, returned to d-luciferin-containing DMEM (900 μl), and imaged before and at the indicated time points after the pulse of TNFα. At tx, TNFα (final concentration 20 ng/ml) or vehicle (colorless DMEM) were again added (100 μl), and imaging was performed before and at the indicated time points after the addition of TNFα.4) TNFα preconditioning (30-s pulse) followed by continuous IL-1β challenge (P + C) was as in method 3, but continuous challenge was performed with IL-1β (10 ng/ml).
TNFα or IL-1β challenge was performed at the following time points: tx = 0 (no preconditioning) and 30, 60, 120, and 240 min postpreconditioning. Assay plates were imaged using an IVIS100 imaging system (Xenogen Caliper, Alameda, CA). Acquisition parameters were as follows: acquisition time, 60 s; binning, 4; field of view, 10 cm; f/stop, 1; filter, open; image-image interval, 5 min; number of acquisitions, 73 (360 min).
To analyze ligand-induced regulation of de novo reporter resynthesis, cells were pretreated with cycloheximide (100 μg/ml) for 1 h before continuous stimulation with TNFα and bioluminescence imaging (as above).
IκBα Responsiveness Assays—HepG2 cells, transfected with pκB5→IκBα-FLuc (as above) or HeLa cells, stably expressing pCMV→IκBα-FLuc (20), were plated in 4 wells of a 6-well plate (one plate per time point) and grown for 48 h. At t0, all wells were washed with prewarmed PBS, pulsed for 30 s with TNFα (20 ng/ml, 1 ml) or vehicle (PBS), washed again with PBS, returned to regular medium (1 ml), and placed in a 37 °C incubator. This procedure was defined as TNFα preconditioning (P). At tx, two wells were treated (continuously) with TNFα (20 ng/ml), and two wells were treated with vehicle only (PBS). This procedure was defined as TNFα challenge (C). Following this TNFα challenge, cells were returned to the incubator. At tx + 25 min (time of maximal IκBα degradation (20); see Fig. 2A for schematic timeline), cells were harvested (by scraping) in reporter lysis buffer (Promega, Madison, WI). Cell lysates were normalized for protein content by a BCA protein assay (Promega), aliquoted, and frozen (-80 °C) for in vitro bioluminescence and Western blot analyses (see below). For in vitro bioluminescence assays, lysates (10 μl, in triplicate) were mixed with luciferase assay buffer (190 μl; 25 mm HEPES, 154 mm NaCl, 5.4 mm MgSO4, 10 mm dithiothreitol, 5 mm ATP, 150 μg/ml d-luciferin, pH 8.0) in a 96-well plate immediately prior to imaging. Assay plates were imaged using the IVIS 100 bioluminescence imager (acquisition time, 10 s; binning, 4; field of view, 10 cm; f/stop, 1; filter, open).
FIGURE 2.

TNFα induced a transient refractory period for IκBα processing. A, dynamic live cell bioluminescence imaging profiles of IκBα-FLuc from TNFα preconditioning plus challenge experiments. The black arrows denote 30-s preconditioning pulse; red arrows denote the beginning of continuous TNFα challenge; black profiles represent cells preconditioned and then challenged at the indicated time points; red profiles represent cells treated at time 0 with continuous TNFα (denoting the maximal possible degradation response of IκBα upon continuous TNFα treatment). Data are presented as -fold initial, -fold TNFα-untreated. B, schematic representation of the experimental timeline as used in C. Cells were preconditioned with TNFα for 30 s and then, at increasing intervals (0-240 min), were continuously challenged with TNFα. The arrowheads represent when cells were harvested and lysates were prepared (25 min post-challenge for quantitative bioluminescence imaging and Western blot analysis). C, IκBα-Fluc and endogenous IκBα levels, 25 min post-TNFα or vehicle challenge, as measured by bioluminescence imaging and Western blot, respectively.
Western Blot Analyses—Whole cell lysates were resolved by 10 or 7.5% SDS-PAGE, transferred to a polyvinylidene difluoride membrane, and probed for the indicated proteins using standard immunoblotting techniques. Primary antibodies against total human IκBα, β-actin, and IKKα were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Anti-phospho-IκBα (Ser-32/36) was from Cell Signaling Technologies (Danvers, MA). Secondary horseradish peroxidase-labeled anti-mouse and anti-rabbit IgG antibodies were from GE Healthcare Biosciences.
IKK Kinase Assay (IKK-KA)—IKK-KA reactions were carried out as per Werner et al. (23) and quantified in a medium throughput manner as per Hastie et al. (24). Briefly, HepG2 cells were grown in 10 cm tissue culture dishes to confluence. Cells were then washed in PBS (once) and treated with 20 ng/ml TNFα using three different treatment regimens: P, C or P + C (see above). To capture the full IKK activity profiles of cells treated with continuous (C) or pulse (P) regimens, cytosolic extracts were prepared at t = 0 (before) or 5, 10, 15, 30, 60, 120, or 240 min post-TNFα treatment. To capture maximal IKK activity of P + C-treated cells, cytosolic extracts were prepared 10 min post-TNFα challenge (given at 10, 30, 60, 120, and 240 min post-preconditioning). Cells were harvested by removing media, washing in ice-cold PBS + EDTA (1 mm), scraping, and pelleting at 2000 × g. To prepare cytosolic extracts, cell pellets were resuspended in 200 μl of cytosolic extract buffer (10 mm HEPES-KOH, pH 7.9, 250 mm NaCl, 1 mm EDTA, 0.5% Nonidet P-40, 0.2% Tween 20, 2 mm dithiothreitol, 20 mm β-glycerophosphate, 10 mm NaF, and 0.1 mm Na3VO4 supplemented with complete protease inhibitor mixture), incubated on ice (2 min), vortexed (1 min), and pelleted at 2000 × g. Supernatants were collected, normalized for protein content by a Bradford assay (Pierce), and stored at -80 °C. To immunoprecipitate IKK complexes, cytoplasmic extracts (100 μl) were incubated with anti-IKKγ antibody (15 μl, overnight, 4 °C with rotation) and then with Protein G 4FF bead slurry (20 μl, 50% (v/v)). Beads were pelleted at 4600 rpm and washed twice with cytosolic extract buffer (500 μl) and once with Kinase Buffer (500 μl; 20 mm HEPES, pH 7.7, 20 mm β-glycerophosphate, 100 mm NaCl, 0.1 mm Na3VO4, 10 mm MgCl2, 2 mm dithiothreitol supplemented with complete protease inhibitor mixture). For the IKK kinase reaction, beads were incubated for 30 min at 30 °C in Kinase Buffer (20 μl) containing 20 μm ATP, 10 μCi of [32P]ATP, and 0.5 μg of GST-IκBα-(1-54). Beads were removed by centrifugation (4600 rpm), and 15 μl of each reaction supernatant was spotted onto a 1-cm2 square of P81 phosphocellulose paper (Millipore, Billerica, MA) and immediately immersed in phosphoric acid (75 mm) for 5 min. Phosphoric acid washes were performed two more times, and papers were rinsed in acetone and then allowed to dry. Each paper was transferred to a scintillation vial, and radioactivity was determined on a β counter (Beckman Coulter, Fullerton, CA). Blank and no-lysate controls were subtracted from the experimental samples. Data were represented as fold-initial (untreated controls).
Calculating Ligand-dependent IKK Responsiveness—IKK responsiveness profiles (i.e. the net kinase capacity of IKK in response to a second challenge of TNFα, as a function of time after initial 30-s preconditioning) were calculated numerically from IKK-KA data using the following formula,
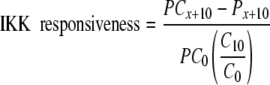 |
where PCx + 10 is IKK activity of preconditioned plus challenged cells, as recorded 10 min postchallenge. Px + 10 is the residual IKK activity of preconditioned but unchallenged cells at this exact time point. C0 and PC0 are initial IKK activities of challenged but unpreconditioned and fully preconditioned and challenged cells, respectively. C10 is the maximal IKK activity of challenged but unpreconditioned cells (recorded 10 min postchallenge). Note that although all parameter units in the nominator and denominator are in counts/min, IKK responsiveness is dimensionless, similar to IκBα responsiveness.
Computational Simulations—To simulate the dynamics of major regulators on the IKK-NF-κB axis, we used a well established computational model generated by Hoffmann et al. (25) and refined by Werner et al. (23). Briefly, an experimentally or hypothetically derived IKK activity profile was fed into the program as an input. Embedded in the model were 24 components, 70 reactions, and 70 parameters or rate constants for these reactions. Differential equations were solved numerically using Matlab 7.0 (Mathworks, Natick, MA) with subroutine Ode15. Interpolated and extrapolated (0-360 min at 5-min intervals) IKK activity profiles were calculated (Origin version 7.5, OriginLab, Northampton, MA) from experimental IKK-KA data (see above). To fit the model, initial steady-state IKK activity (i.e. intracellular concentration of active IKK) was set to be 1 nm. To computationally simulate IκBα dynamics of cells challenged at different times after initial preconditioning, when assuming no upstream IKK or receptor regulation, we used hypothetical IKK activity profiles as inputs, derived from superimposing experimentally acquired IKK activity profiles of 30-s pulsed and continuously treated cells at increasing intervals (30, 60, 120, and 240 min; see Fig. 4, black lines).
FIGURE 4.

Computational simulation of IκBα responsiveness in the absence of upstream receptor or IKK regulation. A, interpolated and extrapolated (0-360 min, at 5-min intervals) IKK activity profiles (right) of cells treated continuously (C, green curve) or by a 30-s pulse (P, blue curve) of TNFα (20 ng/ml) were used as inputs to computationally simulate total IκBα dynamics (right). B-E, left panels, hypothetical IKK activity profiles of preconditioned cells, challenged at the indicated times (denoted by black arrowheads) with a second, continuous dose of TNFα were generated by superimposing the continuous TNFα-induced IKK profiles at increasing intervals after the 30-s pulse TNFα-induced IKK profile. For generating these hypothetical profiles, we assume no preconditioning-induced receptor or IKK regulation. Right panels, the hypothetical IKK profiles were used as inputs into the model to predict IκBα dynamics. Note that challenge-induced IκBα degradation (initiated at the red arrowhead) is recovered in a time-dependent manner.
Immunofluorescence Microscopy—HepG2 cells were seeded into 35-mm glass bottom culture dishes (MatTek Corp., Ashland, MA) and grown to ∼40% confluence. Cells were pulsed for 30 s with TNFα as above and fixed at the indicated time points (by washing once with PBS, followed by fixation (4% paraformaldehyde for at least 15 min) and permeabilization (ice-cold methanol, 10 min at -20 °C)). Cells were washed in PBS, blocked in 5% normal goat serum in 0.3% Triton X-100, PBS (1 h), and then incubated with anti-p65 antibody (Santa Cruz Biotechnology; 1:200 in 0.3% Triton X-100, PBS at 4 °C, overnight with rocking). Cells were next incubated with Alexa-Fluor 635-conjugated goat anti-rabbit antibody (Invitrogen; 1:200 in 0.3% Triton X-100, PBS, 90 min, at room temperature with rocking). Cells were washed three times with PBS before being mounted with VECTASHIELD Mounting medium (Vector Laboratories; Burlingame, CA). Confocal images were captured using the ×40 objective (water immersion) on a Zeiss Axiovert 200 (Zeiss, Thornwood, NY) laser-scanning microscope equipped with the appropriate filter sets and analyzed using a Zeiss LSM Image Browser and Adobe Photoshop CS2.
RESULTS
Real Time Bioluminescence Imaging of pκB5→IκBα-FLuc-expressing Cells Recapitulated IKK-induced Dynamics of Endogenous IκBα—To monitor ligand-induced IκBα rapid dynamics as well as physiologic transcriptionally coupled behavior, we modified our previous IκBα-FLuc fusion reporter (20) to be driven by a synthetic promoter composed of five tandem κB response elements (TGGGGACTTTCCGC) followed by a minimal TATA-box. We hypothesized that this reporter would allow quantitative measurements of IKK-induced degradation as well as NF-κB-induced resynthesis and post-translational stabilization of IκBα from intact living cells (Fig. 1A). To validate use of this reporter, HepG2 cells were transiently transfected with a plasmid encoding the reporter and allowed to recover for 2 days before stimulation with a continuous or 30-s pulse of TNFα (20 ng/ml) to induce IKK activation. Upon the addition of TNFα, a rapid and dramatic decrease in bioluminescence was observed when readouts were normalized to untreated controls (20) under both continuous (C) and 30-s pulse (P) regimens (Fig. 1, B and C). This decrease in normalized bioluminescence, reflecting IKK-induced reporter degradation was followed by a sharp increase in bioluminescence, reflecting NF-κB-dependent reporter resynthesis, reaching maximum values at ∼120 min and then gradually declining toward base line. Note that the rate at which IκBα levels return to base line is steeper under continuous TNFα treatment compared with the 30-s pulse, providing evidence for reactivation of ligand-induced IκBα degradation during continuous stimulation (23). The magnitude of the initial decrease in bioluminescence was greater in continuously treated cells than in 30-s pulsed cells (70% versus 40% of initial decrease, respectively), indicating that a 30-s pulse of TNFα leads to ∼50% depletion of the IκBα·NF-κB pool compared with continuous TNFα exposure (Fig. 1C, 120 min). These data suggested that 1) this reporter construct could report on both IKK-induced IκBα degradation and successive resynthesis of IκBα, 2) a 30-s pulse of TNFα at a saturating concentration (20 mg/ml) elicited robust IKK activity, culminating in IκBα degradation and a full IκBα transcriptional up-regulation, and 3) with the current κB5 synthetic promoter system, there was a nonlinear relationship between IκBα degradation and NF-κB-dependent resynthesis of IκBα (i.e. saturation of IκBα resynthesis even at submaximal IκBα degradation levels).
FIGURE 1.

pκB5→IκBα-FLuc, a transcriptionally coupled reporter for monitoring IκBα dynamics in live cells. A, a schematic representing ligand-induced degradation and transcriptionally coupled resynthesis of the reporter. B, raw bioluminescence images of HepG2 cells transiently expressing pκB5→IκBα-FLuc treated with TNFα (20 ng/ml) continuously (C) or as a 30-s pulse (P) or with vehicle only (V) and imaged for 360 min. Images show pseudocolor-coded photon flux maps superimposed on black-and-white photographs of the assay plate. C, graphical representation of the changes in photon flux from B as a function of time after TNFα addition. Data are plotted as fold-initial, fold-vehicle treated (n = 3 for all points; S.E. ≤5%; representative of three independent experiments). D, Western blot analysis of endogenous IκBα from HepG2 cell lysates prepared at the indicated times after a 30-s pulse or continuous treatment with TNFα (20 ng/ml). E, pretreatment with cycloheximide (CHX;1h, 100 μg/ml) totally abrogated TNFα-induced IκBα-FLuc resynthesis but had no effect on reporter degradation.
Strikingly, Western blot analysis revealed that endogenous IκBα behaved exactly as the reporter under both C and P conditions, recapitulating the degree of degradation, recovery, and return to base line (Fig. 1D). Pretreating pκB5→IκBα-FLuc-expressing HepG2 cells with cycloheximide did not affect degradation of IκBα-FLuc but abolished signal recovery, indicating that this phase was totally dependent upon transcription and translation of new IκBα-FLuc (Fig. 1E).
TNFα Preconditioning Induces a Transient Refractory Period of IκBα Processing—Upon a proinflammatory insult in vivo, effector cells (e.g. circulating macrophages) release TNFα and other activating cytokines in a temporally and spatially discrete manner. As a consequence, stationary target cells (e.g. epithelial cells, endothelial cells, hepatocytes, etc.) will sense a stochastic rise in the levels of such proinflammatory ligands. In such a dynamic environment, as ligand-secreting cells continuously migrate to sites of inflammation, it is anticipated that over time, target cells will experience multiple pulses of activating ligands.
We therefore aimed to elucidate the effects of such ligand pulses on the capacity of hepatocytes to respond to a subsequent challenge of the same ligand. Having shown that 1) pκB5→IκBα-FLuc provided an accurate readout of IκBα processing in intact cells and that 2) a 30-s pulse was sufficient to induce robust IKK activity, we next sought to investigate whether a short 30-s preconditioning pulse with TNFα had a substantial effect on the capacity of cells to process IκBα upon a subsequent continuous TNFα challenge.
HepG2 cells transiently expressing pκB5→IκBα-FLuc were given a 30-s pulse of TNFα (20 ng/ml) or vehicle at t0, washed, replaced in media containing d-luciferin and repeatedly imaged (every 5 min) prior to a TNFα challenge. At t30, t60, t120, or t240 (min) after pulsing, cells were then challenged with a second continuous concentration of TNFα (20 ng/ml), and live cell imaging was continued up to 360 min. To compare the processing dynamics of IκBα-FLuc in naive (unpreconditioned) cells with that of preconditioned cells, the resulting bioluminescence profiles of preconditioned cells (Fig. 2A, black lines) were plotted along with the bioluminescence profile of unpreconditioned cells (i.e. only treated with continuous TNFα at t0, red line, Fig. 2A). The different graphs represent the differential dynamics of IκBα-FLuc processing as the preconditioning pulse-challenge (P-C) intervals temporally increased (0-240 min).
We observed that challenging preconditioned cells with a continuous exposure to TNFα near the time that they had achieved maximal degradation from the preconditioning pulse (i.e. 30 min postpreconditioning) resulted in a small amount of additional IκBα degradation. As the interval between preconditioning and challenge increased, the magnitude of challenge-induced IκBα degradation also increased. These data suggested that the TNFα-NF-κB system possessed a built-in refractory period following TNFα treatment that prevented cells from fully responding to a second exposure to ligand. To quantify this phenomenon independent of confounding factors that may affect dynamic bioluminescence readouts (e.g. d-luciferin, ATP, O2, or pH dynamics) and to verify its existence for endogenous IκBα, we performed a similar experiment, but instead of live cell imaging, we harvested whole cell lysates at tx + 25 min (time of maximal IκBα degradation after a ligand challenge given at tx (Fig. 1C); for a schematic timeline, see Fig. 2B). IκBα-FLuc reporter levels in these lysates were analyzed by bioluminescence imaging (upon the addition of saturating d-luciferin and ATP), and endogenous IκBα levels were determined by Western blot analysis and semiquantitative densitometric analysis (Fig. 2C). From these data, we were then able to calculate responsiveness levels for both IκBα and IκBα-FLuc as a function of time after TNFα preconditioning. Responsiveness at each challenge time was calculated by determining the magnitude of IκBα degradation induced by TNFα challenge divided by the magnitude of IκBα degradation in unpreconditioned cells from the same plate. Specifically, the ratio at tx + 25 min of IκBα in preconditioned cells challenged with TNFα over preconditioned cells challenged with vehicle was divided by the ratio at tx + 25 min of IκBα in unpreconditioned cells challenged with TNFα over unpreconditioned cells challenged with vehicle, the latter ratio representing the maximal possible response. We observed a strong correlation (r = 0.95) between levels of responsiveness for endogenous IκBα and IκBα-FLuc (Table 1). Consistent with our earlier observations derived from live cell dynamic bioluminescence imaging experiments (Fig. 2A), we observed that at 30 min postpreconditioning, cells were approximately half as responsive as naive (i.e. unpreconditioned) cells to a TNFα challenge and had gained full responsiveness by 120 min. Thus, a transient refractory period seemed to exist from 30 to 120 min post-TNFα preconditioning that rendered the cells unable to fully respond (as measured via IκBα degradation) to a second challenge of TNFα, and beyond this period, the cells were able to mount a full response to a second TNFα challenge. Notably, similar experiments performed with HeLa cells stably expressing pCMV→IκBα-FLuc (HeLaIκBα-FLuc (20)) yielded almost identical results (data not shown), suggesting that 1) the TNFα-induced transient refractory period was not limited to hepatocytes and 2) this effect was independent of both NF-κB-induced IκBα transcription and the initial levels of IκBα-FLuc (substantially higher in HeLaIκBα-FLuc (20)).
TABLE 1.
Percentage responsiveness of IκBα processing
Quantification of IκBα-FLuc and IκBα responsiveness to a second continuous challenge of TNFα at the indicated interval following a 30-s preconditioning pulse of TNFα was determined from the bioluminescence imaging and Western blot data shown in Fig. 2C. Responsiveness at each challenge time was calculated by determining the percentage of challenge-specific IκBα degradation divided by the percentage of IκBα degradation in unpreconditioned cells from the same plate. The responsiveness of IKK was determined by an IKK kinase assay.
|
Responsiveness of IκBα processing
|
||||
|---|---|---|---|---|
| 30 min after TNFα preconditioning | 60 min after TNFα preconditioning | 120 min after TNFα preconditioning | 240 min after TNFα preconditioning | |
| % | % | % | % | |
| IκBα (Western blot) | 49 | 80 | 100 | 82 |
| IκBα-FLuc (bioluminescence) | 43 | 74 | 90 | 94 |
| IKK activity | 35 | 56 | 75 | 69 |
The Ligand-induced Transient Refractory Period for IκBα Processing Correlated in Part with Temporal Down-regulation of IKK but Not Receptor Dynamics—Hypothetically, this loss and regain of the capacity of cells to process IκBα can be explained by 1) internalization or shedding of TNFα receptors, followed by their recycling to the cell membrane (26, 27), 2) transient down-regulation of IKK activity as previously reported (15, 28), or, alternatively, 3) by a yet unknown mechanism of regulation, downstream of IKK. We therefore sought to establish the relative contributions of receptor dynamics and IKK regulation to this refractory period.
To determine the extent of receptor dynamics in governing the observed loss and regain of IκBα processing, we took advantage of a discovery, made 20 years ago (14), that IL-1β induces transient down-regulation of TNFα receptors but not vice versa (i.e. TNFα has no effect on either the affinity or the number of IL-1β surface receptors), as tested in a variety of cell lines and primary cells. Hence, we aimed to determine IκBα responsiveness to an IL-1β challenge as a function of time after TNFα preconditioning in HepG2 cells. Cells expressing pκB5→IκBα-FLuc were treated with a 30-s pulse of TNFα (20 ng/ml), followed by a continuous challenge with IL-1β, initiated at increasing P-C intervals (0-240 min). IκBα processing was analyzed by live cell dynamic bioluminescence imaging (Fig. 3A). Using this experimental setup, we again observed a transient refractory period (from 30 to 120 min post-TNFα preconditioning) during which HepG2 cells exhibited decreased IκBα responsiveness. The magnitude of the ligand-induced degradation increased as the interval to the IL-1β challenge increased, becoming fully responsive again by 120 min (Fig. 3A). These data suggested that even in the absence of ligand-induced receptor desensitization or cross-regulation, the capacity of cells to process IκBα was compromised within the first 2 h after a short TNFα stimulation.
FIGURE 3.

Impact of receptor dynamics and IKK regulation on IκBα responsiveness. A, dynamic live cell bioluminescence imaging profiles of IκBα-FLuc from TNFα preconditioning, IL-1β challenge experiments. The black arrows denote the 30-s preconditioning pulse of TNFα; blue arrows denote the beginning of continuous IL-1β (10 ng/ml) challenge; black profiles represent cells preconditioned and then challenged with IL-1β at the indicated time points; blue profiles represent cells treated at time 0 with continuous IL-1β (denoting the maximal possible degradation response of IκBα upon continuous IL-1β treatment). Data are presented as -fold initial. B, IKK kinase activity was measured at the indicated time points after initiation of a continuous (blue curve) or a 30-s pulse (red curve) of TNFα (20 ng/ml). Results are presented as background-normalized, -fold initial (untreated) controls. C, Western blot analysis of IKKα in cytoplasmic fractions, used as inputs for immunoprecipitation and kinase reactions presented in B.
We next aimed at deciphering whether transient down-regulation of IKK activity could explain the observed loss and regain in IκBα responsiveness. We therefore performed a series of IKK kinase assays in order to directly measure the temporal activity profile of IKK, a central junction of the TNFα and IL-1β pathways that integrates signals from a myriad of upstream regulators (e.g. TNF receptor-associated factors, mitogen-activated protein/extracellular signaling-regulated kinase kinase kinase, TGFβ-activated kinase-binding protein, TGFβ-activated kinase, NF-κB-inducing kinase, receptor-interacting protein, A20, protein kinase Cζ, etc. (2, 7, 29)). HepG2 cells were treated with TNFα (20 ng/ml) either as a 30-s pulse or continuously. At the indicated time points, cells were harvested, and IKK complexes were immunoprecipitated and assayed for their capacity to phosphorylate exogenous GST-IκBα-(1-54) (23). We found that for both 30-s pulses and continuous TNFα exposure, temporal profiles of IKK activity were almost identical, with both peaking at 10 min. However, consistent with our earlier findings that continuous TNFα treatment elicits greater IκBα degradation than a 30-s pulse (Fig. 1C), continuous TNFα treatment exhibited slightly elevated and more sustained levels of IKK activity compared with pulsed TNFα treatment (Fig. 3B). Importantly, Western blot analysis showed that IKK complex levels (as determined by IKKα protein) did not change over the experimental time course (Fig. 3C), confirming that the increase in net kinase activity was due specifically to IKK activation.
IKK-KA data were also collected from preconditioned cells, 10 min post-challenge (at the time of maximal IKK activity; see Fig. 3B) at increasing P-C intervals (0-240 min). Using these data together with the IKK activity profiles generated for 30-s pulse and continuous TNFα treatment regimens (Fig. 3B), we were able to calculate the net capacity of IKK to phosphorylate IκBα as a function of time after TNFα preconditioning (i.e. IKK responsiveness (Table 1); see “Experimental Procedures” for details on this calculation). Based on this calculation, we noted that the capacity of IKK to respond to a second challenge of TNFα was significantly compromised at 30 min post-TNFα preconditioning and then gradually increased, reaching ∼75% responsiveness by 120 min. Up to 240 min, IKK activity did not fully recover to initial levels, consistent with other reports indicating that upon TNFα stimulation, IKK activity rapidly and transiently declines due to autocatalytic C-terminal hyperphosphorylation (15) and protein phosphatase 2C-dependent dephosphorylation (16), followed by late NF-κB-dependent down-regulation, a process attributed, in part, to A20, an IKK-inhibitory protein (29). Hence, these data suggested that 1) the observed ligand-induced transient refractory period of IκBα processing (Figs. 2 and 3 and Table 1) correlated only in part with ligand-induced transient down-regulation of IKK activity and that 2) the level to which cells are able to degrade IκBα was not linear with the capacity of IKK to phosphorylate IκBα (i.e. full IκBα responsiveness was observed as soon as 120 min post-TNFα preconditioning (Figs. 2 and 3A), a time point where IKK responsiveness was still compromised (Table 1)). These data indicated that either submaximal IKK activity could now fully support ligand-induced IκBα degradation following the refractory period or that additional ligand-responsive elements existed that converged on IκBα to induce a full response.
Computational Modeling of NF-κB Signaling Suggested an Additional Layer of Regulation, Downstream of IKK, Governing the Observed Refractory Period for IκBα Processing—The NF-κB pathway provides an excellent example of a complex signaling system employing numerous temporally distinct autoregulatory mechanisms and negative feedback loops. IKK enzymatic activity, which is both endogenously and exogenously regulated, controls the degradation of its own substrate (IκBα), which is later strongly up-regulated in an NF-κB-dependent manner (Fig. 1A). Rapid changes in substrate availability, conformation, and subcellular localization imply that alternative mechanisms of regulation might exist other than changes in enzymatic activity. Although a ligand-induced transient refractory period of IκBα processing could be explained in part by down-regulation of IKK activity, we were intrigued to examine whether an alternative regulatory mechanism, based on substrate (IκBα) dynamics, might exist to complement or “back up” IKK regulation. Obviously, inhibition of IKK was not a viable option for analyzing downstream regulation, since such inhibition will result in complete loss of responsiveness in the absence or presence of preconditioning. We therefore decided to undertake a computational approach and explore IκBα dynamics in silico, assuming no down-regulation of IKK activity. We used a well accepted computational model that used experimentally or hypothetically driven IKK activity profiles as inputs and, in return, calculated ligand-induced dynamics of 24 different subpopulations of mediators on the IKK-NF-κB axis.
As a first step, to test the robustness of the model, we sought to compare our IκBα-FLuc bioluminescence imaging data for 30-s pulsing and continuous TNFα treatments (Fig. 1C) with the dynamics of IκBα, as predicted by the computational model. To accomplish this, we used as inputs the IKK activity profiles generated for 30-s pulse and continuous TNFα treatment regimens (Fig. 4A, left; see “Experimental Procedures” for details on numerical processing of the raw data to fit the model). The dynamics of six different free and complexed IκBα subpopulations could be predicted by the model (i.e. free IκBαcyt, IκBα·IKKcyt, IκBα·NF-κBcyt, IκBα·IKK·NF-κBcyt, free IκBαnuc, and IκBα·NF-κBnuc). Since live cell bioluminescence imaging of IκBα-FLuc could not distinguish between these populations, we summed up the predicted concentrations of all IκBα subpopulations and plotted the predicted total IκBα levels as a function of time (Fig. 4A, right). For both treatment regimens, we noted an excellent correlation between the predicted profiles of IκBα and the experimentally generated profiles of IκBα-FLuc (Fig. 1C). The timing and extent of IκBα degradation as well as the overall dynamic behavior were highly similar. However, differences in the amplitude and timing of resynthesis (experimental: ∼8-fold initial at ∼120 min; computational: 1.2-1.5-fold initial at ∼90 min) were observed and could be explained by dynamic differences between the endogenous IκBα promoter and the synthetic κB5-TATA promoter driving IκBα-FLuc (i.e. differences in binding affinity and cooperativity toward NF-κB).
We next generated hypothetical IKK profiles representing IKK activities from preconditioned/challenged cells, assuming no upstream receptor or IKK regulation (i.e. experimentally derived challenge-specific IKK activity was overlaid on top of experimentally derived precondition-specific residual IKK activity). These hypothetical IKK activity profiles (Fig. 4, B-E, left, each generated with a different P-C interval) were used as inputs for computing total IκBα dynamics (Fig. 4, B-E, right). Surprisingly, the computational model predicted that even in the absence of receptor dynamics or IKK regulation, IκBα processing would be transiently compromised (compare, for example, the second, challenge-induced degradation phase at 120 or 240 min with the ones at 30 or 60 min). These data suggested that although IKK down-regulation partially correlated with the ligand-induced transient refractory period for IκBα processing, an additional regulatory mechanism was present down-stream of IKK. Importantly, IκBα availability per se was not sufficient to explain changes in IκBα responsiveness, because, as confirmed experimentally and computationally, at 60 min post-preconditioning, the IκBα concentration had already recovered, whereas degradation potential was still low (compare Fig. 2, A and C, Table 1, and Fig. 4C).
Nuclear Export of IκBα—NF-κB complexes may also control the capacity of cells to process IκBα. Having demonstrated experimentally the phenomenon of a ligand-induced transient refractory period for IκBα processing and after dissecting biochemically and computationally the origins of this observation, we next sought to more closely examine the components of the computational model in order to identify candidates, down-stream of IKK, capable of regulating IκBα responsiveness. While examining the rate constants of a variety of reactions used by the model, we noticed that free versus NF-κB-bound IκBα differed tremendously in their capacity to associate with IKK (1.35 versus 11.1 μm-1 min-1, respectively) and to be degraded in an IKK-dependant manner (0.12 versus 0.00006 min-1, respectively). These differences in IKK association and ligand-induced degradation were experimentally established by Zandi et al. (30).
This led us to put forward the following model (Fig. 5A). 1) free IκBα and NF-κB-bound IκBα represent “protected” and “unprotected” populations with respect to ligand-induced, IKK-dependent proteasomal degradation. 2) Under steady-state conditions, there is a stoichiometric excess of IκBα over NF-κB in the cytoplasm (∼0.7 NF-κB per IκBα according to the model). This may explain our observations that even at saturating concentrations of TNFα or IL-1β, IκBα degradation never exceeded 70-80% of initial level (e.g. Fig. 1C). 3) Upon ligand stimulation, NF-κB-bound IκBα is degraded, NF-κB translocates to the nucleus, and IκBα is resynthesized. 4) At this point, although IκBα is highly abundant, its capacity to be degraded in response to a second stimulus is still severely compromised, because NF-κB is in the nucleus. 5) IκBα can freely shuttle between the cytoplasm and the nucleus, pulling NF-κB molecules (that lack nuclear export signals (31)) back to the cytoplasm. This step may be the rate-limiting step for acquisition of full responsiveness. 6) Newly synthesized IκBα molecules uncomplexed with NF-κB are rapidly degraded (32), and only after all NF-κB molecules are recovered back to the cytosol and the NF-κB-bound IκBα over free IκBα ratio returns to prestimulation levels (∼0.7) are cells able to mount a full response again.
FIGURE 5.

Nuclear export of NF-κB may regulate IκBα sensitivity to ligand-induced degradation. A, a schematic illustrating subcellular localization and levels of free IκBα, free NF-κB, and NF-κB-bound IκBα in response to a pulse of TNFα. B, computationally predicted profile of all cytoplasmic populations of NF-κB following a pulse of TNFα. Note the exceedingly small free NF-κB population. C, HepG2 cells were stimulated with a 30-s TNFα pulse. At the indicated time points, cells were fixed, permeabilized, and immunostained for p65 NF-κB. Shown are representative immunofluorescence confocal photomicrographs.
To experimentally examine the nuclear export hypothesis, we sought to analyze ligand-induced changes in cytoplasmic IκBα·NF-κB complexes. However, the computational model predicted that ligand-induced changes of cytoplasmic IκBα·NF-κB and total cytoplasmic NF-κB were essentially the same (i.e. at any given time, virtually all cytoplasmic NF-κB was bound to IκBα; Fig. 5B), suggesting that monitoring cytoplasmic total NF-κB was an excellent approximation for following cytoplasmic IκBα·NF-κB complexes. We therefore pulsed HepG2 cells for 30 s with TNFα (20 ng/ml), and at various times after stimulation, we fixed, permeabilized, and immunostained the cells for p65 NF-κB (Fig. 5C). We found that upon a 30-s TNFα pulse, p65 rapidly translocated to the nucleus (maximal by 30 min) but by 60-120 min was back in the cytoplasm. The excellent temporal correlation between the levels of cytoplasmic NF-κB (as derived computationally or experimentally; Fig. 5, B and C, respectively) and the competence of cells to degrade IκBα in response to a proinflammatory ligand (i.e. Table 1) strongly suggested that nuclear transport of NF-κB provided a potential alternative mechanism to transiently desensitize IκBα processing (refractory period), in addition to the mechanism of IKK down-regulation (Fig. 3, B and C, and Table 1).
DISCUSSION
Ligand-induced desensitization is a common theme in many biological systems (13), thereby allowing cells to mount an appropriate response independently of ligand exposure time. Thus, prolonged exposures will not result in excessive responses, but instead, cells are enabled to build up a downstream response while being unable to perceive a second activating cue. Desensitization and resensitization are traditionally perceived to be linked to receptor dynamics (internalization, shedding, and recycling); however, any mediator or regulator along a signaling pathway can be hypothetically desensitized, therefore transiently blocking signal transduction (13).
In this work, we demonstrated that although cells can efficiently activate NF-κB in response to a TNFα exposure as short as 30 s, such stimulation was followed by a refractory period during which the capacity of cells to respond to a second homologous or heterologous stimulus was severely compromised. We further found that this transient refractory period correlated in part with a temporal down-regulation of IKK activity but not with receptor desensitization. Computational modeling enabled us to identify an additional layer of regulation, downstream of IKK, controlling the capacity of cells to respond to a second challenge. Ligand-induced dynamic changes in substrate (IκBα) availability, conformation, and subcellular localization form the basis for this mechanism. Further analysis led us to conclude that nuclear export of NF-κB may be a rate-limiting step in controlling IκBα homeostatic metabolism, a term recently coined by O'Dea et al. (33).
Our study highlights the multifaceted regulation of NF-κB signaling (Fig. 6) and sheds light on the refractory nature of IκBα processing as a route to transiently desensitize NF-κB activity upon subsequent rounds of stimulation. Rapid and transient deactivation of IKK activity as well as temporal reduction in its capacity to respond to a subsequent challenge (IKK responsiveness) seems to play a crucial role in this process. Previous studies indicated that both the amplitude and the timing of IKK activation affect not only the intensity of NF-κB-dependent transcription but also the specificity of the transcriptional response (23, 34). This indicated that besides resolution of the inflammatory response and induction of a refractory period (temporally preventing subsequent rounds of IκBα degradation upon restimulation), rapid down-regulation of IKK activity (28) plays a pivotal role in determining the type of elicited transcriptional program.
FIGURE 6.

Refractory period in NF-κB signaling. Shown is a schematic representation of the different ligand-induced autoregulatory mechanisms that control responsiveness in the NF-κB signaling pathway.
In addition to IKK regulation, our work demonstrated that nuclear export of IκBα·NF-κB complexes may have also regulated IκBα responsiveness (Figs. 4 and 5). This suggested that NF-κB positively controls IκBα both transcriptionally and post-translationally. Such double-layered feedback regulation ensures that NF-κB transcriptional activity will fully resume only after reconstitution of the cytosolic pool of NF-κB. Two other IκB isoforms, IκBβ and IκBε, are degraded more slowly under both TNFα-induced and unstimulated conditions (23, 25) and have been implicated in dampening IκBα-mediated oscillations of NF-κB activity (25, 35, 36). IκBε has been shown to be highly NF-κB-inducible in mouse embryo fibroblasts and to contribute to nuclear export of NF-κB but only at times greater than 3 h poststimulation (37). Thus, it is seems unlikely that IκBε contributes substantially to the export of NF-κB over the 2 h of the refractory period observed in the present study. It may be interesting to determine whether similar transient refractory periods exist for processing of other IκB isoforms.
Although TNFα-induced resynthesis of endogenous IκBα peaks at ∼60-90 min after onset of stimulation (as validated both experimentally and computationally (Fig. 1D and 4A, respectively)), maximum levels of newly synthesized IκBα-FLuc reporter were observed ∼120 min after TNFα stimulation (Figs. 1, C and E, and 2A). This discrepancy may be explained by differences likely to be present in affinity and cooperativity of binding of NF-κB to endogenous versus synthetic promoters (the endogenous promoter contains three distant κB sites, whereas the synthetic promoter contains five tandem high affinity κB sites). Nevertheless, since both endogenous IκBα and IκBα-FLuc exhibit similar half-life times (20), differences in the timing of resynthesis cannot be explained by differences in turnover rate. Following the peak of IκBα resynthesis, both endogenous IκBα and our IκBα-Fluc reporter begin returning to base-line levels faster under continuous TNFα treatment, suggesting that ligand-induced reactivation of IκBα degradation is occurring under continuous TNFα exposure, as expected (23).
In the present and previous studies (20), we demonstrated that dynamic bioluminescence imaging of IκBα-FLuc reporters in live cells provides robust and accurate readouts of ligand-induced IκBα dynamics. In effect, real time bioluminescence imaging was equivalent to performing continuous on-line Western blots of IκBα at 5-min intervals. An analogous transcriptionally coupled reporter (kB5→IκBα-EGFP) was generated by Nelson et al. (35) for monitoring IκBα dynamics in single cells by live cell fluorescence microscopy. Although such a system provides the means to monitor ligand-induced translocations and oscillations in IκBα levels, temporal resolution of this reporter is limited by the long maturation time of EGFP (>1 h) (38, 39). This notion and the fact that Nelson et al. (35, 36) co-overexpressed p65-red fluorescent protein may explain the vast difference between the observed period of IκBα-EGFP oscillations (∼300 min) and the period of endogenous IκBα oscillations, as predicted computationally (∼90-120 min) (25). Although longer term IκBα oscillatory behavior was not the focus of the present study, we did observe single oscillations within ∼150-180 min. Because FLuc is active immediately upon translation, our reporter should afford greater temporal resolution, enabling accurate readouts of IκBα dynamics and oscillations in live cells for such studies as well as the multistimulation protocols as described herein.
Of note, a previous study aimed at analysis of IκBα stabilization indicated a role for p38 in IκBα stabilization and, in some cell lines, in prevention of sequential degradation of IκBα upon concurrent exposure to TNFα following continuous pretreatment with IL-1β (40). However, since IL-1β has been shown to induce rapid and dramatic down-regulation of TNFα receptors (but not vice versa) (14), inhibition of TNFα-induced IκBα processing, as observed by Place et al. (40), could be attributed directly to receptor dynamics rather than IκBα stabilization. This confounding factor highlights the importance of asymmetric receptor cross-desensitization, a phenomenon that remains poorly understood but has far reaching physiological consequences.
In conclusion, TNFα preconditioning protocols and dynamic imaging revealed a transient suppression of the capacity of cells to process IκBα. This refractory period for IκBα processing was controlled both by IKK activity and NF-κB distribution. In particular, the data suggested that nuclear export of NF-κB may provide additional rate-limiting regulation governing the refractory period machinery. These regulatory mechanisms provide a “molecular timer” controlling the amplitude, timing, and specificity of the NF-κB-mediated transcriptional program.
Acknowledgments
We thank Prof. Alexander Hoffmann, Shannon Werner, and Derren Barken (University of California, San Diego) for providing the computational model software. We also thank Dr. Yun-Feng Feng and Prof. Greg Longmore (Washington University) and Prof. Phillip Cohen (University of Dundee) for advice with the IKK kinase assays and Dr. Dustin Maxwell (Washington University) for assistance with immunofluorescence microscopy.
This work was supported in part by National Institutes of Health Molecular Imaging Center Grant P50 CA94056. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: NF-κB, nuclear factor-κB; IκB, inhibitor of NF-κB; IKK, IκB kinase; IL-1, interleukin-1; TNFα, tumor necrosis factor α; FLuc, firefly luciferase; GST, glutathione S-transferase; PBS, phosphate-buffered saline; DMEM, Dulbecco/Vogt modified Eagle's minimal essential medium; IKK-KA, IKK kinase assay; C, continuous; P, pulse; P + C, pulse followed by continuous; EGFP, enhanced green fluorescent protein.
References
- 1.Han, J., and Ulevitch, R. J. (2005) Nat. Immunol. 6 1198-1205 [DOI] [PubMed] [Google Scholar]
- 2.Winsauer, G., and de Martin, R. (2007) Thromb. Haemostasis 97 364-369 [PubMed] [Google Scholar]
- 3.Hoffmann, A., and Baltimore, D. (2006) Immunol. Rev. 210 171-186 [DOI] [PubMed] [Google Scholar]
- 4.Perkins, N. D. (2007) Nat. Rev. Mol. Cell. Biol. 8 49-62 [DOI] [PubMed] [Google Scholar]
- 5.Karin, M., and Greten, F. R. (2005) Nat. Rev. Immunol. 5 749-759 [DOI] [PubMed] [Google Scholar]
- 6.Karin, M. (2006) Nature 441 431-436 [DOI] [PubMed] [Google Scholar]
- 7.Hayden, M. S., and Ghosh, S. (2004) Genes Dev. 18 2195-2224 [DOI] [PubMed] [Google Scholar]
- 8.Bunting, K., Rao, S., Hardy, K., Woltring, D., Denyer, G. S., Wang, J., Gerondakis, S., and Shannon, M. F. (2007) J. Immunol. 178 7097-7109 [DOI] [PubMed] [Google Scholar]
- 9.Naamane, N., van Helden, J., and Eizirik, D. L. (2007) BMC Bioinformatics 8 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim, H. J., Hawke, N., and Baldwin, A. S. (2006) Cell Death Differ. 13 738-747 [DOI] [PubMed] [Google Scholar]
- 11.Karin, M., Cao, Y., Greten, F. R., and Li, Z. W. (2002) Nat. Rev. Cancer 2 301-310 [DOI] [PubMed] [Google Scholar]
- 12.Karmann, K., Min, W., Fanslow, W. C., and Pober, J. S. (1996) J. Exp. Med. 184 173-182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alberts, B., Bray, D., Lewis, J., Raff, M., Roberts, K., and Watson, J. D. (1994) in Molecular Biology of the Cell, 3rd Ed., pp. 771-785, Garland Publishing, New York
- 14.Holtmann, H., and Wallach, D. (1987) J. Immunol. 139 1161-1167 [PubMed] [Google Scholar]
- 15.Delhase, M., Hayakawa, M., Chen, Y., and Karin, M. (1999) Science 284 309-313 [DOI] [PubMed] [Google Scholar]
- 16.Prajapati, S., Verma, U., Yamamoto, Y., Kwak, Y. T., and Gaynor, R. B. (2004) J. Biol. Chem. 279 1739-1746 [DOI] [PubMed] [Google Scholar]
- 17.Trinchieri, G., and Sher, A. (2007) Nat. Rev. Immunol. 7 179-190 [DOI] [PubMed] [Google Scholar]
- 18.Gloire, G., Legrand-Poels, S., and Piette, J. (2006) Biochem. Pharmacol. 72 1493-1505 [DOI] [PubMed] [Google Scholar]
- 19.Blanque, R., Meakin, C., Millet, S., and Gardner, C. R. (1998) Gen. Pharmacol. 31 301-306 [DOI] [PubMed] [Google Scholar]
- 20.Gross, S., and Piwnica-Worms, D. (2005) Nat. Methods 2 607-614 [DOI] [PubMed] [Google Scholar]
- 21.Nejjari, M., Hafdi, Z., Dumortier, J., Bringuier, A. F., Feldmann, G., and Scoazec, J. Y. (1999) Int. J. Cancer 83 518-525 [DOI] [PubMed] [Google Scholar]
- 22.Wullaert, A., van Loo, G., Heyninck, K., and Beyaert, R. (2007) Endocr. Rev., 28 365-386 [DOI] [PubMed] [Google Scholar]
- 23.Werner, S. L., Barken, D., and Hoffmann, A. (2005) Science 309 1857-1861 [DOI] [PubMed] [Google Scholar]
- 24.Hastie, C. J., McLauchlan, H. J., and Cohen, P. (2006) Nat. Protoc. 1 968-971 [DOI] [PubMed] [Google Scholar]
- 25.Hoffmann, A., Levchenko, A., Scott, M. L., and Baltimore, D. (2002) Science 298 1241-1245 [DOI] [PubMed] [Google Scholar]
- 26.Higuchi, M., and Aggarwal, B. B. (1994) J. Immunol. 152 3550-3558 [PubMed] [Google Scholar]
- 27.Dri, P., Gasparini, C., Menegazzi, R., Cramer, R., Alberi, L., Presani, G., Garbisa, S., and Patriarca, P. (2000) J. Immunol. 165 2165-2172 [DOI] [PubMed] [Google Scholar]
- 28.Cheong, R., Bergmann, A., Werner, S. L., Regal, J., Hoffmann, A., and Levchenko, A. (2006) J. Biol. Chem. 281 2945-2950 [DOI] [PubMed] [Google Scholar]
- 29.Liu, Y. C., Penninger, J., and Karin, M. (2005) Nat. Rev. Immunol. 5 941-952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zandi, E., Chen, Y., and Karin, M. (1998) Science 281 1360-1363 [DOI] [PubMed] [Google Scholar]
- 31.Huxford, T., Huang, D. B., Malek, S., and Ghosh, G. (1998) Cell 95 759-770 [DOI] [PubMed] [Google Scholar]
- 32.Pando, M. P., and Verma, I. M. (2000) J. Biol. Chem. 275 21278-21286 [DOI] [PubMed] [Google Scholar]
- 33.O'Dea, E. L., Barken, D., Peralta, R. Q., Tran, K. T., Werner, S. L., Kearns, J. D., Levchenko, A., and Hoffmann, A. (2007) Mol. Syst. Biol. 3 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Covert, M. W., Leung, T. H., Gaston, J. E., and Baltimore, D. (2005) Science 309 1854-1857 [DOI] [PubMed] [Google Scholar]
- 35.Nelson, D. E., Ihekwaba, A. E., Elliott, M., Johnson, J. R., Gibney, C. A., Foreman, B. E., Nelson, G., See, V., Horton, C. A., Spiller, D. G., Edwards, S. W., McDowell, H. P., Unitt, J. F., Sullivan, E., Grimley, R., Benson, N., Broomhead, D., Kell, D. B., and White, M. R. (2004) Science 306 704-708 [DOI] [PubMed] [Google Scholar]
- 36.Barken, D., Wang, C. J., Kearns, J., Cheong, R., Hoffmann, A., and Levchenko, A. (2005) Science 308 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kearns, J. D., Basak, S., Werner, S. L., Huang, C. S., and Hoffmann, A. (2006) J. Cell Biol. 173 659-664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sniegowski, J. A., Lappe, J. W., Patel, H. N., Huffman, H. A., and Wachter, R. M. (2005) J. Biol. Chem. 280 26248-26255 [DOI] [PubMed] [Google Scholar]
- 39.Zhang, L., Patel, H. N., Lappe, J. W., and Wachter, R. M. (2006) J. Am. Chem. Soc. 128 4766-4772 [DOI] [PubMed] [Google Scholar]
- 40.Place, R. F., Haspeslagh, D., and Giardina, C. (2003) J. Cell. Physiol. 195 470-478 [DOI] [PubMed] [Google Scholar]