

Abstract
The Shaker family voltage-dependent potassium channels (Kv1) assemble with cytosolic β-subunits (Kvβ) to form a stable complex. All Kvβ subunits have a conserved core domain, which in one of them (Kvβ2) is an aldoketoreductase that utilizes NADPH as a cofactor. In addition to this core, Kvβ1 has an N terminus that closes the channel by the N-type inactivation mechanism. Point mutations in the putative catalytic site of Kvβ1 alter the on-rate of inactivation. Whether the core of Kvβ1 functions as an enzyme and whether its enzymatic activity affects N-type inactivation had not been explored. Here, we show that Kvβ1 is a functional aldoketoreductase and that oxidation of the Kvβ1-bound cofactor, either enzymatically by a substrate or non-enzymatically by hydrogen peroxide or NADP+, induces a large increase in open channel current. The modulation is not affected by deletion of the distal C terminus of the channel, which has been suggested in structural studies to interact with Kvβ. The rate of increase in current, which reflects NADPH oxidation, is ∼2-fold faster at 0-mV membrane potential than at -100 mV. Thus, cofactor oxidation by Kvβ1 is regulated by membrane potential, presumably via voltage-dependent structural changes in Kv1.1 channels.
Voltage-dependent potassium channels (Kv) are activated and eventually inactivated by depolarization. Hyperpolarizing potassium currents control the timing and frequency of action potentials and therefore are essential for inter- and intracellular electrical signaling (1, 2). The duration of the open states of potassium channels is determined by the rates of deactivation and inactivation. One of several mechanisms of inactivation is N-type inactivation, in which an N-terminal segment of the channel-forming subunit, called the inactivation ball or inactivation gate, blocks the ion conduction pathway (3, 4). Alternatively, the inactivation gate can be formed by an N-terminal segment of an auxiliary β-subunit, as in the complex of Kvβ1 and Kv1 family channels (5, 6) or in the complex of the large conductance voltage- and calcium-activated potassium channels and their β-subunits (7, 8).
Kvβ subunits assemble with Kv1 channels in the endoplasmic reticulum early in biogenesis (9), and the two form a stable complex with a stoichiometry of (Kv1)4:(Kvβ)4 that is preserved during purification (10-14). In mammalian neurons, reciprocal co-immunoprecipitation experiments have shown that all Kv1 channels are associated with Kvβ subunits and vice versa (15).
There are three mammalian Kvβ genes, Kvβ1-3 (16), and all Kvβ subunits have a highly conserved core domain consisting of ∼330 amino acid residues. By sequence analysis, this domain is homologous to aldoketoreductases (AKRs)2 (17); furthermore, high resolution x-ray crystal structures of the Kvβ2 conserved core show a canonical AKR structural fold with an NADPH cofactor tightly bound (11, 13, 18). We have shown recently that Kvβ2 is a functional AKR that converts an aldehyde to an alcohol, utilizing NADPH as cofactor (19).
Like Kvβ2, Kvβ1 has the core domain, but unlike Kvβ2, it has an N-type inactivation gate. A previous study showed that mutations in the putative catalytic site of Kvβ1 change the rate of channel inactivation (20), but did not show that Kvβ1 functions as a redox enzyme or that its activity affects channel activity. We now demonstrate the catalytic activity of Kvβ1 and its effect on channel inactivation. Furthermore, we demonstrate that the AKR activity of Kvβ1 is affected by membrane potential, presumably through the voltage-dependent structural transitions of Kv1.1.
EXPERIMENTAL PROCEDURES
Protein Expression—The cDNA of the conserved core of rat Kvβ1 (NCBI accession number NM_017303; residues 71-401) was cloned into a modified pQE70 vector (Qiagen Inc.) between the SphI and BamHI sites with a C-terminal 6-histidine tag cleavable by thrombin. XL1-Blue cells were used to express protein. Cells were grown in Luria broth at 37 °C to an absorbance of ∼1.2 and induced with 0.5 mm isopropyl β-d-thiogalactopyranoside (final concentration). Immediately after induction, the temperature was reduced to 20 °C, and cells were harvested 16 h after induction. The cell pellet was resuspended in Buffer A (20 mm Tris (pH 8.0), 300 mm KCl, 1 mm β-mercaptoethanol, and 10% glycerol by volume). Cells were broken by a high pressure homogenizer (EmulsiFlex-C3, Avestin Inc.), and cell debris was cleared by centrifugation at 40,000 × g for 40 min at 4 °C. The supernatant was then loaded onto a Talon Co2+ affinity column (Clontech). Nonspecifically bound protein was washed away with Buffer A supplemented with 20 mm imidazole, and Kvβ1 protein was eluted with Buffer A plus 300 mm imidazole. The 6-histidine tag was removed by incubation with thrombin (Roche Diagnostics) at a Kvβ1/enzyme ratio of 2 mg to 1 unit overnight at 4 °C. The protein was then loaded onto a Superdex 200 column (GE Healthcare) for final purification. The column was equilibrated with Buffer B (150 mm KCl and 20 mm Tris (pH 8.0)). The column volume was ∼25 ml, ∼7.8 ml of which was the void volume. The Kvβ1 core elutes as a single peak at ∼11.3 ml (RF ≈ 0.8). Protein concentration was determined using the BCA kit (Pierce). Compared with the Kvβ2 core, the Kvβ1 core is less stable and tends to aggregate. A large fraction of the overexpressed Kvβ1 core protein was not in the supernatant after the ultracentrifugation (see Fig. 1A, inset, compare lanes 2 and 3).
FIGURE 1.

Enzymatic properties of Kvβ1. A, the conserved core (amino acids 71-401) of Kvβ1 elutes as a single peak on a size exclusion gel filtration column. The inset shows an SDS gel of samples taken at various steps during the purification process. Lane 1, protein standard markers with molecular masses indicated; lane 2, whole-cell lysate; lane 3, supernatant of the lysate after ultra-centrifugation; lane 4, flow-through from the cobalt affinity column; lane 5, elution with 20 mm imidazole; lane 6, elution with 300 mm imidazole; lane 7, thrombin digestion to remove the 6-histidine tag; lane 8, elution peak from the fast protein liquid chromatography size exclusion column. mAu, milli-absorbance units. B, fluorescence emission spectra of the purified Kvβ1 core before (0 min) and at different time points after mixing with 5 mm 4-CY. The excitation wavelength was 360 nm, at which protein-bound NADPH has the highest absorption. C, fraction of the fluorescence peak plotted versus time for the wild-type (WT; •) and K152M mutant (○) Kvβ1 core after mixing with 4-CY. The smooth curve for wild-type Kvβ1 is a single-component exponential function fit to the data. Error bars are S.E. from three independent experiments. D, rate constants of the hydride transfer reaction plotted versus 4-CY concentrations for the single-turnover reaction, i.e. the consumption of protein-bound NADPH, for Kvβ1 (▪) and Kvβ2 (○). Error bars are S.E. from three to five independent experiments.
Single-turnover Enzymatic Reaction Measurement—Single-turnover hydride transfer reaction rates were measured on a FluoroMax-3 spectrofluorometer (HORIBA Jobin Yvon Inc.) at room temperature (20-24 °C). The reaction mixture contained 2 μm Kvβ1 protein and different concentrations of substrates in a final volume of 150 μl. 4-Cyanobenzaldehyde (4-CY) was first prepared in ethanol as a 0.5 m stock solution and then diluted in Buffer B to the desired concentrations. The excitation wavelength was set at 360 nm with a 1-nm band-pass slit size, and the emission was measured at 454 nm with a 5-nm band-pass slit size at various time points after the reaction was initiated with the addition of 4-CY. The K152M mutant Kvβ1 protein has much lower NADPH fluorescence compared with the wild-type protein, and therefore, an equal molar concentration (2 μm) of free NADPH was supplied in the single-turnover experiment.
Channel Expression and Electrophysiology—Full-length cDNA of rat Kv1.1 (NCBI accession number NM_173095) or that of rat Kvβ1 was cloned into a modified pBluescript vector (a gift from Dr. Mark Sonders, Columbia University) between the KpnI and EcoRI sites for in vitro transcription. Kv1.1-inact was generated by splicing the DNA sequence encoding residues 1-70 of Kvβ1 into that of Kv1.1 (encoding residues 2 to 495-stop) by the overlapping PCR method, and the PCR product was then inserted into the same modified pBluescript vector. Kv1.1ΔC was generated by amplifying the DNA sequence encoding residues 1 to 435-stop using primers CCGGTACCATGTATCCGGAATCAACC and CCGAATTCTCATAAGTTAGGAGAACTAAC, and the PCR product was then inserted into the same modified pBluescript vector. Several shorter constructs were also tested but failed to generate sufficiently high expression levels for patch clamp studies.
Point mutations were made using the QuikChange kit (Stratagene). The sequences of all constructs were verified by DNA sequencing through the entire coding region.
mRNA was prepared by in vitro T7 polymerase transcription after DNA was linearized with NotI. mRNAs were purified using TRIzol reagent (Invitrogen) and injected into Xenopus oocytes for channel expression. For coexpression, mRNAs of Kv1.1 and Kvβ1 were mixed and injected together into oocytes. Different ratios of the two were tested to achieve a sufficiently high level of Kvβ1 expression so that most of the channels assembled with Kvβ1, as indicated by the almost complete inactivation of channel current. Wild-type Kvβ1 has a cysteine residue at position 7 that can be oxidized on inside-out patches to affect channel inactivation, and this can be prevented by the addition of reducing reagents such as dithiothreitol (5). To eliminate concerns that the change in inactivation could be due to cysteine oxidation, we mutated the cysteine to alanine and used Kvβ1(C7A) as the “wild type” throughout this work. In separate experiments, 4-CY was found to induce similar changes in Kv1.1 coexpressed with wild-type Kvβ1 in the presence of 5 mm dithiothreitol (data not shown).
Patch clamp currents were recorded on inside-out patches pulled from oocytes 3-5 days after injection. Electrodes were drawn from patch glass (G85150T-4, Warner Instruments) and polished (MP-803, Narishige Co.) to a resistance of 0.6-1 megaohms. The pipette solution contained 130 mm KCl, 2 mm MgCl2, and 10 mm KH2PO4 (pH 7.4). The bath solution contained 80 mm KCl, 5 mm EGTA, and 50 mm KH2PO4 (pH 7.4). The pH was adjusted with KOH. K+ currents were elicited by holding the patch at -100 mV for at least 30 s and stepping to +60 mV for 200 ms. The analog signals were amplified by an Axon 200B patch clamp amplifier (Molecular Devices Inc.), filtered at 1 kHz using the built-in Bessel filter, digitized at 100 μs by Digidata 1322a (Molecular Devices Inc.), and recorded to a computer hard disk. The volume of the recording chamber (Warner Instruments) was ∼200 μl, and complete exchange of solution was achieved by perfusing at least 1 ml of solution by gravity flow at a flow rate of 2-3 ml/min.
Data Analysis—The Kvβ1 subunit confers fast inactivation to otherwise non-inactivating Kv1.1 channels by a “ball-and-chain” mechanism (5). Inactivation time constants were obtained by fitting the current decay with exponential functions in Clampfit software (Molecular Devices Inc.). The current decay was best fit with a two-component exponential function: a predominant and fast component and a minor and slower component (see Table 1). The inverse of the fast time constant was defined as the on-rate of the N-type inactivation and is plotted in Fig. 6B. The slower component was likely contributed by the C-type inactivation, and its effect was minimized in the high K+ recording solution. Recovery from inactivation was measured with a paired-pulse protocol (21). Briefly, from a holding potential of -100 mV, inactivation was induced by a first pulse with a depolarization to +60 mV for 200 ms; the patch was then repolarized back to the holding potential; and a second pulse to +60 mV was applied after a delay of 0.01-1 s. The inactivating portion of the current, i.e. peak current subtracted by steady-state current (at the end of the 200-ms pulse), was normalized and is plotted versus the delay time in Fig. 6C. Recovery from inactivation was well fit by a single-component exponential function.
TABLE 1.
Parameters of channel activation and inactivation
τfast and τslow are the time constants obtained from fitting the current decay with a two-component exponential function, and the fraction of each component is ffast and fslow, respectively. ΔIss is the increase in steady-state current normalized by the initial inactivating current. V½ is the voltage at which the relative Popen of the channel reaches 0.5. τrec is the time constant for recovery from inactivation. Data are presented as means ± S.E., and n indicates the number of patches. A one-way ANOVA test was used to assess whether changes in a parameter were statistically significant after perfusion of an oxidant. The 4-CY effects on Kv1.1 + Kvβ1, Kv1.1-inact, Kv1.1 + Kvβ1(K152M), and Kv1.1ΔC + Kvβ1 were compared with the 1% ethanol vehicle control effects, and the results are shown (**, p<0.01; ***, p<0.001; NS, p>0.05). The NADP+ effects on Kv1.1 + Kvβ1 and Kv1.1ΔC + Kvβ1 were compared with those on Kv1.1-inact, and the results are shown (Δ, p<0.05; ΔΔ, p<0.01; ΔΔΔ, p<0.001; NS, p>0.05). The H2O2 effects on Kv1.1 + Kvβ1, Kv1.1 + Kvβ1 (K152M), and Kv1.1ΔC + Kvβ1 were compared with those on Kv1.1-inact, and the results are shown (#, p<0.05; ##, p<0.01; ###, p<0.001; NS, p>0.05). For τfast, τslow, ffast, and fslow, the -fold change after perfusion of an oxidant was assessed in the ANOVA test.
| τfast | ffast | τslow | fslow | ΔIss | V½ | τrec | |
|---|---|---|---|---|---|---|---|
| ms | ms | mV | ms | ||||
| 4-CY | |||||||
| Kv1.1 + Kvβ1 | |||||||
| Before | 3.3 ± 0.1 | 0.83 ± 0.01 | 35.1 ± 2.2 | 0.17 ± 0.01 | −59.1 ± 1.6 | 71.7 ± 6.1 | |
| After | 11.2 ± 0.5*** | 0.42 ± 0.02*** | 103.3 ± 0.3** | 0.58 ± 0.02*** | 0.50 ± 0.04*** | −65.6 ± 0.9 | 96.0 ± 10.9 |
| n | 30 | 30 | 30 | 30 | 30 | 10 | 11 |
| Kv1.1 + Kvβ1 (vehicle) | |||||||
| Before | 3.3 ± 0.2 | 0.85 ± 0.01 | 35.7 ± 2.3 | 0.15 ± 0.01 | |||
| After | 3.9 ± 0.07 | 0.79 ± 0.01 | 43.5 ± 2.8 | 0.21 ± 0.01 | 0.07 ± 0.003 | ||
| n | 7 | 7 | 7 | 7 | 7 | ||
| Kv1.1-inact | |||||||
| Before | 3.3 ± 0.1 | 0.88 ± 0.01 | 16.8 ± 1.8 | 0.12 ± 0.01 | |||
| After | 4.2 ± 0.2NS | 0.79 ± 0.01NS | 27.9 ± 4.7NS | 0.21 ± 0.01NS | 0.06 ± 0.01NS | ||
| n | 8 | 8 | 8 | 8 | 8 | ||
| Kv1.1 + Kvβ1(K152M) | |||||||
| Before | 4.2 ± 0.1 | 0.80 ± 0.02 | 56.4 ± 6.4 | 0.20 ± 0.02 | |||
| After | 4.7 ± 0.2NS | 0.75 ± 0.02NS | 55.2 ± 6.4NS | 0.25 ± 0.05NS | 0.06 ± 0.01NS | ||
| n | 7 | 7 | 7 | 7 | 7 | ||
| Kv1.1ΔC + Kvβ1 | |||||||
| Before | 4.4 ± 0.1 | 0.74 ± 0.01 | 44.0 ± 2.2 | 0.26 ± 0.01 | −61.5 ± 0.9 | 75.8 ± 7.1 | |
| After | 10.9 ± 0.5*** | 0.33 ± 0.03*** | 91.8 ± 5.4NS | 0.67 ± 0.03*** | 0.52 ± 0.03*** | −66.1 ± 1.1 | 91.8 ± 5.1 |
| n | 13 | 13 | 13 | 13 | 13 | 3 | 3 |
| NADP+ | |||||||
| Kv1.1 + Kvβ1 | |||||||
| Before | 3.1 ± 0.1 | 0.84 ± 0.01 | 36.7 ± 3.0 | 0.16 ± 0.01 | −59.7 ± 1.8 | 78.4 ± 4.6 | |
| After | 8.6 ± 0.8Δ | 0.52 ± 0.03ΔΔΔ | 75.1 ± 5.3Δ | 0.48 ± 0.03ΔΔΔ | 0.36 ± 0.02ΔΔΔ | −64.4 ± 2.5 | 92.7 ± 8.0 |
| n | 18 | 18 | 18 | 18 | 18 | 3 | 3 |
| Kv1.1-inact | |||||||
| Before | 3.3 ± 0.04 | 0.88 ± 0.01 | 19.1 ± 2.1 | 0.12 ± 0.01 | |||
| After | 3.6 ± 0.1 | 0.84 ± 0.02 | 24.8 ± 3.7 | 0.16 ± 0.02 | 0.03 ± 0.01 | ||
| n | 3 | 3 | 3 | 3 | 3 | ||
| Kv1.1ΔC + Kvβ1 | |||||||
| Before | 4.4 ± 0.1 | 0.75 ± 0.01 | 44.5 ± 3.8 | 0.25 ± 0.01 | −62.6 ± 1.1 | 73.8 ± 4.8 | |
| After | 8.5 ± 0.4Δ | 0.46 ± 0.04NS | 73.8 ± 3.4NS | 0.54 ± 0.04ΔΔ | 0.44 ± 0.03ΔΔΔ | −66.6 ± 0.7 | 97.4 ± 7.4 |
| n | 11 | 11 | 11 | 11 | 11 | 3 | 3 |
| H2O2 | |||||||
| Kv1.1 + Kvβ1 | |||||||
| Before | 3.2 ± 0.1 | 0.85 ± 0.02 | 33.7 ± 3.0 | 0.15 ± 0.02 | −59.9 ± 1.4 | 76.8 ± 8.2 | |
| After | 8.3 ± 0.9### | 0.51 ± 0.05## | 81.8 ± 10.4## | 0.49 ± 0.05## | 0.40 ± 0.06### | −64.9 ± 1.5 | 93.2 ± 5.5 |
| n | 5 | 5 | 5 | 5 | 5 | 3 | 3 |
| Kv1.1-inact | |||||||
| Before | 3.3 ± 0.1 | 0.89 ± 0.01 | 19.5 ± 1.2 | 0.11 ± 0.01 | |||
| After | 3.9 ± 0.1 | 0.82 ± 0.01 | 27.2 ± 2.4 | 0.18 ± 0.01 | 0.04 ± 0.01 | ||
| n | 4 | 4 | 4 | 4 | 4 | ||
| Kv1.1 + Kvβ1(K152M) | |||||||
| Before | 4.4 ± 0.2 | 0.76 ± 0.01 | 54.6 ± 7.7 | 0.24 ± 0.01 | |||
| After | 8.5 ± 0.7NS | 0.44 ± 0.06## | 102.1 ± 12.4## | 0.56 ± 0.06NS | 0.35 ± 0.05## | ||
| n | 5 | 5 | 5 | 5 | 5 | ||
| Kv1.1ΔC + Kvβ1 | |||||||
| Before | 4.5 ± 0.1 | 0.73 ± 0.02 | 41.6 ± 2.2 | 0.27 ± 0.02 | −62.3 ± 0.7 | 73.1 ± 4.5 | |
| After | 8.9 ± 0.4# | 0.48 ± 0.02## | 78.3 ± 5.3## | 0.52 ± 0.02NS | 0.46 ± 0.03### | −67.2 ± 1.2 | 92.1 ± 9.1 |
| n | 8 | 8 | 8 | 8 | 8 | 3 | 3 |
FIGURE 6.

Modulation of the N-type inactivation. A, paired-pulse measurement of recovery from inactivation for Kv1.1 coexpressed with Kvβ1 before (upper panel) and after (lower panel) 4-CY modulation (on a single patch). The patch was held at -100 mV, and inactivation was first induced by a pulse to +60 mV for 200 ms (time 0). The patch was then repolarized to -100 mV, and a second pulse to +60 mV was applied after different time intervals. Example traces recorded after 20, 75, 125, and 225 ms are shown. B, on-rate of N-type inactivation before (○) and after (•) 4-CY modulation plotted versus membrane potentials. Error bars are S.E. from 10 patches. Paired Student's t test was used to assess the values before and after 4-CY perfusion, and the results are shown (**, p < 0.01; ***, p < 0.001). C, fraction of recovery from inactivation plotted versus interpulse duration before (○) and after (•) 4-CY modulation. The solid curves are a single-component exponential fit. Error bars are S.E. from 11 patches. The time constants are 71.7 ± 6.1 ms (before 4-CY) and 96.0 ± 10.9 ms (after 4-CY).
Voltage-dependent channel activation was measured using an instantaneous tail current protocol. Briefly, a patch was held at -100 mV and stepped to different voltages between -90 and +60 mV for 200 ms and then stepped back to -100 mV. When the voltage was reversed to -100 mV, the high extracellular potassium concentration (∼150 mm) produced a pronounced “tail current,” shown in Fig. 5C. The peak of the current is a measure of the fraction of channels activated by the preceding 200-ms pulse. The normalized peaks of the tail currents were plotted versus membrane potential and fit with the following Boltzmann function in Clampfit (Equation 1),
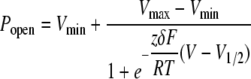 |
(Eq.1 |
where R is the universal gas constant; T is the absolute temperature; F is the Faraday constant; and Vmin and Vmax are the minimum and maximum voltages used for channel activation, respectively. The two parameters obtained from curve fitting are zδ and V½, where zδ is the nominal amount of charges that move across the membrane voltage field, and V½ is the voltage at which the relative Popen is 0.5.
FIGURE 5.

Modulation of channel activation gating. A, K+ currents recorded on the same inside-out patch pulled from an oocyte expressing both Kv1.1 and Kvβ1 before (left panel) and after (right panel) perfusion of 5 mm 4-CY. The holding potential was -100 mV, and currents were elicited by depolarizing pulses to voltages between -90 and +60 mV in 10-mV increments. B, I-V plot for the peak (Ip; left panel) and steady-state (Is; right panel) currents before (○) and after (•) 4-CY modulation. Currents were normalized to the peak or steady-state amplitude at +60 mV before 4-CY perfusion. Error bars are S.E. of 10 independent patches. C, left panel, inward tail K+ currents elicited by a voltage step to -100 mV after a depolarizing pulse to potentials between -90 and +60 mV before 4-CY perfusion. The gray vertical line indicates where the current amplitudes were measured for analysis. The inset shows the complete recording traces, and the dashed box demarcates the part of the traces that were magnified. Right panel, peak of the tail current normalized and plotted versus the activation voltage before (○) and after (•) 4-CY modification. The solid curves are Boltzmann functions fit to the data. The V½ values are -59.1 ± 1.6 mV (before 4-CY) and -65.6 ± 0.9 mV (after 4-CY). Error bars represent S.E. from 10 different patches.
The rate of channel modulation at -100 mV was measured by first perfusing a patch with either 4-CY or NADP+, holding the patch at -100 mV, and depolarizing the patch to +60 mV for 200 ms every 60 s. The normalized current amplitudes were then plotted versus the accumulative exposure time and fit with a single-component exponential function. The inverse of the time constant was defined as the rate of modulations.
Because significant channel inactivation occurs at 0 mV, the rate of channel modulation at 0 mV was measured with a slightly different protocol to allow channels to recover from inactivation at -100 mV. After a stable inside-out patch was obtained, the patch was held at 0 mV, and 4-CY or NADP+ was perfused for 2 min. At the end of the 2-min perfusion, 4-CY or NADP+ was washed away. The holding potential was then stepped to -100 mV for 30 s to allow complete channel recovery from inactivation, and current was elicited by stepping the membrane potential to +60 mV. This process was then repeated until the modulation reached steady state. Similar to the measurement at -100 mV, the normalized current amplitude versus the accumulative exposure time was well fit with a single-component exponential function.
Data Statistics—The Origin 7.5 software package was used for statistical analysis of the data. The results are expressed as means ± S.E. Student's t tests (both paired and unpaired) and one-way analysis of variance (ANOVA) were used to assess changes in a mean value.
Chemical Reagents—Chemical reagents were purchased from Sigma, unless indicated otherwise. 4-CY was first dissolved in ethanol and then diluted to the desired final concentration. NADP+ was purchased as a sodium salt, and because sodium ion blocks potassium channels from the intracellular side, we exchanged the sodium with potassium ion using a size exclusion column. Isopropyl β-d-thiogalactopyranoside, dithiothreitol, and kanamycin were purchased from LabScientific, Inc.
RESULTS
Kvβ1 Is a Functional AKR—To test the enzymatic activity of Kvβ1, its conserved core (residues 71-401) was expressed and purified to homogeneity (Fig. 1A). The purified protein elutes as a single peak on a Superdex 200 size exclusion column at ∼11.3 ml (Fig. 1A), identical to Kvβ2, which is known to form a tetramer (18). Also similar to Kvβ2, the purified Kvβ1 protein has a prominent fluorescence emission peak at 454 nm when excited at 360 nm (Fig. 1B), indicative of a co-purified NADPH cofactor. These properties suggest that the purified Kvβ1 core has the correct structural fold and likely has AKR activities. Because NADP+ does not have fluorescence emission, an enzymatic redox reaction, i.e. transfer of a hydride from the Kvβ-bound NADPH to a substrate, can be monitored by a reduction in the 454 nm fluorescence.
A known Kvβ2 substrate (4-CY, 5 mm) was then mixed with purified Kvβ1, and the fluorescence intensity at 454 nm was monitored over time. The NADPH fluorescence decreased over time following a single-component exponential function with a time constant of 3.6 ± 0.3 min (n = 3) (Fig. 1, B and C). The single-component exponential kinetics is consistent with the single-step hydride transfer reaction. The inverse of the exponential time constant was defined as the hydride transfer rate, and the rate was measured at different 4-CY concentrations and is plotted in Fig. 1D, along with the same measurement obtained with Kvβ2 (19). Compared with Kvβ2, Kvβ1 has a slightly higher enzymatic rate.
Lysine 152 at the catalytic site was mutated to methionine because the same mutation to the conserved lysine at the equivalent position in Kvβ2 and 7α-hydroxysteroid dehydrogenase showed significantly reduced enzymatic activity (19, 22). Kvβ1(K152M) was expressed, purified, and mixed with 5 mm 4-CY, and the NADPH fluorescence remained essentially the same after 20 min (Fig. 1C). We conclude that Kvβ1 is a functional AKR and that our fluorescence assay measured its activity.
Kvβ1 Substrate Increases Kv1.1 Current—To examine whether the AKR activity of Kvβ1 modulates channel function, Kv1.1 was coexpressed with Kvβ1 in Xenopus oocytes, and channel activity was recorded in inside-out patches. Kvβ1 with cysteine 7 mutated to alanine was used throughout this study as the wild type to eliminate a known cysteine oxidation effect at this position (see “Experimental Procedures” and Ref. 5). The presence of fast inactivation indicates that Kv1.1 and Kvβ1 are co-assembled because Kv1.1 expressed alone has non-inactivating current (Fig. 2D). 4-CY was perfused to the intracellular side of the channel until the current reached steady state and was then replaced with normal inside solution. After the 4-CY perfusion, the on-rate of channel inactivation at +60 mV slowed 3.4-fold from 316 ± 12 to 93 ± 4 s-1 (n = 30), and as a result, potassium current increased significantly (Fig. 2A and Table 1). The 4-CY effect was highly reproducible and was observed in all 30 inside-out patches from seven batches of oocytes. To quantify current increase, we defined the parameter ΔIss, i.e. increase in steady-state current expressed as a fraction of the initial inactivating current (see “Experimental Procedures”), and the ΔIss is 50 ± 4% (n = 30) (Fig. 2G). In a vehicle control of 1% ethanol, a much smaller current increase was observed with a ΔIss of 7 ± 0.3% (n = 7) (Fig. 2, C and G). Therefore, 1% ethanol has a small potentiation effect on open channel current, but the large changes in channel inactivation and current level are due mostly to the presence of 4-CY (p < 0.001 in one-way ANOVA test; see Table 1). Channel modulation reached steady state in ∼16 min and was only slightly reversed in the presence of 2 mm dithiothreitol (Fig. 2B), indicating that the modulation is not the result of the oxidation of free cysteines.
FIGURE 2.

A functional AKR core mediates channel modulation. Shown are current traces of Kv1.1 coexpressed with wild-type Kvβ1 (A and C), Kv1.1 only (D), Kv1.1-inact (E), and Kv1.1 coexpressed with Kvβ1(K152M) (F). In each case, the black trace was recorded before 4-CY or vehicle (1% ethanol) perfusion. After perfusion, a patch was held at -100 mV, and its current level was monitored every 30 s with a 200-ms pulse to +60 mV until the current level reached steady state. The solution was then exchanged with the normal inside buffer, and the red trace was recorded. This protocol was also used for measuring H2O2 and NADP+ modulation shown in Figs. 3 and 4. Scale bars in Figs. 2, 3, 4, 5, 6, 7 and 8 represent 300 pA and 10 ms. B, steady-state current from an inside-out patch expressing Kv1.1 and Kvβ1 plotted versus time. Perfusion of 4-CY and dithiothreitol (DTT) is indicated by horizontal bars. G, change in steady-state current (ΔIss) after 4-CY modulation. Error bars are S.E., and n is the number of patches. One-way ANOVA test was used to assess the effects of 4-CY, and the results are shown (***, p < 0.001; n. s., not significantly different with p > 0.05).
To test whether modulation of N-type inactivation requires the presence of the conserved core of Kvβ1, we produced a chimeric channel (Kv1.1-inact) in which amino acid residues 1-70 of Kvβ1 were spliced to the N terminus of Kv1.1. Therefore, Kv1.1-inact is a channel that has an N-terminal inactivation gate from Kvβ1 but does not have the conserved AKR core. As expected, Kv1.1-inact produced inactivating K+ current similar to that produced by coexpression of Kv1.1 and Kvβ1 (Fig. 2E). When 4-CY was perfused to the intracellular side of Kv1.1-inact, only a small change in current was observed with a ΔIss of 6 ± 1% (n = 8) (Fig. 2, E and G, and Table 1). The small ΔIss is not significantly different from that of the vehicle control (p > 0.05 in one-way ANOVA test) (Fig. 2, C and G, and Table 1). Thus, the 4-CY effect is dependent upon the presence of the conserved AKR core.
Next, we examined whether the AKR activity is required for channel modulation. We made the K152M mutation in Kvβ1 and coexpressed the mutant with Kv1.1. Kvβ1(K152M) induced channel inactivation, and the on-rate of channel inactivation was 238 ± 6 s-1 (n = 7), which is significantly slower than that of the wild type (p < 0.001 in unpaired t test) (Fig. 2F and Table 1). The slower inactivation rate is consistent with a previous study that also reported a slower rate when the lysine was mutated to alanine (20). Here, we focused on the 4-CY effect, and we found that 4-CY slightly increased the current with a ΔIss of 6 ± 1% (n = 7) (Fig. 2, F and G, and Table 1). The slight change in current is not significantly different compared with the vehicle control and Kv1.1-inact (p > 0.05 in one-way ANOVA test). This result suggests that the 4-CY effect requires not just the presence but also the catalytic activity of the AKR core.
Oxidation of Kvβ-bound NADPH Induces Channel Modulation—That a substrate induces channel modulation through a functional AKR suggests that NADPH oxidation is required. To find out whether NADPH oxidation is sufficient to induce channel modulation, we next investigated whether reagents (other than substrates) that oxidize Kvβ1-bound NADPH also induce similar changes in channel current. Two non-substrate oxidizing reagents, hydrogen peroxide (H2O2) and NADP+, were tested.
H2O2 is a physiologically relevant compound because its concentration changes during inflammation and oxidative stress (23). When 10 mm H2O2 was mixed with purified Kvβ1 protein, NADPH fluorescence was eliminated, indicating that the bound cofactor was oxidized (Fig. 3A). In a parallel experiment, H2O2 (100 μm) was perfused to inside-out patches expressing both Kv1.1 and Kvβ1, and an increase in channel current (similar to that induced by a substrate) was observed with a ΔIss of 40 ± 6% (n = 5) (Fig. 3, B and F, and Table 1). By contrast, as high as 10 mm H2O2 induced only a rather small change in Kv1.1-inact and Kv1.1 (Fig. 3, C, D, and F) with ΔIss values of 4 ± 1% (n = 4) and 5 ± 1% (n = 3), respectively. Combined, these results indicate that the H2O2 effect is mediated by the AKR core.
FIGURE 3.

Channel modulation by H2O2. A, fluorescence spectra of purified Kvβ1 core before (black trace) and after (red trace) mixing with 10 mm H2O2. Shown are K+ currents recorded from inside-out patches expressing both Kv1.1 and Kvβ1 (B), Kv1.1-inact only (C), Kv1.1 only (D), and Kv1.1 coexpressed with Kvβ1(K152M) (E) before (black traces) and after (red traces) perfusion of H2O2. 100 μm H2O2 was perfused in B and E, whereas 10 mm H2O2 was perfused in C and D. F, ΔIss after H2O2 modulation. Error bars are S.E., and n is the number of patches. One-way ANOVA test was used to assess the H2O2 effect, and the results are shown (##, p < 0.01; n. s., not significantly different with p > 0.05).
Although H2O2 itself can directly oxidize Kvβ-bound NADPH, it can also react with membrane lipids to produce aldehydic metabolic intermediates (24) that could be Kvβ substrates. Thus, the H2O2 effect could potentially have two pathways. We therefore tested H2O2 on Kvβ1(K152M). We found that H2O2 induced a large increase in current with a ΔIss of 35 ± 5% (n = 5) (Fig. 3, E and F, and Table 1), which is not significantly different from that of Kv1.1 coexpressed with Kvβ1 (p > 0.05 in ANOVA test). We conclude that the H2O2 effect is mostly, if not completely, due to its direct oxidation of the bound cofactor.
We next tested NADP+. When purified Kvβ1 protein was mixed with 0.2 mm NADP+, the fluorescence peak at 454 nm decreased to 35 ± 1% of the original intensity (n = 3) (Fig. 4A). The decrease in fluorescence is likely due to the displacement of NADPH from a relatively non-polar binding site environment to an aqueous environment. We confirmed that NADPH had been replaced by NADP+ by separating Kvβ1 from the free cofactor after the fluorescence no longer changed (Fig. 4A). Although it is possible that the bound cofactor was oxidized by NADP+ instead of being exchanged, it is clear that NADP+ converted Kvβ-bound NADPH to NADP+.
FIGURE 4.

Channel modulation by NADP+. A, fluorescence spectra of the purified Kvβ1 core before (black trace) and after (red trace) mixing with 0.2 mm NADP+. After the fluorescence no longer changed, Kvβ1 was purified from the reaction mixture, and its spectrum was recorded (blue trace). Shown are K+ currents recorded from inside-out patches expressing both Kv1.1 and Kvβ1 (B) and Kv1.1-inact only (C) before (black traces) and after (red traces) perfusion of 0.2 mm NADP+. D, ΔIss after NADP+ modulation. Error bars are S.E., and n is the number of patches. Unpaired Student's t test was used to assess the NADP+ effect on Kv1.1 + Kvβ1 and Kv1.1-inact (ΔΔΔ, p < 0.001).
It was observed previously that NADP+ reduces inactivation in whole-cell patch-clamped cells coexpressing Kv1.5 and Kvβ1 (25). A potential complication of the whole-cell patch clamp experiment is that NADP+ could generate metabolic intermediates that react with Kvβ through other oxidoreductases. To eliminate this concern, we perfused 0.2 mm NADP+ directly to inside-out patches expressing both Kv1.1 and Kvβ1, and we observed an increase in channel current with a ΔIss of 36 ± 2% (n = 18) (Fig. 4, B and D, and Table 1), which is similar to that induced by H2O2. The AKR core is required for this effect because when NADP+ was perfused to the intracellular side of Kv1.1-inact, the current was almost unchanged with a ΔIss of 3 ± 1% (n = 3) (Fig. 4C and Table 1). Combined, the H2O2 and NADP+ effects indicate that oxidation of Kvβ-bound NADPH is sufficient to potentiate Kv1 current.
Modulation of Both Activation and Inactivation Gatings by Oxidation—To further dissect the 4-CY effect, we next studied the 4-CY effect on both channel activation and inactivation processes. 4-CY induced a shift in the midpoint of the current-voltage relationship (V½) of the Kv1.1-Kvβ1 complex from -59.1 ± 1.6 to -65.6 ± 0.9 mV (n = 10) (Fig. 5, A-C). Although the shift in V½ is modest, the increase in channel current is large and is ∼6-fold at -60 mV (Fig. 5B, right panel). Because many cells have resting membrane potentials at around that voltage, a large change in current level may significantly affect the excitability of a cell.
One prominent feature of the effect of exposure to 4-CY is the almost complete loss of channel inactivation. To further quantify the change, the on-rate of channel inactivation was measured at voltages between -20 and +60 mV, where significant channel inactivation was observed. After 4-CY perfusion, a reduction in the on-rate was observed at all voltages (Fig. 6B). In contrast to the large change in the on-rate, the rate of recovery from inactivation (measured using a paired-pulsed protocol) was unchanged (Fig. 6, A and C, and Table 1).
Because the V½ for Kv1.1 coexpressed with Kvβ1 is approximately -60 mV, all channels are maximally activated for voltages more positive than -20 mV (Fig. 5C), and as a result, the large increase in current at positive membrane potentials is due mainly to the slower on-rate of inactivation. In excitable cells, oxidation of NADPH bound to Kvβ1 increases K+ current at positive voltages, shortens the action potential, and speeds up repolarization of a cell.
Deletion of the Kv1.1 Distal C Terminus Does Not Affect Current Potentiation—It has been suggested in two structural studies that the C terminus of a Kv1 channel interacts with Kvβ (13, 14), which naturally led us to hypothesize that the C terminus is involved in functional coupling. To test this, we constructed Kv1.1ΔC by deleting the distal C-terminal 60 amino acid residues of the channel. Several other constructs with more C-terminal residues deleted were also generated, but none expressed at sufficiently high levels for patch clamp studies. When aligned with the Kv1.2 channel, the high resolution atomic structure of which was solved by x-ray crystallography (Protein Data Bank code 2A79) (13), Kv1.1ΔC has 15 amino acid residues beyond the last structurally resolved C-terminal residue in the Kv1.2 structure. Because the last structurally resolved residue in Kv1.2 is ∼51 Å away from the closest residue in Kvβ in its crystallized complex, the C-terminal 15 amino acids in Kv1.1ΔC are unlikely to interact with Kvβ, even in their most extended conformation.
When paired with Kvβ1, the on-rate of Kv1.1ΔC inactivation at +60 mV is 225 ± 4 s-1 (n = 32), which is slightly slower than that of the wild type, and the rate of recovery from inactivation is 14 ± 1 s-1 (n = 9), which is similar to that of the wild type. It seems that deleting the C terminus has only a modest effect on channel inactivation. 4-CY, NADP+, and H2O2 all induced large potentiations of channel current (Fig. 7, A-D) with ΔIss values of 52 ± 3% (n = 13), 44 ± 3% (n = 11), and 46 ± 3% (n = 8), respectively. All three ΔIss values are significantly different from that of Kv1.1-inact (p < 0.001 in unpaired t tests). We conclude that the C terminus of Kv1.1 is not required for its modulation by Kvβ1 observed here.
FIGURE 7.

Modulation of channel inactivation on Kv1.1ΔC + Kvβ1. Shown are current traces recorded on inside-out patches coexpressing Kv1.1ΔC and Kvβ1 before (black traces) and after (red traces) perfusion of 5 mm 4-CY (A), 0.2 mm NADP+ (B), or 0.1 mm H2O2(C). D, ΔIss after NADPH oxidation in Kv1.1ΔC + Kvβ1 (black bars) and Kv1.1-inact (white bars). Error bars are S.E., and n is the number of patches. Unpaired Student's t test was used to assess the values from Kv1.1ΔC + Kvβ1 and Kv1.1-inact (see also Table 1; ***, p < 0.001).
Modulation of Enzymatic Reaction Rates by Membrane Potentials—The tight association between Kv1 and Kvβ has inspired the hypothesis that AKR function is modulated by membrane potentials because of the different conformations of a channel, i.e. Kvβ could be a voltage-dependent enzyme (18). Although the physiological utility of such an AKR is not obvious, an example of a voltage-dependent enzyme has emerged recently. A voltage-dependent phosphatase has a membrane-embedded voltage-sensing domain covalently linked to a cytosolic phosphatase, the activity of which is different at different membrane potentials (26).
We do not have a direct measure of enzymatic activity for Kvβ coexpressed with Kv1.1 on cell membranes. However, because we know that NADPH oxidation induces channel modulation, we could measure the rate of channel modulation as an indirect readout for the rate of NADPH oxidation. After 4-CY was perfused to the intracellular side of Kv1.1 coexpressed with Kvβ1, channel current increased over time and reached a steady state (Fig. 8A). When the normalized current amplitude was plotted against time, the data points could be fit with a single-component exponential function (Fig. 8B). The inverse of the exponential time constant is defined as the rate of channel modulation.
FIGURE 8.

Rate of channel modulation is voltage-dependent. A, current traces from an inside-out patch coexpressing Kv1.1 and Kvβ1 recorded at the indicated 4-CY exposure times. The gray vertical line indicates the time point (17 ms after the pulse) at which the current amplitudes were taken for further analysis in B. After 16 min, channel current essentially reached a steady state. B, fractions of total current change plotted versus substrate exposure time for 4-CY (left panel) and NADP+ (right panel) at -100 mV (○ and □) or 0 mV (• and ▪). The smooth curves are single-component exponential functions fit to the data points. Error bars are S.E. The time constants and number of patches are as follows: 7.9 ± 0.7 min (-100 mV for 4-CY, n = 29), 4.9 ± 0.6 min (0 mV for 4-CY, n = 14), 12.2 ± 0.9 min (-100 mV for NADP+, n = 13), and 5.9 ± 0.9 min (0 mV for NADP+, n = 10).
We measured the rate of channel modulation at two different membrane potentials: -100 mV, at which most of the channels are closed, and 0 mV, at which most of the channels are open and inactivated. Both the 4-CY and NADP+ effects occurred ∼2-fold faster at 0 mV than at -100 mV (p < 0.01 in unpaired t test) (Fig. 8B), consistent with the notion that membrane potentials affect NADPH oxidation or exchange.
DISCUSSION
By identifying reagents that oxidize Kvβ1-bound NADPH and studying in parallel the effect of these reagents on channel current, we observed a correlation between NADPH oxidation and potentiation of channel current. These properties suggest that the Kv1-Kvβ complex is capable of transducing a change in cellular metabolic redox state into a change in channel activity and hence cell excitability. The correlation between cofactor oxidation and channel modulation also provides a measure of enzymatic activity on inside-out patches, and we found that the rates of both hydride transfer and cofactor exchange are significantly faster at 0-mV membrane potential than at -100 mV, providing the first evidence of a voltage-regulated aldoketoreductase.
The Kv1-Kvβ complex is widely expressed in excitable cells, such as neurons and muscle cells (27, 28), and in non-excitable cells, such as lymphocytes and alveolar cells (29, 30). In the central nervous system, Kv1 family channels are preferentially targeted to axons (31) and likely play an important role in controlling the invasion and propagation of an action potential. In humans, mutations of the Kv1 channel have been linked to episodic ataxia (32), and loss of the Kvβ gene has been associated with seizures (33). In non-excitable T-lymphocytes, the activity of Kv1 channels changes significantly during the T-cell maturation process (34). However, how the redox-sensing capability of the Kv1-Kvβ complex would serve cellular functions remains to be explored.
In a native cell, multiple members of the Kv1 and Kvβ families express at the same time, so the (Kv1)4-(Kvβ)4 complexes may be formed by heterotetramers of Kv1 and Kvβ subunits. In such complexes, Kv1 modulation induced by oxidation of Kvβ-bound NADPH would no doubt be more complicated than we have observed in the heterologous oocyte expression system. From the work presented here and a previous study (19), we know that both Kvβ1 and Kvβ2 are functional AKRs and that both mediate substrate-induced channel modulation. Even though Kvβ2 does not have an N-type inactivation gate, it reduces the on-rate of N-type inactivation inherent to Kv1.4 channels (19) and, as a result, potentiates K+ current. It is very likely that conditions in a cell that lead to oxidation of bound NADPH would increase Kv1 current, whether it is through the leftward shift of voltage-dependent channel activation or through the reduction of channel inactivation.
Different forms of redox modulation of Kv channels have been reported, including methionine oxidation by methionine-sulfoxide reductase or reactive oxygen species (35-37) and cysteine oxidation by reactive oxygen species (38). This study adds NADPH to the targets of oxidation, and in this case, because it is mediated by an aldoketoreductase, it has the potential of being highly selective for a specific substrate. The challenge now is to identify the physiological substrate of Kvβ.
Because the inactivation gate is on Kvβ1, the change in channel inactivation could be due to the dissociation of Kvβ1 after oxidation of the bound cofactor. This is not the mechanism, however, because the V½ measured after oxidation is -65.6 ± 0.9 mV, which is significantly different from the V½ of Kv1.1 expressed alone (-56.8 ± 0.8 mV, p < 0.001 in unpaired t test). We speculate that oxidation of the bound cofactor induces a structural change in Kvβ, which may propagate to Kv1.1 to affect channel inactivation. We have ruled out the C terminus of Kv1.1, which is not required for the modulation. A likely prospect is the intracellular T1 domain of Kv1.1, which serves as a docking site for Kvβ.
In conclusion, we have found that Kvβ1 responds to a variety of redox signals and modulates channel function. We have also found that Kvβ1 aldoketoreductase is voltage-dependent: depolarization speeds up the rate of oxidation. Thus, the Kv1.1-Kvβ1 complex exhibits bidirectional allosteric modulation, which we are starting to understand at the molecular level.
Acknowledgments
We thank Dr. R. MacKinnon for advice and generous help throughout the project, Drs. A. Karlin and H. Colecraft for critical comments on the manuscript, and J. Riley for expert technical assistance.
This work was supported in part by March of Dimes Birth Defects Foundation Research Grant 5-FY06-20, American Heart Association Grant SDG 0630148N, and National Institutes of Health Grant HL086392 (to M. Z.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
This article was selected as a Paper of the Week.
Footnotes
The abbreviations used are: AKR, aldoketoreductase; 4-CY, 4-cyanobenzaldehyde; ANOVA, analysis of variance.
References
- 1.Connor, J. A., and Stevens, C. F. (1971) J. Physiol. (Lond.) 213 31-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aldrich, R. W., Getting, P. A., and Thompson, S. H. (1979) J. Physiol. (Lond.) 291 531-544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zagotta, W. N., Hoshi, T., and Aldrich, R. W. (1990) Science 250 568-571 [DOI] [PubMed] [Google Scholar]
- 4.Hoshi, T., Zagotta, W. N., and Aldrich, R. W. (1990) Science 250 533-538 [DOI] [PubMed] [Google Scholar]
- 5.Rettig, J., Heinemann, S. H., Wunder, F., Lorra, C., Parcej, D. N., Dolly, J. O., and Pongs, O. (1994) Nature 369 289-294 [DOI] [PubMed] [Google Scholar]
- 6.Heinemann, S. H., Rettig, J., Wunder, F., and Pongs, O. (1995) FEBS Lett. 377 383-389 [DOI] [PubMed] [Google Scholar]
- 7.Wallner, M., Meera, P., and Toro, L. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 4137-4142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xia, X. M., Ding, J. P., and Lingle, C. J. (1999) J. Neurosci. 19 5255-5264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nagaya, N., and Papazian, D. M. (1997) J. Biol. Chem. 272 3022-3027 [DOI] [PubMed] [Google Scholar]
- 10.Scott, V. E., Rettig, J., Parcej, D. N., Keen, J. N., Findlay, J. B., Pongs, O., and Dolly, J. O. (1994) Proc. Natl. Acad. Sci. U. S. A. 91 1637-1641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gulbis, J. M., Zhou, M., Mann, S., and MacKinnon, R. (2000) Science 289 123-127 [DOI] [PubMed] [Google Scholar]
- 12.Parcej, D. N., and Eckhardt-Strelau, L. (2003) J. Mol. Biol. 333 103-116 [DOI] [PubMed] [Google Scholar]
- 13.Long, S. B., Campbell, E. B., and MacKinnon, R. (2005) Science 309 897-903 [DOI] [PubMed] [Google Scholar]
- 14.Sokolova, O., Accardi, A., Gutierrez, D., Lau, A., Rigney, M., and Grigorieff, N. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 12607-12612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rhodes, K. J., Keilbaugh, S. A., Barrezueta, N. X., Lopez, K. L., and Trimmer, J. S. (1995) J. Neurosci. 15 5360-5371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pongs, O., Leicher, T., Berger, M., Roeper, J., Bahring, R., Wray, D., Giese, K. P., Silva, A. J., and Storm, J. F. (1999) Ann. N. Y. Acad. Sci. 868 344-355 [DOI] [PubMed] [Google Scholar]
- 17.McCormack, T., and McCormack, K. (1994) Cell 79 1133-1135 [DOI] [PubMed] [Google Scholar]
- 18.Gulbis, J. M., Mann, S., and MacKinnon, R. (1999) Cell 97 943-952 [DOI] [PubMed] [Google Scholar]
- 19.Weng, J., Cao, Y., Moss, N., and Zhou, M. (2006) J. Biol. Chem. 281 15194-15200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bahring, R., Milligan, C. J., Vardanyan, V., Engeland, B., Young, B. A., Dannenberg, J., Waldschutz, R., Edwards, J. P., Wray, D., and Pongs, O. (2001) J. Biol. Chem. 276 22923-22929 [DOI] [PubMed] [Google Scholar]
- 21.MacKinnon, R. (1991) Nature 350 232-235 [DOI] [PubMed] [Google Scholar]
- 22.Schlegel, B. P., Jez, J. M., and Penning, T. M. (1998) Biochemistry 37 3538-3548 [DOI] [PubMed] [Google Scholar]
- 23.Veal, E. A., Day, A. M., and Morgan, B. A. (2007) Mol. Cell 26 1-14 [DOI] [PubMed] [Google Scholar]
- 24.Lee, S. H., and Blair, I. A. (2000) Chem. Res. Toxicol. 13 698-702 [DOI] [PubMed] [Google Scholar]
- 25.Tipparaju, S. M., Saxena, N., Liu, S. Q., Kumar, R., and Bhatnagar, A. (2005) Am. J. Physiol. 288 C366-C376 [DOI] [PubMed] [Google Scholar]
- 26.Murata, Y., Iwasaki, H., Sasaki, M., Inaba, K., and Okamura, Y. (2005) Nature 435 1239-1243 [DOI] [PubMed] [Google Scholar]
- 27.Nerbonne, J. M. (2000) J. Physiol. (Lond.) 525 285-298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trimmer, J. S., and Rhodes, K. J. (2004) Annu. Rev. Physiol. 66 477-519 [DOI] [PubMed] [Google Scholar]
- 29.Lewis, R. S., and Cahalan, M. D. (1995) Annu. Rev. Immunol. 13 623-653 [DOI] [PubMed] [Google Scholar]
- 30.Lee, S. Y., Maniak, P. J., Ingbar, D. H., and O'Grady, S. M. (2003) Am. J. Physiol. 284 C1614-C1624 [DOI] [PubMed] [Google Scholar]
- 31.Gu, C., Jan, Y. N., and Jan, L. Y. (2003) Science 301 646-649 [DOI] [PubMed] [Google Scholar]
- 32.Adelman, J. P., Bond, C. T., Pessia, M., and Maylie, J. (1995) Neuron 15 1449-1454 [DOI] [PubMed] [Google Scholar]
- 33.Heilstedt, H. A., Burgess, D. L., Anderson, A. E., Chedrawi, A., Tharp, B., Lee, O., Kashork, C. D., Starkey, D. E., Wu, Y. Q., Noebels, J. L., Shaffer, L. G., and Shapira, S. K. (2001) Epilepsia 42 1103-1111 [DOI] [PubMed] [Google Scholar]
- 34.Cahalan, M. D., Wulff, H., and Chandy, K. G. (2001) J. Clin. Immunol. 21 235-252 [DOI] [PubMed] [Google Scholar]
- 35.Ciorba, M. A., Heinemann, S. H., Weissbach, H., Brot, N., and Hoshi, T. (1999) FEBS Lett. 442 48-52 [DOI] [PubMed] [Google Scholar]
- 36.Ciorba, M. A., Heinemann, S. H., Weissbach, H., Brot, N., and Hoshi, T. (1997) Proc. Natl. Acad. Sci. U. S. A. 94 9932-9937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoshi, T., and Heinemann, S. (2001) J. Physiol. (Lond.) 531 1-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ruppersberg, J. P., Stocker, M., Pongs, O., Heinemann, S. H., Frank, R., and Koenen, M. (1991) Nature 352 711-714 [DOI] [PubMed] [Google Scholar]