

Abstract
Recent studies have identified a novel family of oxidized phosphatidylcholines (oxPCCD36) that serve as highly specific ligands for scavenger receptor CD36. oxPCCD36 accumulate in vivo and mediate macrophage foam cell formation as well as promote platelet hyper-reactivity in hyperlipidemia via CD36. The structural basis of oxPCCD36 binding to CD36 has not been elucidated. We used liquid-phase binding to glutathione S-transferase fusion proteins containing various regions of CD36 to initially identify the region spanning CD36 amino acids 157-171 to contain a major binding site for oxPCCD36. A bell-shaped pH profile and salt concentration dependence suggest an electrostatic mechanism of the binding. Two conserved, positively charged amino acids in the region 157-171 (lysines at positions 164 and 166) were identified as critical for oxPCCD36 and oxidized low density lipoprotein (oxLDL) binding to CD36. Lysine neutralization with chemical modifier or site-directed mutagenesis of lysine 164/166 to alanine or glutamate, but not to arginine, abolished binding. Cells expressing full-length CD36 with mutated lysines (164 and 166) failed to recognize oxPCCD36 and oxLDL. Synthetic peptides mimicking the CD36 binding site, but not mutated or scrambled peptides, effectively prevented: (i) oxLDL binding to CD36, (ii) macrophage foam cell formation induced by oxLDL, and (iii) platelet activation by oxPCCD36. These data indicate that CD36 (160-168) represents the core of the oxPCCD36 binding site with lysines 164/166 being indispensable for the binding.
CD36 is a 472-amino-acid, 88-kDa heavily glycosylated transmembrane protein that is expressed in various cell types including macrophages, platelets, microvascular endothelial cells, and adipocytes (1, 2). CD36 has been shown to play a significant role in a number of physiological and pathological processes in vivo including atherogenesis, lipid sensing and metabolism, innate immune responses, angiogenesis, uptake of apoptotic cells, and diabetes (2-4). CD36 involvement in such a variety of processes can be partially explained by its capacity to recognize a number of various distinct ligands. Examples of CD36 ligands include thrombospondin-1 (5), oxidized low density lipoproteins (oxLDL)2 (6), oxidized phospholipids (7-9), fatty acids (10), microbial diacylglycerides (4), hexarelin (3), collagen (11), and malarial parasite-infected erythrocytes (12).
That CD36 can function as a multiligand receptor is conceivable assuming that it has multiple ligand binding domains. Several studies suggest, for example, that the binding sites of thrombospondin-1 and oxLDL on CD36 are different (13, 14). Two distinct binding sites are proposed for oxLDL on CD36. Studies using a monoclonal antibody have shown that domain 155-183 of CD36 plays a critical role in the binding of LDL oxidized by copper (15). Solid phase binding assays using recombinant fusion proteins spanning various regions of CD36, however, implicate the domain 28-93 as the major binding site for oxLDL (16).
We have recently identified a novel family of oxidized choline glycerophospholipids (oxPCCD36) that mediate CD36-dependent recognition of LDL oxidized by various pathways. The structural aspect of oxPCCD36 essential for high affinity binding to CD36 is an sn-2 acyl group that incorporates a terminal γ-hydroxy(or oxo)-α,β-unsaturated carbonyl. A characteristic feature of oxPCCD36 conformation is a negatively charged distal end of the sn-2 acyl chain residue that partitions into the aqueous phase (17). oxPCCD36 are formed during the oxidation of LDL by multiple distinct pathways, serve as specific high affinity ligands for CD36 (9), and are present in vivo at sites of enhanced oxidative stress (18-20). OxPCCD36 mediate foam cell formation induced by oxidized LDL via macrophage CD36 and induce a prothrombotic phenotype in hyperlipidemia via platelet CD36 (18, 20).
In this current study, we investigated the structural basis for the recognition of oxPCCD36 by CD36 using a combination of site-directed mutagenesis and ligand binding analyses of various GST-CD36 fusion proteins bound to glutathione-Sepharose beads. We demonstrate that amino acids 160-168 of CD36 represent the core of the binding site for oxPCCD36 and oxidized LDL and that the electrostatic interaction between evolutionary conserved lysines 164 and 166 and oxidized phospholipid moieties is crucial in this binding.
EXPERIMENTAL PROCEDURES
General Materials—Tissue culture media and supplements were purchased from Invitrogen. 125I-labeled sodium iodide was supplied by ICN Pharmaceuticals, Inc. (Costa Mesa, CA), and [3H]1,2-dihexadecanoyl-sn-glycerol-3-phosphocholine was from American Radiolabel Chemicals, Inc., (St. Louis, MO). 9-Keto-10-dodecendioic acid (KDdiA-PC) and 5-keto-6-octendioic acid esters of 2-lysoPC (KOdiA-PC) were purchased from Cayman, Inc. (Ann Arbor, MI). CD36 blocking antibody FA6-152 was obtained from Immunotech-Beckman Coulter. All oligonucleotide primers used for PCR, mutagenesis, or sequencing were made by Integrated DNA Technologies (Coralville, IA). Peptides CD36 long, VQMILNSLINKSKSS (CD36L), CD36 short, SLINKSKSSMF (CD36S), CD36 minimal, SLINKSKSS (CD36M), CD36 164E166E, VQMILNSLINESESS (CD36EE), and CD36 scrambled, SILKVNLSQMKILNSI (CD36Scr) were synthesized by Sigma-Genosys (The Woodlands, TX) and purified to >95% as determined by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. All other reagents were obtained from Sigma unless otherwise specified.
Cloning and Expression of Full-length CD36—For transient expression in mammalian cells, pCGCG-CD36 with a C-terminal HA tag was used. For stable expression in HEK 293T, the CD36 insert with HA tag was cloned into pIRES2-EGFP vector (Clontech).
Cloning, Mutagenesis, Expression, and Purification of GST Fusion Proteins—An array of recombinant GST-CD36 fusion constructs was generated in the bacterial expression vector pGEX3T (16). All the fusion proteins were made in Rosetta™(DE3)pLacI strain of Escherichia coli (EMD Biosciences-Novagen, San Diego, CA) and purified by using glutathione-Sepharose 4B beads (GE Healthcare). The size, amount, and purity of the fusion proteins were examined by SDS-PAGE. The molecular weight was found to be close to the predicted value, and purity was typically >95%. PCR-based site-directed mutagenesis of the pGEX3T-CD36 fusion construct or pCGCG-CD36 was carried out by using the QuikChange XL kit (Invitrogen).
Lipoprotein Isolation, Oxidation, and Radiolabeling—LDL was isolated from fresh human plasma by sequential centrifugation, labeled with 125I, and oxidized as described (8).
Phospholipid Vesicle Preparation and Modification—Small unilamellar vesicles of PAPC were prepared by repeated extrusion through a 0.1-μm polycarbonate filter (9). For direct binding experiments, 25 μCi/mg of phospholipids of [3H]1,2-dihexadecanoyl-sn-glycerol-3-phosphocholine was added as a tracer.
Foam Cell Formation—Mouse peritoneal macrophages were
isolated and cultured in RPMI 1640/10% fetal bovine serum containing oxidized
(by
myeloperoxidase/H2O2/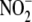 system) LDL (50 μg/ml) that was preincubated with the indicated peptide.
After 24 h, cells were fixed with 4% formaldehyde and stained with hematoxylin
and Oil Red-O. Images were acquired using a bright field microscope (Leica)
and quantification of lipid droplets was performed by using Image Pro Plus
software (Media Cybernetics, Bethesda, MD). The original magnifications were
×400.
system) LDL (50 μg/ml) that was preincubated with the indicated peptide.
After 24 h, cells were fixed with 4% formaldehyde and stained with hematoxylin
and Oil Red-O. Images were acquired using a bright field microscope (Leica)
and quantification of lipid droplets was performed by using Image Pro Plus
software (Media Cybernetics, Bethesda, MD). The original magnifications were
×400.
Flow Cytometry—Human platelets were isolated and purified by gel filtration as described earlier (20). 5 μm oxPCCD36 was preincubated with CD36S, CD36L, or CD36Scr peptide (final concentration 50 μm) for 30 min at room temperature. 1.0 × 106 platelets in Tyrode's buffer containing 2 mm Ca2+ and 1 mm Mg2+ were incubated with oxPCCD36 (5 μm) or a preincubated mixture of oxPCCD36 and peptide for 15 min at room temperature. The activated platelets were detected with fluorescein isothiocyanate-conjugated anti-P-selectin antibody (1:1000) using a FACSCalibur instrument (BD Biosciences) and analyzed by FlowJo software.
Binding Assays—Ligand binding to GST fusion proteins was assessed by incubating proteins bound on the glutathione-Sepharose beads (typically 1.5 μg of protein/tube) with radio-labeled ligands in phosphate-buffered saline containing 0.4% bovine serum albumin for 3 h at 25 °C with gentle rocking. Unbound ligands were removed by repeated washing of the beads with phosphate-buffered saline using low speed centrifugation; bound radioactivity was quantified. In competition experiments, unlabeled competitors were added at 20-fold excess. Ligand binding to full-length CD36 was performed using confluent monolayers of cells in 24-well plates. The indicated amounts of 125I-labeled native or modified LDL or high density lipoprotein or 3H-labeled unilamellar phospholipids vesicles were added in 250 μl of medium containing 10% fetal bovine serum. Following2hof incubation at 4 °C, cells were washed three times with medium and lysed by adding 0.1 m NaOH, and then cell-associated radioactivity was quantified.
Statistics and Image Analysis—Data represent the mean ± S.D. for the indicated number of samples. Statistical analyses were made using the Student's t test. Binding parameters for different ligands of CD36 were obtained from non-linear regression analyses in Prism 4 software (GraphPad Software Inc). Images were captured in TIF format, white points were adjusted, and the final images were sharpened by unsharp masking in PhotoShop CS2.
RESULTS
Mapping of the CD36 Binding Site for oxPCCD36 with GST-CD36 Deletion Constructs—An array of recombinant GST-CD36 fusion constructs was generated as described (16) and tested in fluid-phase assays for the binding of specific oxidized phospholipids (oxPCCD36) as well as lipoproteins oxidized by various pathways. The construct that spans CD36 amino acids 118-182 reproducibly showed maximum binding of oxPCCD36 vesicles and lipoproteins oxidized by various pathways (data for copper-oxidized LDL are shown, Fig. 1A). Two other constructs, GST-CD365-143 and GST-CD3667-157, possessing an alternative binding site for oxidized LDL and having significant overlapping regions with GST-CD36118-182, showed only about 20% of the binding when compared with CD36118-182. OxPCCD36 and oxLDL binding to the remaining constructs containing other regions of CD36 was found to be significantly lower (data not shown). This result suggests that the major binding site on CD36 for oxPCCD36-containing ligands lies between amino acids 157 and 182.
FIGURE 1.

Localization of OxPCCD36 and oxLDL binding domain. A, binding of 125I-labeled oxLDL to GST-fusion proteins spanning various regions of CD36. Binding was performed at 10 μg/ml 125I-labeled oxLDL with 1.5 μg each of the bead-immobilized purified GST-CD36 fusion proteins. B, in competition assays, 1.5 μg GST-CD36118-182 protein was pretreated either with 20-fold excess cold oxLDL or 3 μg of FA6-152 antibody or non-immune IgG (Non-Im IgG), and then binding was performed as in panel A. Alternatively, binding was performed in the presence of collagen type I or thrombospondin 1 (TSP) each at 100 μg/ml). NA, no addition. C, binding of 3H-labeled small unilamellar vesicles containing the indicated phospholipids to GST-CD36 fusion proteins was performed as described under “Experimental Procedures.” D, binding of 125I-labeled oxLDL to GST-CD36118-182 protein and its sequential C-terminal deletion constructs was performed as in panel A. Binding to GST alone was subtracted as a background, and the values are presented as the percentage of the binding to GST-CD36118-182 protein.
To further characterize GST-CD36118-182 and compare it with full-length CD36, we examined its binding properties. oxLDL binding to GST-CD36118-182 was saturable, having half-maximal binding at 11.2 ± 1.4 μg/ml (≅22 nm), demonstrating high affinity binding (data not shown). Twenty-fold excess unlabeled oxLDL, but not native LDL, significantly inhibited binding, indicating the specificity of the binding (Fig. 1B). Importantly, anti-CD36 antibody FA6-152 (but not isotype-matched non-immune IgG), which effectively blocks binding of various forms of oxLDL and oxPCCD36 to CD36-overexpressing cells, significantly reduced binding of oxLDL to GST-CD36118-182 (Fig. 1B). Thrombospondin and collagen type I were reported to be ligands for CD36. However, their binding sites differ from one reported for oxLDL (13, 14, 21, 22). Both failed to compete with oxLDL binding to GST-CD36118-182. These results further demonstrated the specificity of the binding and showed that the same binding site for oxPCCD36 operates in full-length CD36 and GST-CD36118-182. This was further supported by direct binding experiments. Similar to full-length CD36, GST-CD36118-182 bound both PAPC vesicles oxidized by myeloperoxidase-generated reactive nitrogen species and vesicles containing synthetic ligand oxPCCD36 (data shown for KDdiA-PC, a prototypic member of oxPCCD36 family (9)) significantly more effectively than vesicles containing native PAPC (Fig. 1C). Under the same conditions, binding of oxPAPC- or KDdiA-PC-containing vesicles by GST-CD365-143, GST-CD3667-153, and even GST-CD36118-160 was significantly lower (Fig. 1C and data not shown). Taken together, these experiments demonstrate that the binding properties of CD36118-182 mirror those of the full-length cell-expressed CD36.
To further delineate the oxPCCD36 and oxLDL binding domain of CD36, the GST-CD36118-182 construct was mutated to sequentially delete 9-11 amino acids at a time from the C terminus. These constructs were then used in binding assays performed with ligands containing oxPCCD36 (data for Cu-oxLDL shown, Fig. 1D). The binding activity of the GST fusion protein was unaffected when amino acids 172-182 were deleted but dropped dramatically upon further deletion of amino acids 161-171. This, together with the previous experiment (Fig. 1A), suggests that the binding domain for oxPCCD36 and oxidized LDL on CD36 lies between amino acids 157 and 171.
Nature of the Binding Interaction—To investigate the nature of the ligand binding interaction of oxPCCD36 and CD36, KDdiA-PC binding was performed at varying pH and salt concentrations (Fig. 2A). Under the experimental conditions employed, there was no loss in binding of GST-CD36118-182 to glutathione-Sepharose beads. Thus, the decrease in bead-associated radioactivity can only be attributed to the actual loss in binding of ligand. A bell-shaped pH profile of the binding (peak at approximately pH 6.5) and a gradual reduction with salt concentration increasing above 0.4 m were observed, indicating involvement of electrostatic interaction.
FIGURE 2.

CD36 lysine 164 and lysine 166 are required for oxLDL binding. A, pH profile and salt effect of [3H]KDdiA-PC binding to GST-CD36118-182 fusion protein. All data are expressed as mean ± S.D. B, alignment of various mammalian CD36 and synthetic peptide amino acid sequences. C and D, relative binding of 125I-labeled oxLDL to GST fusion protein CD36118-182 or HEK-293T cells overexpressing full-length CD36 without or with point mutations shown. For binding with GST fusion proteins and CD36-overexpressing cells, GST- and vector-transfected cells were used as controls, and the values for binding to these were subtracted as background. WT, wild type.
Aligning various mammalian CD36 protein sequences (Fig. 2B) revealed that the putative binding domain contains two conserved lysine residues at positions 164 and 166. Thus, we hypothesized that these two positively charged residues interact with the negatively charged sn-2 acyl residues of oxPCCD36 and play a critical role in ligand binding. To test this hypothesis, we first used an amine-reactive chemical modifier, N-hydroxysuccinimide. Pretreatment of either GST-CD36118-182- or CD36-overexpressing HEK 293T cells with N-hydroxysuccinimide resulted in about an 80% reduction in oxLDL binding. This result suggests that lysines are critical in the CD36-mediated oxLDL binding process both in vitro and in vivo.
Mutagenesis of the Putative Binding Site—To further test our hypothesis stated above, we induced systematic mutagenesis of lysines 164 and 166, individually or both, by substitution of the positively charged amino acid to neutral, negative, or positive amino acids, i.e. to alanine, glutamate, or arginine, respectively. We performed mutagenesis both in GST-CD36118-182 as well as in full-length CD36, and binding assays were subsequently carried out using both purified mutated proteins and HEK 293T cells expressing the corresponding full-length mutated CD36. The surface expression of all the mutants in HEK 293T cells was similar as analyzed by surface biotinylation followed by Western blots for HA (data not shown). We observed that substitution of either of the two lysines to negatively charged glutamic acid or neutrally charged alanine effectively reduced binding to the nonspecific level both in GST-CD36118-182 and in full-length CD36 (Fig. 2C). At the same time, when the positive charge was retained by a lysine to arginine mutation, binding was affected only slightly (Fig. 2C). Importantly, when other residues in the region 160-168 were mutated individually to alanine, oxLDL binding was not altered (Fig. 2D), strongly suggesting that lysines 164-166 are the only primary requirement for binding.
Peptides Mimicking the CD36 Binding Domain—Four peptides were synthesized to see whether the putative binding domain described above is sufficient for direct interaction to oxLDL (Fig. 2B). Three small overlapping peptides (CD36L, CD36S, and CD36M) that contain the human CD36 binding domain were found to inhibit oxLDL binding to GST-CD36118-182 in a dose-dependent fashion, whereas peptides either with substitution of the 164/166 lysines to glutamates (CD36EE) or with a scrambled amino acid sequence (CD36Scr) were far less effective in inhibition (Fig. 3A). The half-maximal inhibition by peptides CD36L, CD36S, and CD36M was achieved at a peptide:GST-CD36 molar ratio of 1 (peptide concentration ∼2 μm), implying that they have affinity to oxLDL similar to that of GST-CD36118-182 and hence that of full-length CD36. Also, since these three peptides have similar inhibition curves, the amino acid sequence (160-168), which is common to them, seems to be the minimal oxLDL binding domain of CD36.
FIGURE 3.

Synthetic peptides containing the CD36 binding domain sequence inhibit oxLDL binding to both GST-CD36 fusion protein and full-length CD36 overexpressed in cells. A, concentration-dependent inhibition of oxLDL binding to GST-CD36118-182 by peptides: CD36L (CD36154-168), CD36S (CD36160-170), and CD36M (CD36160-168). Two other peptides, CD36154-168Scramble and CD36154-168164E/166E, were used as controls. B, oxLDL binding to full-length CD36 overexpressed in HEK-293T in the presence of peptides at 4 μm.
The CD36 mimicking peptides are a potent inhibitor of oxLDL binding to cells expressing CD36 (Fig. 3B). Interestingly, scrambled peptide (CD36Scr) had no significant effect on the binding while possessing two lysines (Fig. 3B), suggesting that CD36L peptide contains additional required structural features that are lost upon amino acid rearrangement.
High affinity binding of oxidized LDL to CD36 has long been implicated in
the formation of lipid-laden foam cells, a critical early cellular event in
the atherosclerotic process. In a final set of experiments, we examined
whether CD36L can prevent foam cell formation. LDL was oxidized by the
physiologically relevant
myeloperoxidase/H2O2/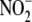 system (8) to generate
NO2-LDL, a specific ligand for macrophage CD36 but not for
macrophage scavenger receptor type A
(23). NO2-LDL was
then preincubated with CD36L, CD36S, or CD36M peptides and incubated for 24 h
with thioglycollate-elicited mouse peritoneal macrophages. Cells were
microscopically examined following neutral lipid staining with Oil Red-O.
Control NO2-LDL induced significant cholesterol deposition in
macrophages (Fig. 4A),
as anticipated. In contrast, cells incubated with NO2-LDL exposed
either to CD36L or to CD36S or CD36M but not to CD36Scr or CD36EE were devoid
of Oil Red-O positive droplets (Fig.
4A and data not shown). A parallel experiment
demonstrated the lack of effect by either peptide on foam cell formation
induced by acetylated LDL, a specific ligand for scavenger receptor type A
(data not shown), demonstrating the specificity of the effect of CD36L
peptide. Finally, we also tested whether CD36L can prevent the recently
described activation of platelets by oxPCCD36, an event responsible
for platelet hyper-reactivity in dyslipoproteinemia
(20). OxPCCD36
(data for KOdiA-PC shown) induced platelet P-selectin expression and
activation of the platelet fibrinogen receptor integrin
αIIbβ3, as anticipated. Preincubation of
oxPCCD36 with CD36L or CD36S peptide prevented platelet activation
by either ligand (Fig.
4C). Preincubation with CD36Scr peptide did not prevent
activation, indicating the specificity of the effect. Importantly, CD36L,
CD36S, and CD36Scr had no effect on platelet activation induced by such
agonists as ADP and thrombin (Fig.
4C and data not shown). Collectively, these results
demonstrate that CD36L and CD36S peptides specifically inhibit the biological
activity of oxPCCD36 and of oxLDL mediated by oxPCCD36
in vitro and further.
system (8) to generate
NO2-LDL, a specific ligand for macrophage CD36 but not for
macrophage scavenger receptor type A
(23). NO2-LDL was
then preincubated with CD36L, CD36S, or CD36M peptides and incubated for 24 h
with thioglycollate-elicited mouse peritoneal macrophages. Cells were
microscopically examined following neutral lipid staining with Oil Red-O.
Control NO2-LDL induced significant cholesterol deposition in
macrophages (Fig. 4A),
as anticipated. In contrast, cells incubated with NO2-LDL exposed
either to CD36L or to CD36S or CD36M but not to CD36Scr or CD36EE were devoid
of Oil Red-O positive droplets (Fig.
4A and data not shown). A parallel experiment
demonstrated the lack of effect by either peptide on foam cell formation
induced by acetylated LDL, a specific ligand for scavenger receptor type A
(data not shown), demonstrating the specificity of the effect of CD36L
peptide. Finally, we also tested whether CD36L can prevent the recently
described activation of platelets by oxPCCD36, an event responsible
for platelet hyper-reactivity in dyslipoproteinemia
(20). OxPCCD36
(data for KOdiA-PC shown) induced platelet P-selectin expression and
activation of the platelet fibrinogen receptor integrin
αIIbβ3, as anticipated. Preincubation of
oxPCCD36 with CD36L or CD36S peptide prevented platelet activation
by either ligand (Fig.
4C). Preincubation with CD36Scr peptide did not prevent
activation, indicating the specificity of the effect. Importantly, CD36L,
CD36S, and CD36Scr had no effect on platelet activation induced by such
agonists as ADP and thrombin (Fig.
4C and data not shown). Collectively, these results
demonstrate that CD36L and CD36S peptides specifically inhibit the biological
activity of oxPCCD36 and of oxLDL mediated by oxPCCD36
in vitro and further.
FIGURE 4.

CD36 peptides inhibit macrophage foam cell formation induced by NO2 LDL and platelet activation induced by OxPCCD36. A, thioglycollate-elicited mouse peritoneal macrophages were cultured in the presence of NO2LDL (50 μg/ml) for 24 h in the absence or presence of 4 μm peptides. Cells were fixed with 4% formaldehyde, stained with Oil Red-O and hematoxylin, and analyzed by bright field microscopy. Representative images of cells are shown. Original magnification, ×400. B, lipid droplets in A were quantified using Image Pro-plus software and expressed as the percentage of the cell area occupied by lipid droplets. C, human platelets isolated by gel filtration were incubated either with KOdiA-PC (5 μm) or with a mixture of KOdiA-PC (5 μm) and peptides (50 μm) for 15 min at room temperature. Platelets were then incubated with fluorescein isothiocyanate-conjugated anti-P-selectin antibody and analyzed by flow cytometry. NA, no addition.
DISCUSSION
CD36 is a major contributor to the macrophage uptake of lipoproteins modified by various oxidative pathways and subsequently to foam cell formation in vitro and in vivo (23, 24). In platelets, CD36 serves as a sensor of specific oxidized phospholipids that accumulate in plasma in hyperlipidemia and mediates platelet hyper-reactivity and a prothrombotic phenotype associated with hyperlipidemia (20). Thus, elucidation and characterization of the binding site on CD36 for oxLDL and specific oxidized phospholipids are important for understanding the molecular mechanisms of cardiovascular disease as well as for the development of preventive therapies.
In this study, we used GST fusion proteins to identify a short linear stretch of amino acids in CD36 that specifically recognizes the novel family of specific oxidized phospholipids and mediates recognition of oxLDL by CD36. We then demonstrated that electrostatic attraction between conserved positively charged lysines in CD36 and the negatively charged oxidized phospholipids is a mechanism of the recognition. We then showed that soluble short synthetic peptides of CD36 block proatherogenic and prothrombotic properties of oxLDL and specific oxidized phospholipids.
The elucidation of a binding site for oxidized LDL in this work was aided by previous identification of a ligand in oxLDL that is recognized by CD36, specifically the novel family of oxidized phospholipids oxPCCD36 (9, 18). A characteristic feature of oxPCCD36 conformation is a negatively charged distal end of the sn-2 acyl chain residue, which was shown recently to be exposed into the aqueous phase (17). We have previously demonstrated that binding of oxPCCD36 to CD36 generally increased with increased negative charge of the sn-2 residue (9). In this study, the apparent decrease in binding with increasing salt concentration and a bell-shaped binding curve over a pH range implied that the interaction is primarily electrostatic in nature. The apparent half-maximal binding was observed at pH ∼5.2 and 8.9, represented by pKa of titratable opposing charge groups. Thus, an electrostatic interaction, presumably between the negatively charged groups in the lipids and the conserved positively charged lysines in CD36, is the primary mechanism of the ligand recognition.
When the lysine 164 and 166 residues of human CD36 were mutated to arginine, binding of oxLDL was unaffected, indicating that the size of the positively charged amino acid is less critical for effective binding. On the other hand, mutagenesis to alanine or glutamic acid completely abolished binding. Taken together these results demonstrate that positive charges at positions 164 and 166 are absolutely critical for CD36 binding ability and that the specific amino acid is less important. Control experiments demonstrated that alanine substitution of amino acids other then Lys-164 and Lys-166 in the region 160-168 resulted in only slight changes in binding property. Previously, similar alanine scanning mutagenesis of another oxLDL receptor, lectin-like oxLDL receptor-1 (LOX-1), showed involvement of basic residues (25). In vitro binding assays using LOX-1 mutants revealed a “basic spine” structure, consisting of linearly aligned arginine residues, that is responsible for ligand binding (26, 27). Since no crystal structure is available for CD36, it is hard to predict further similarities between the oxLDL binding motifs of these two receptors.
Previous studies using a monoclonal antibody directed against the domain 155-183 have shown that this domain of CD36 plays the critical role in the binding of LDL oxidized by copper (15). The same domain has also been implicated in platelet activation, cell adhesion of malaria parasite-infected erythrocytes (1), phagocytosis of apoptotic neutrophils (28), and binding of growth hormone-releasing peptide (29). A later report using recombinant fusion proteins spanning various regions of CD36 and solid phase binding assays, however, implicated the domain 28-93 as the major binding site for oxLDL (16). Taking into account that competition with monoclonal antibody (15) provides only indirect evidence and that solid-phase binding studies (16), on the other hand, may have restrictions due to potential protein conformation issues, we have used a combination of approaches to characterize the binding site of CD36 that recognizes oxPCCD36. In the present study, ligand binding to CD36-GST fusion proteins attached to glutathione-Sepharose beads in solution, coupled with mutation analysis and protein overexpression in mammalian cells, identified a small, linear domain of CD36 that binds oxPCCD36. Similar values for half-maximal concentration for GST-CD36118-182 and full-length CD36 expressed in cells (8, 20) indicate that oxLDL binding by GST-CD36 fusion proteins in vitro is representative of binding to full-length CD36.
Demonstration of a critical role of CD36 in atherosclerotic lesion formation and a prothrombotic phenotype in the settings of dyslipidemia suggests that CD36 may be a novel target for treatment and/or prevention of cardiovascular disease. One approach has been reported recently where CD36 was blocked in vivo by EP 80317, a novel ligand derived from the growth hormone-releasing peptide family (30). EP 80317 induced significant reduction of atherosclerosis in apoE-deficient mice fed Western diet. The disadvantage of such an approach may be inhibition of important physiological functions of CD36, such as delivery of long chain fatty acids to cardiomyocytes and brain (31, 32). Another approach is to specifically inhibit pathological ligands for CD36. Previously, a recombinant, soluble form of CD36 was shown to compete for the binding of oxLDL to membrane-expressed CD36 and to prevent oxLDL-induced adhesion of monocytes to endothelial cells (33). In this study, a short synthetic peptide representing the identified binding domain inhibited biological activity of oxPCCD36 and oxidized LDL in macrophage foam cell assays and in a platelet activation assays.
Acknowledgments
We are grateful to Dr. Roy L. Silverstein and Dr. Maria Febbraio for GST-CD36 fusion constructs and to Dr. Michael E. Greenberg for the full-length CD36 clone. We thank Valentina Verbovetskaya for technical assistance.
This work was supported by National Institutes of Health Grants HL077213 and HL053315 (to E. A. P.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: LDL, low density lipoprotein; oxLDL, oxidized LDL; GST, glutathione S-transferase; KDdiA-PC and KOdiA-PC, 9-keto-10-dodecendoioic acid and 5-keto-6-octendioic acid esters of 2-lyso-PC; HA, influenza hemagglutinin epitope tag; HDL, high density lipoprotein; PAPC, 1-hexa-docanoyl-2eicosa-tetra-5′, 8′, 11′, 14′-enoyl-sn-glycero-3-phosphocholine.
References
- 1.Daviet, L., and McGregor, J. L. (1997) Thromb. Haemostasis 78 65-69 [PubMed] [Google Scholar]
- 2.Febbraio, M., Hajjar, D. P., and Silverstein, R. L. (2001) J. Clin. Investig. 108 785-791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bodart, V., Febbraio, M., Demers, A., McNicoll, N., Pohankova, P., Perreault, A., Sejlitz, T., Escher, E., Silverstein, R. L., Lamontagne, D., and Ong, H. (2002) Circ. Res. 90 844-849 [DOI] [PubMed] [Google Scholar]
- 4.Hoebe, K., Georgel, P., Rutschmann, S., Du, X., Mudd, S., Crozat, K., Sovath, S., Shamel, L., Hartung, T., Zahringer, U., and Beutler, B. (2005) Nature 433 523-527 [DOI] [PubMed] [Google Scholar]
- 5.Asch, A. S., Barnwell, J., Silverstein, R. L., and Nachman, R. L. (1987) J. Clin. Investig. 79 1054-1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Endemann, G., Stanton, L. W., Madden, K. S., Bryant, C. M., White, R. T., and Protter, A. A. (1993) J. Biol. Chem. 268 11811-11816 [PubMed] [Google Scholar]
- 7.Boullier, A., Gillotte, K. L., Horkko, S., Green, S. R., Friedman, P., Dennis, E. A., Witztum, J. L., Steinberg, D., and Quehenberger, O. (2000) J. Biol. Chem. 275 9163-9169 [DOI] [PubMed] [Google Scholar]
- 8.Podrez, E. A., Schmitt, D., Hoff, H. F., and Hazen, S. L. (1999) J. Clin. Investig. 103 1547-1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Podrez, E. A., Poliakov, E., Shen, Z., Zhang, R., Deng, Y., Sun, M., Finton, P. J., Shan, L., Gugiu, B., Fox, P. L., Hoff, H. F., Salomon, R. G., and Hazen, S. L. (2002) J. Biol. Chem. 277 38503-38516 [DOI] [PubMed] [Google Scholar]
- 10.Abumrad, N. A., el-Maghrabi, M. R., Amri, E. Z., Lopez, E., and Grimaldi, P. A. (1993) J. Biol. Chem. 268 17665-17668 [PubMed] [Google Scholar]
- 11.Tandon, N. N., Kralisz, U., and Jamieson, G. A. (1989) J. Biol. Chem. 264 7576-7583 [PubMed] [Google Scholar]
- 12.Ockenhouse, C. F., Tandon, N. N., Magowan, C., Jamieson, G. A., and Chulay, J. D. (1989) Science 243 1469-1471 [DOI] [PubMed] [Google Scholar]
- 13.Asch, A. S., Liu, I., Briccetti, F. M., Barnwell, J. W., Kwakye-Berko, F., Dokun, A., Goldberger, J., and Pernambuco, M. (1993) Science 262 1436-1440 [DOI] [PubMed] [Google Scholar]
- 14.Nicholson, A. C., Frieda, S., Pearce, A., and Silverstein, R. L. (1995) Arterioscler. Thromb. Vasc. Biol. 15 269-275 [DOI] [PubMed] [Google Scholar]
- 15.Puente Navazo, M. D., Daviet, L., Ninio, E., and McGregor, J. L. (1996) Arterioscler. Thromb. Vasc. Biol. 16 1033-1039 [DOI] [PubMed] [Google Scholar]
- 16.Pearce, S. F., Roy, P., Nicholson, A. C., Hajjar, D. P., Febbraio, M., and Silverstein, R. L. (1998) J. Biol. Chem. 273 34875-34881 [DOI] [PubMed] [Google Scholar]
- 17.Li, X. M., Salomon, R. G., Qin, J., and Hazen, S. L. (2007) Biochemistry 46 5009-5017 [DOI] [PubMed] [Google Scholar]
- 18.Podrez, E. A., Poliakov, E., Shen, Z., Zhang, R., Deng, Y., Sun, M., Finton, P. J., Shan, L., Febbraio, M., Hajjar, D. P., Silverstein, R. L., Hoff, H. F., Salomon, R. G., and Hazen, S. L. (2002) J. Biol. Chem. 277 38517-38523 [DOI] [PubMed] [Google Scholar]
- 19.Sun, M., Finnemann, S. C., Febbraio, M., Shan, L., Annangudi, S. P., Podrez, E. A., Hoppe, G., Darrow, R., Organisciak, D. T., Salomon, R. G., Silverstein, R. L., and Hazen, S. L. (2006) J. Biol. Chem. 281 4222-4230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Podrez, E. A., Byzova, T. V., Febbraio, M., Salomon, R. G., Ma, Y., Valiyaveettil, M., Poliakov, E., Sun, M., Finton, P. J., Curtis, B. R., Chen, J., Zhang, R., Silverstein, R. L., and Hazen, S. L. (2007) Nat. Med. 13 1086-1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frieda, S., Pearce, A., Wu, J., and Silverstein, R. L. (1995) J. Biol. Chem. 270 2981-2986 [DOI] [PubMed] [Google Scholar]
- 22.Matsuno, K., Diaz-Ricart, M., Montgomery, R. R., Aster, R. H., Jamieson, G. A., and Tandon, N. N. (1996) Br. J. Haematol. 92 960-967 [DOI] [PubMed] [Google Scholar]
- 23.Podrez, E. A., Febbraio, M., Sheibani, N., Schmitt, D., Silverstein, R. L., Hajjar, D. P., Cohen, P. A., Frazier, W. A., Hoff, H. F., and Hazen, S. L. (2000) J. Clin. Investig. 105 1095-1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Febbraio, M., Podrez, E. A., Smith, J. D., Hajjar, D. P., Hazen, S. L., Hoff, H. F., Sharma, K., and Silverstein, R. L. (2000) J. Clin. Investig. 105 1049-1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen, M., Inoue, K., Narumiya, S., Masaki, T., and Sawamura, T. (2001) FEBS Lett. 499 215-219 [DOI] [PubMed] [Google Scholar]
- 26.Park, H., Adsit, F. G., and Boyington, J. C. (2005) J. Biol. Chem. 280 13593-13599 [DOI] [PubMed] [Google Scholar]
- 27.Ohki, I., Ishigaki, T., Oyama, T., Matsunaga, S., Xie, Q., Ohnishi-Kameyama, M., Murata, T., Tsuchiya, D., Machida, S., Morikawa, K., and Tate, S. (2005) Structure (Camb.) 13 905-917 [DOI] [PubMed] [Google Scholar]
- 28.Navazo, M. D., Daviet, L., Savill, J., Ren, Y., Leung, L. L., and McGregor, J. L. (1996) J. Biol. Chem. 271 15381-15385 [DOI] [PubMed] [Google Scholar]
- 29.Demers, A., McNicoll, N., Febbraio, M., Servant, M., Marleau, S., Silverstein, R., and Ong, H. (2004) Biochem. J. 382 417-424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marleau, S., Harb, D., Bujold, K., Avallone, R., Iken, K., Wang, Y., Demers, A., Sirois, M. G., Febbraio, M., Silverstein, R. L., Tremblay, A., and Ong, H. (2005) FASEB J. 19 1869-1871 [DOI] [PubMed] [Google Scholar]
- 31.Tanaka, T., Nakata, T., Oka, T., Ogawa, T., Okamoto, F., Kusaka, Y., Sohmiya, K., Shimamoto, K., and Itakura, K. (2001) J. Lipid Res. 42 751-759 [PubMed] [Google Scholar]
- 32.Abumrad, N. A., Ajmal, M., Pothakos, K., and Robinson, J. K. (2005) Prostaglandins Other Lipid Mediat. 77 77-83 [DOI] [PubMed] [Google Scholar]
- 33.Stewart, B. W., and Nagarajan, S. (2006) Mol. Immunol. 43 255-267 [DOI] [PubMed] [Google Scholar]