

Abstract
Long-term exposure to antivirals is associated with serious cellular toxicity to the kidney and other tissues. Organic anion transporters (OATs) are believed to mediate the cellular uptake, and hence cytotoxicity, of many antivirals. However, a systematic in vitro and ex vivo analysis of interactions between these compounds with various OAT isoforms has been lacking. To characterize substrate interactions with mOat1, mOat3, and mOat6, a fluorescence-based competition assay in Xenopus oocytes as well as wild-type and knock-out whole embryonic kidney (WEK) organ culture systems was developed using 6-carboxyfluorescein, 5-carboxyfluorescein, and fluorescein. Of nine common antiviral drugs assessed in oocytes, many manifested higher affinity for SLC22a6 (mOat1), originally identified as NKT (e.g. adefovir and cidofovir), two (ddC and ddI) manifested significantly higher affinity for mOat3, while mOat6 had comparatively low but measurable affinity for certain antivirals. A live organ staining approach combined with fluorescent uptake in WEK cultures allowed the visualization of OAT-mediated uptake ex vivo into developing proximal tubule-like structures, as well as quantification of substrate interactions of individual OAT isoforms. In general, antiviral specificity of SLC22a6 (Oat1) (in Oat3-/- WEK culture) and SLC22a8 (Oat3) (in Oat1-/- WEK culture) was consistent with the Xenopus oocyte data. The combined observations suggest SLC22a8 (Oat3) is the major transporter interacting with ddC and ddI. Finally, quantitative structure-activity relationship analysis of the nine antivirals' physicochemical descriptors with their OAT affinity indicates that antiviral preferences of mOat1 are explained by high polar surface areas (e.g. phosphate groups), whereas mOat3 prefers hydrogen bond acceptors (e.g. amines, ketones) and low rotatable bond numbers. In contrast, hydrogen bond donors (e.g. amides, alcohols) diminish binding to mOat6. This suggests that, despite sharing close overall sequence homology, Oat1, Oat3, and Oat6 have signficantly different binding pockets. Taken together, the data provide a basis for understanding potential drug interactions in combination antiviral therapy, as well as suggesting structural mdifications for drug design, especially in the context of targeting toward or away from specific tissues.
The organic anion transporters (OATs)2 mediate the uptake of structurally diverse substrates: endogenous compounds (steroids, odorants, cyclic nucleotides, neurotransmitters), drugs (non-steroidal anti-inflammatory drugs, diuretics, and antibiotics), environmental toxins (ochratoxin A and mercuric chlorides), and other organic wastes (1, 2). It has recently been hypothesized that OATs and other SLC22 family members participate in a remote sensing mechanism for small molecules between tissues and also between organisms (1, 23). The prototype, now called Oat1, was initially identified as NKT (3-5). Since then, at least six OATs have been identified, all of which are members of the larger SLC22 family of transporters. Most closely related to SLC22a6 (Oat1) are SLC22a8 (Oat3), originally identified as Roct (6), and the recently identified SLC22a20 (Oat6), expressed in olfactory mucosa (1, 7). Importantly, the OATs have been postulated to constitute the initial step in the uptake of antiviral nucleoside and nucleobase analog antiretroviral, uptake (8, 9). To combat chronic disease because of HIV, herpes, smallpox, and other viruses, antiviral drugs are often administered, with HIV also requiring prolonged combination therapies as either triple or quadruple drug “cocktails” (10). Unfortunately, long-term exposure to antivirals is often associated with nephrotoxicity, lactic acidosis, hepatic failure, and skeletal myopathy (11, 12), problems that can be further exacerbated by additional, as yet undefined, drug interactions.
Despite their plausible role via OAT transport in cytotoxicity, data on the relative affinity of antiviral drugs for OATs (Km or IC50) is limited mostly to Oat1 (13, 14), with some data indicating the involvement of Oat3 (15-17). However, the relative contribution of each OAT to the uptake of antivirals in epithelial and other tissues is unknown. The analysis is further confounded by the presence of multiple, simultaneously expressed transporters; varying degrees of substrate specificity at the (individual) transporter level; as well as a complex pattern of tissue expression and cellular localization. These issues have yet to be addressed in a single system or in a single set of experiments; unfortunately, widely used heterologous systems for addressing OAT affinity do not take these issues into account.
In the original description of NKT/Oat1, it was shown that the gene is expressed during kidney development (4). Whole organ culture could conceivably provide a system for studying substrate interactions in a complete environment, providing native cellular machinery recapitulating the whole tissue response ex vivo. Rat whole embryonic kidney (WEK) cultures have previously been shown to accumulate fluorescein in a probenecid-sensitive fashion (18). Exploiting OATs expressed in WEKs, OAT isoform-specific fluorescent anion “trackers” capable of visualizing OAT-mediated uptake in organ systems could conceivably dissect the role of individual Oats in intact tissue. In fact, it has been shown that 6-carboxyfluorescein is an effective fluorescent tracer for studying hOat1-mediated translocation in transfected Chinese hamster ovary cells (19). In general, carboxyfluoresceins (CF) have better kinetic properties for studying cellular OAT-mediated uptake than fluorescein, because the extra negative charge on the molecule allows for better cell retention (20). In light of this, we developed and characterized a fluorescence-based assay for Xenopus oocytes and WEKs for determining OAT isoform substrate interactions using differently substituted carboxyfluoresceins.
Additionally, we have utilized the uptake of fluorescent OAT substrates in the in vitro and the whole organ system to determine the affinity values for a subset of nucleotide reverse transcriptase inhibitors (NRTI) and acyclic nucleotide/nucleoside antivirals interacting with mOat1, mOat3, and mOat6: adefovir, cidofovir, tenofovir, acyclovir, ddC, ddI, 3TC, d4T, and AZT (21). We demonstrate that mouse WEKs can be used for quantitative analyses and, using a novel live organ lectin staining procedure, localize distinct proximal tubule-like uptake patterns in WEK. By using mOat1 and mOat3 WEK knock-out strains, we confirm that adefovir and cidofovir interact predominantly with mOat1, although ddC and ddI seem to primarily interact with mOat3. The ex vivo data are generally consistent with our in vitro oocyte transport results obtained in this study.
Finally, quantitative structure-activity relationship (QSAR) analysis was used to correlate the physicochemical properties of the nine antiviral molecules with their relative binding interactions to mOat1, mOat3, and mOat6. These characteristics include polar surface area, presence of hydrogen bond donors and acceptors, and the number of rotatable bonds. Thus, we have quantitatively and systematically analyzed the interaction of commonly used antiviral drugs with OATs on multiple levels. These results should aid in the design of antivirals directed toward or away from OAT expressing tissues with the goal of enhancing or diminishing transport (and presumably toxicity) into tissue compartments, and help in predicting drug-drug interactions and cytotoxicity during combination therapy. The quantitative multilevel analysis should also help provide a “systems” basis for understanding antiviral handling via OATs.
EXPERIMENTAL PROCEDURES
Organic Anions
The radiolabeled tracers [3H]p-aminohippurate (PAH) (specific activity 4.2 Ci/mmol) and [3H]estrone-3-sulfate (ES) (57 Ci/mmol) were purchased from PerkinElmer Life Sciences. [3H]ddC (30 Ci/mmol) was obtained from Moravek Biochemicals (Brea, CA). Fluorescent anions, TRITC-conjugated lectins (Dolichos biflorus lectin and Lotus lectin (Sigma)), and prototypical OAT compounds (ES, PAH), and fluorescent dyes (4-carboxyfluorescein dimethylester (4-CFDM), 5-carboxyfluorescein (5-CF), 6-carboxyfluorescein (6-CF), fluorescein, eosin Y, and resorufin) were obtained from Sigma. Antivirals (adefovir, cidofovir, tenofovir, acyclovir, zidovudine (AZT), lamivudine (3TC), stavudine (d4T), zalcitabane (ddC), and didanosine (ddI)) were obtained from Moravek Biochemicals or Sigma.
Xenopus Oocyte Uptake
Xenopus oocyte assays were performed as described previously (1, 22). Capped RNA was synthesized from linearized plasmid DNA for SLC22a6 (mOat1) (Image clone 4163278), SLC22a8 (mOat3) (Image clone 4239544), and SLC22a20 (mOat6) (Image clone 6309674;) using the mMessage mMachine in vitro transcription kit (Ambion, Austin, TX). Briefly, oocytes were isolated and maintained in Barths buffer growth medium, injected with cRNA solution (0.5 μg/μl, 23 nl/oocyte), and allowed to mature for 3-4 days post-injection prior to transport assays.
Experimental groups of 16-20 oocytes were placed in a 24-well plate with 1 ml of Barths buffer containing 1 μCi of a 3H-labeled organic anion tracer or a fluorescent anion tracer: 20 μm 6-CF (for mOat1-injected oocytes), 100 μm 5-CF (mOat3), or 30 μm fluorescein (mOat6). Samples contained different concentrations of an unlabeled organic anion. After a 1-h incubation at 25 °C, oocytes were washed 3 times with 4 °C Barths buffer and each experimental group was divided into four samples of 4-5 oocytes. Radioactivity was measured by scintillation counting (Rackbeta, Beckman-Coulter) or by fluorescence (PolarStar plate reader, BMG Labtechnologies, Durham, NC). To calculate the OAT-mediated component, the background tracer uptake was measured in uninjected oocytes and was subtracted from the uptake of OAT-injected oocytes in all experimental samples.
Calculations and Statistics
Substrate activity was calculated for fluorescent tracers as the fluorescence clearance (CL) from the incubation medium (CL = Vtransport/S), by dividing the fluorescence (relative fluorescence units) absorbed per oocyte per unit time (Vtransport, relative fluorescence units/oocyte/h) by the tracer concentration in the incubation medium (S, relative fluorescence units/ml). [3H] transport rates utilize counts/min instead of the relative fluorescence unit term. Affinity of an inhibitor organic anion (IC50, Ki, or Km) was determined by measuring tracer uptake in the presence of 3-4 inhibitor concentrations in 10-fold increments. Each data point is the average of 2-3 experiments. Inhibition data were curve fitted using nonlinear regression in Prism software 5.0 (GraphPad Inc., San Diego, CA) to calculate the IC50 ± S.E., the Michaelis-Menten equation for Km, and the Ki value was determined as shown previously (23). Maximum uptake (Vmax) was calculated as Vmax = CL · ([S]+ Ki), where CL values were measured in a single oocyte experiment with all fluorescent compounds tested at the concentration, S = 100 μm, and Ki values were from Table 1.
TABLE 1.
OAT-binding affinities and transport characteristics of fluorescent substrates
| OAT | Fluorescent substrate | Kia | CLb | Vmaxc |
|---|---|---|---|---|
| μm | nanoliter/oocyte/h | pmol/oocyte/h | ||
| mOat1 (SLC22a6) | Fluorescein | 3.0 ± 0.1 | 12.7 ± 1.5 | 1.3 ± 0.2 |
| 5-Carboxyfluorescein | 523.4 ± 80 | 3.6 ± 1.0 | 2.2 ± 1.4 | |
| 6-Carboxyfluorescein | 56.6 ± 3 | 26.7 ± 5.5 | 4.2 ± 1.1 | |
| 4-Carboxyfluorescein-DM | 34.8 ± 4 | 8.7 ± 2.6 | 1.2 ± 0.4 | |
| Eosin-Y | 0.1 ± 0.03 | 1.5 ± 1.7 | 0.2 ± 0.2 | |
| Resorufin | 4.7 ± 0.2 | NDd | ND | |
| mOat3 (SLC22a8) | Fluorescein | 35.0 ± 2 | 81.6 ± 32.7 | 11.0 ± 4.8 |
| 5-Carboxyfluorescein | 373.3 ± 28 | 95.5 ± 21.5 | 45.2 ± 16.9 | |
| 6-Carboxyfluorescein | 10.7 ± 0.1 | 33.5 ± 9.2 | 3.7 ± 1.0 | |
| 4-Carboxyfluorescein-DM | 17.6 ± 0.9 | 30.3 ± 8.8 | 3.6 ± 1.0 | |
| Eosin-Y | 0.3 ± 0.04 | 0.6 ± 0.4 | 0.06 ± 0.04 | |
| Resorufin | 8.3 ± 0.1 | ND | ND | |
| mOat6 (SLC22a20) | Fluorescein | 93.3 ± 6.2 | 26.4 ± 12.3 | 5.1 ± 2.6 |
| 5-Carboxyfluorescein | 3581.0 ± 582 | 1.4 ± 1.8e | NDe | |
| 6-Carboxyfluorescein | 3162.3 ± 241 | 1.4 ± 2.1e | NDe | |
| 4-Carboxyfluorescein-DM | 4456 ± 117 | 3.8 ± 0.5 | 2.1 ± 0.9 | |
| Eosin-Y | 4.8 ± 0.7 | 37.4 ± 26.6 | 3.9 ± 2.9 | |
| Resorufin | >3000 | ND | ND |
Calculated from data of Fig. 1, the mean ± S.E.
For each of the transporters, OAT-mediated clearance values were determined for all fluorescent tracers in a singie preparation of OAT-injected oocytes. For each transporter, data of a typical single experiment is presented (data obtained with different oocyte preparations are not comparable to each other).
Calculated as Vmax = CL · (S + Kd), with all fluorescent compounds tested at a concentration, S = 100 μm.
ND, uptake of resorufin was difficult to measure because it was rapidly decolorized (apparently metabolized) after being taken up by oocytes.
mOat6-mediated transport rate was not measurable for 5-CF and 6-CF because their uptake in OAT-injected oocytes was not significantly different from uninjected oocytes.
In Vitro and Ex Vivo Tissue Systems
WEK Culture/Tissue Preparation—Knockouts of mOat1 and mOat3 were bred as described previously (22, 24). As described previously (18), whole kidneys were microdissected from embryonic day 13.5 mice (E13.5) and grown on polycarbonate Transwell filters with Dulbecco's modified Eagle's medium/F-12 (Mediatech, Herndon, VA) supplemented with 10% fetal bovine serum (Mediatech) and 1% penicillin/streptomycin (Invitrogen) at 37 °C for 4 days. All animal protocols conformed to NIH guidelines for acceptable animal use.
Whole Embryonic Kidney Uptake Assay and Confocal Microscopy—After 4 days of culture, E13.5 kidneys were washed once in phosphate-buffered saline, and WEKs (on the filter) were incubated for 1 h at 25 °C in a minimal saline solution (phosphate-buffered saline, 0.1 mm CaCl2, and 1 mm MgCl2) with 1 μm fluorescent tracker (5-CF, 6-CF, or fluorescein) and with or without tracker uptake inhibitor (1-2 mm probenecid: PAH, ES, ddC, ddI, adefovir, or cidofovir). WEKs were also incubated with 20 μg/ml D. biflorus lectin, a collecting duct marker, or Lotus lectin, a proximal tubule marker (25). Following the 1-h uptake, WEKs were washed 3 times in 4 °C phosphate-buffered saline, cut from the Transwell filter, and mounted on a glass slide with fluormount (Southern Biotech, Birmingham, AL). To prevent fluorescent tracker diffusion out of the cell, samples were kept on ice until imaged. Confocal images were taken with the Nikon D-eclipse C1 confocal microscope (A.G. Heinze, Lake Forest, CA). In each individual experiment images were taken using the same settings (e.g. gain, saturation) for intensity comparisons.
Quantitative Image Analysis—Image analysis was carried out using the ImageJ software (NIH, Bethesda, MD) using pixel intensity measurements. Images were manually outlined twice to measure intensity, and then the average value obtained was taken as the total intensity of a kidney. Four representative background areas were traced in interstitial spaces. Average background measurements were then subtracted from the total intensity. Significance values were determined using the t test and all values represent triplicate kidney samples.
QSAR Analysis—The antiviral affinity data for mOat1, mOat3, and mOat6 were correlated with physicochemical descriptors using multiple linear regression. IC50 values were converted to Ki before conversion to the -log value (pKi). The descriptors, molecular weight, number of rotatable bonds (RB), LogP, and the calculated molar refractivity were calculated using Sybyl software (Tripos Discovery Informatics, St. Louis, MO) where as number of hydrogen bond donor, number of hydrogen bond acceptor, and polar surface area (PSA) were calculated using the Chemaxon package. The affinity data (pKi) and various descriptors were used as dependent and independent variables, respectively. The final model for each OAT contains statistically significant descriptors (p < 0.05).
RESULTS
Fluorescent Tracers for OAT-mediated Uptake—To identify fluorescent compounds with the potential to serve as tracers of OAT-mediated transport, a panel of six fluorescent anions was tested for suitable characteristics. Key criteria included a high Vmax (translocation rate) and a suitable Ki (affinity constant) to minimize the chance of tracer competition with the test compound. The affinity values for each fluorescent anion for mOat1, mOat3, and mOat6 were determined by inhibiting the uptake of a radiolabeled tracer, either PAH or ES (Fig. 1 and Table 1). The fluorescent compounds (fluorescein, 4-CFDM, 5-CF, 6-CF, eosin Y, and resorufin) were all found to have significant interaction with all OATs tested. Because this does not necessarily indicate they are substrates, the uptake rates (Vmax) of the fluorescent compounds were also determined for each transporter (Fig. 1 and Table 1). The uptake of 5 of the compounds (simultaneously tested at 100 μm and used to calculate the Vmax) were obtained in a single typical experiment. All compounds tested proved to be translocated by (i.e. to be substrates for) at least one of the three transporters.
FIGURE 1.

Inhibition of radiolabeled tracer uptake by fluorescent organ anions. To determine the affinity of a particular fluorescent anion for a particular OAT, varying concentrations of the fluorescent anion were used to inhibit the uptake of a prototypical radiolabeled tracer ([3H]PAH for mOat1 and [3H]ES for mOat3 and mOat6) in Xenopus oocytes. Values are plotted here as percent inhibition of control uptake (mean ± S.E., n = 4). Curves were fit using nonlinear regression and affinity values are presented in Table 1.
The data from Table 1 was then used to determine the best “tracers” (i.e. probes for cellular accumulation) and trackers (i.e. visualization of transport in organ culture systems) for the different transporters. Based on the Vmax values determined, 6-CF, 5-CF, and fluorescein had the best characteristics for being used as probes of the transport activities of, respectively, mOat1, mOat3, and mOat6.
6-CF had the best characteristics for mOat1 transport compared with other compounds tested exhibiting a Ki of 57.3 μm and a Vmax of 4.2 pmol/oocyte/h (Table 1). Although 6-CF binding affinity for mOat1 proved to be lower than that of fluorescein, 4-CFDM, eosin Y, and resorufin, the translocation rate, Vmax, was the highest (Table 1). Similarly, whereas 5-CF showed the poorest affinity for mOat3 (Ki, of 373.3 μm, compared with the second poorest, fluorescein, which was 35.1 μm), it had the highest Vmax value, 45.2 pmol/oocyte/h. Therefore, 5-CF has the best characteristics for use as a fluorescent mOat3 substrate. For mOat6, fluorescein had the second strongest binding affinity (Ki, 93.2 μm, compared with eosin Y, 5 μm) as well as the highest Vmax (5.1 pmol/oocyte/h), qualities that suggest its use as an effective tracer for mOat6.
OAT Antiviral Affinities—Whereas mOat1 and mOat3 are coexpressed in many tissues, the relative proportion, as well as functionality, can be variable. For example, mOat1 is expressed in mouse olfactory mucosa (along with mOat6), whereas in retina, mOat3 expression appears higher than that of mOat1 (7). Thus, expression patterns are likely to be important determinants of tissue penetration for various antivirals. Using the identified fluorescent probes (5-CF, 6-CF, and fluorescein), binding affinities for a diverse group of antiviral drugs were determined for mOat1, mOat3, and mOat6 in Xenopus oocytes. The uptake of many of these compounds has been determined for hOat1 (8, 9, 12), although not side by side in the same study. Due to structural homology, Oat3 and Oat6 might translocate them as well, although this remains to be determined. Because of this, and specifically for these compounds, IC50 (half-maximal inhibition of transport) should be a good approximation of transport affinity (Km), because translocation by a SLC22a family member has been established.
Adefovir, tenofovir, cidofovir, and acyclovir all demonstrated significantly greater affinity for mOat1 than for its homolog mOat3, with cidofovir showing the highest mOat1 selectivity (Table 2 and supplemental Fig. S1). This conclusion is consistent with previous findings that suggest mOat3 has only low affinity for these four antivirals, whereas mOat1 makes a significantly higher contribution to their uptake (17, 26).
TABLE 2.
IC50 of antivirals against mOat1, mOat3, and mOat6
|
IC50a±
S.E.b
|
|||
|---|---|---|---|
| Antiviral compound | mOatl (SLC22a6) | mOat3 (SLC22a8) | mOat6 (SLC22a20) |
| μm | |||
| NRTI | |||
| Didanosine (ddI) | 600 ± 23 | 136 ± 9 | >3000 |
| Stavudine (d4T) | 628 ± 66 | 2113 ± 274 | 1671 ± 181 |
| Zalcitabane (ddC) | 1479 ± 67 | 203 ± 30 | 729 ± 30 |
| Lamivudine (3tC) | 104 ± 3 | 140 ± 7 | >3000 |
| Zidovudine (AZT) | 78 ± 3 | 38 ± 5 | 218 ± 17 |
| Acyclic nucleotide analogs | |||
| Adefovir | 36 ± 2 | 597 ± 46 | >3000 |
| Tenofovir | 81 ± 3 | 384 ± 45 | >3000 |
| Cidofovir | 25 ± 7 | >3000 | >3000 |
| Acyclic nucleoside analogs | |||
| Acyclovir | 209 ± 23 | 729 ± 75 | >3000 |
Calculated from data in supplemental Fig. 1 Fluorescent tracers determined in Fig. 1 and Table 1 were used as probes in competition with antivirals as listed. The data represents multiple 10-fold increases in antiviral concentration from two experiments plotted using nonlinear regression (see “Experimental Procedures” and supplemental Fig. 1) to determine the IC50.
S.E. was calculated as the deviation of the inhibitor points to the fitted nonlinear curve.
The studies of OAT interactions with antivirals revealed unexpectedly striking differences in binding and transport, especially given the strong overall sequence homology between these three transporters. These results offer important insights into the binding sites of these transporters as well as important information (computationally analyzed below) into antiviral drug design. The results indicate that some NRTI antivirals, such as ddI and ddC have a substantially higher affinity for mOat3 than for mOat1. As an exception, d4T was the sole NRTI that exhibited poor affinity for mOat3 compared with mOat1. Because the data suggests ddC predominantly interacts with mOat3, radiolabeled ddC was assessed in mOat3 injected oocytes (Fig. 2). The estimated Km (125.9 μm) closely resembles the IC50 determined in Table 2 (203 μm). This confirms that mOat3 is a high affinity transporter of ddC and because the IC50 and Km are similar, strengthens the conclusion, that at least for these compounds, inhibition mirrors Km. The compounds 3TC and AZT showed equal specificity for both mOat1 and mOat3 (Table 2).
FIGURE 2.

Kinetics of dideoxycytidine (ddC) uptake by mOat3. 30 nCi/mmol [3H]ddC was incubated with mOat3-injected Xenopus oocytes along with increasing concentrations of unlabeled ddC. Uptake of ddC by mOat3 was saturable and concentration dependent with a Km of 125.9 μm. These values are consistent with the IC50 determinations and suggest that for these groups of compounds, IC50 is an appropriate indicator of Km. Background ddC accumulation in uninjected oocytes was linear, suggesting non-mediated mechanisms of uptake. The Km was determined in GraphPad using the Michaelis-Menten equation. Values are plotted as mean ± S.E. (n = 4).
With respect to the olfactory transporter, for all compounds tested, only ddC, d4T, and AZT exhibited significant interaction with mOat6, which had no detectable interaction with ddI, 3TC, cidofovir, and acyclovir. This is in contrast to its two closest homologs, mOat1 and mOat3, which, although differing in specificity have the ability to interact with all antivirals tested.
Localization of Carboxyfluoresceins in Whole Embryonic Kidneys—Different CF were used as trackable molecules in WEK for visualizing OAT-mediated uptake. This ex vivo system is an alternative to the use of renal slices and has the unique advantage that the tissue not only survives in culture, but continues to grow and develop. WEKs were incubated in a minimal saline solution containing CF before analysis under confocal microscopy. In this study, both 5-CF and 6-CF uptake reveals punctate cellular accumulation in convoluted structures (Fig. 3). Inhibition of uptake by the addition of 2 mm probenecid was sufficient to completely eliminate the fluorescence patterns, strongly suggesting OAT-mediated uptake. Additionally, high magnification images of the tubules (Fig. 3, C and F) indicated that fluorescence accumulates within tubular cells. Whereas the lack of strong luminal fluorescence suggests that the CF compounds enter through a basolateral step in tubular epithelial cells, there appears to be rate-limiting apical excretion based on significantly higher fluorescence in the cytosol than the lumen. These observations are in contrast to fluorescein uptake, which was found to rapidly accumulate within the lumen of the developing tubules (18), and would prove problematic for determining OAT uptake.
FIGURE 3.

Comparison of carboxyfluorescent dyes in E13.5 WEK. A, WT WEK + 5CF. B, WTWEK + 5CF in presence of 2 mm probenecid. C, high power view of single tubule in A. D, WTWEK + 6CF. E, WTWEK + 6CF in presence of 2 mm probenecid. F, zoom of single tubule in D. Both 5-CF and 6-CF show discrete patterns that resemble proximal tubule structures. Accumulation of 5-CF and 6-CF are in punctuate cellular sites (C and F), with overaccumulation in the cytosol. This would suggest the apical step is rate-limiting. Both dyes also show complete probenecid sensitivity, indicating an OAT-dependent mechanism of tubular accumulation (B and E).
Whereas the structures displaying CF accumulation appear to be proximal tubules, it was not clear if there was any uptake in the collecting duct. Using a live staining method, we performed co-imaging of CF uptake with D. biflorus staining, a collecting duct marker (25), which demonstrates that uptake is separate from the collecting duct structures (Fig. 4, A and B). Sites of CF accumulation appear to begin at the tips of collecting ducts, sites in the developing kidney where nephrogenesis is highest and where the distal portion of the nephron connects to the collecting system. As expected from an OAT-mediated process, CF uptake begins at the proximal portion of the nephron-like structure.
FIGURE 4.

Localization of 5-CF and 6-CF uptake in E13.5 WEKs grown for 4 days. A, 6-CF uptake in D. biflorus stained WT WEK. B, high power view of collecting duct in A. C, 5-CF uptake in Lotus lectin stained WT WEK. D, high power view of tubule structure in C. Red staining is the specified lectin marker. Green structures represent accumulation of CF. Co-staining of CF uptake with D. biflorus demonstrates localization is separate from the collecting duct structures. Staining with Lotus lectin suggests CF is in proximal tubule-like structures. Note that accumulation begins from the tubule tips and abruptly ends approximately halfway along the convoluted tubule (C and D). CD, collecting duct; CF, carboxyfluorescein; PT, proximal tubule; DT, distal tubule.
To further define these structures, colocalization with Lotus lectin, a proximal tubule marker (25), was performed. Lotus lectin staining was not as specific as D. biflorus because it marked all epithelial structures; however, it more intensely stained the convoluted tubules (Fig. 4, C and D). Therefore, the relative intensity could be used to define tubule structures. Structures intensely stained by Lotus lectin showed distinct CF accumulation that appeared three-dimensionally arranged around a lumen (Fig. 4, C and D). Of note, uptake was not seen along the entire length of the convoluted tubule (including the presumed loop of Henle) but only along half the tubule, suggesting the OAT-mediated uptake of CF is limited to distinct portions of the developing proximal tubule.
Antiviral Interaction with Whole Embryonic Kidneys—Oat1 and Oat3 knock-out mice have previously been described in detail (22, 24) and here we have used WEK from these mice to identify substrate interactions for single OAT isoforms within an intact organ system. This approach proved to be quantitative for ligand interactions and was consistent with data obtained by other methods. By measuring the intensity of inhibited samples in comparison to control images (see “Experimental Procedures”), it was possible to ascertain the inhibition potency (Figs. 5F, 6F, and supplemental Fig. S2F).
FIGURE 5.

Isolation of mOat1 uptake and inhibition patterns using mOat3-/- WEK. A, positive control, 1 μm 6-CF. B, +1 mm adefovir. C, +1mm cidofovir. D, +2mm ddC. E, +1mm ddI. F, quantitative image analysis. To isolate Oat1-mediated uptake, WEK of Oat3-/- mice were used with 6-CF. As predicted from Table 2, adefovir and cidofovir (B and C) had a significant effect on 6-CF uptake. ddC and ddI were less potent but inhibition was statistically significant compared with control. The negative control (1 mm PAH; image not shown) reduced 6-CF uptake to negligible levels. All images are representative of triplicate WEKs from the same experiment and pregnant female. F, values are presented as mean ± S.E., percent of control. *, p < 0.01; **, p < 0.005; ***, p < 0.0005.
FIGURE 6.

Isolation of mOat3 uptake and inhibition pattern using mOat1-/- WEK. A, positive control, 1 μm 6-CF. B, +1mm adefovir. C, +1mm cidofovir. D, +2mm ddC. E, +1mm ddI. F, quantitative image analysis. Oat3 mediated uptake was isolated using WEK from Oat1-/- mice using 6-CF (for comparison to Fig. 5). Adefovir and cidofovir (B and C) showed poor inhibition of Oat3-mediated uptake. ddC showed significant inhibition of Oat3-mediated uptake, in contrast to Oat1-mediated uptake seen in Fig. 7. 1 mm ES is sufficient to eliminate 6-CF uptake (not shown). Images are representative of triplicate WEKs. F, values are presented as mean ± S.E., percent of control. *, p < 0.05; **, p < 0.001.
Before testing the knock-out WEKs, the differences between 5-CF and 6-CF in wild-type (WT) tissue was determined, because they express both Oat1 and Oat3. The inhibitory effects of cidofovir, ddC, and ES on WT WEK uptake were determined to define the differences between using 5-CF versus 6-CF. This was used to create an inhibitory profile. Both fluorescent compounds are translocated by the two major transporters (mOat1 and mOat3), but with very different affinities and rates. The data are summarized in supplemental Fig. S2; left sides of the panels contain 5CF-tracked WEKs, whereas the right side shows 6-CF as a tracker.
Data already presented from oocyte studies suggest 5-CF may be more selective for mOat3, predicting that 1 mm cidofovir should not significantly reduce 5CF uptake (Table 2), whereas ddC at 2 mm and ES at 100 μm should almost completely inhibit 5-CF uptake (IC50 ES = 9.1 μm (8)). Cidofovir inhibition was statistically indistinguishable from the control (left supplemental Fig. S2, A versus B). In contrast, both ddC and ES blocked 5-CF uptake to below 15% of control levels (left supplemental Fig. S2, A versus C + D). This would indicate that using 5-CF as a tracker in WEK may mostly reflect the action of mOat3 (the predicted inhibitory profile), with little contribution from mOat1.
On the other hand, 6-CF as a tracker may reflect more complex contributions from both mOat1 and mOat3. This is suggested by the similar Vmax shown in Table 2. All three compounds had intermediary effects on 6-CF uptake, with values showing 50-70% levels of fluorescence compared with the controls (right supplemental Fig. S2). These values roughly reflect the combined inhibitory profiles of mOat1 and mOat3 (Table 3). Therefore, 6-CF may be used to track both mOat1- and mOat3-mediated translocation in an intact organ system.
TABLE 3.
Experimental, calculated pKi, and residuals from multiple linear regression studies
|
Compounds
|
Experimental
pKia
|
Calculated
pKib
|
Residual
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| mOat1 | mOat3 | mOat6 | mOat1 | mOat3 | mOat6 | mOat1 | mOat3 | mOat6 | |
| Didanosine | 3.22 | 3.87 | 3.25 | 4.10 | −0.03 | −0.23 | |||
| Stavudine | 3.2 | 2.68 | 2.78 | 3.05 | 2.90 | 3.19 | 0.15 | −0.22 | −0.41 |
| Zalcitabane | 2.83 | 3.69 | 3.14 | 3.24 | 3.23 | 3.19 | −0.41 | 0.46 | −0.05 |
| Lamivudine | 3.98 | 3.85 | 3.74 | 4.10 | 0.24 | −0.25 | |||
| Zidovudine | 4.11 | 4.42 | 3.66 | 3.77 | 4.10 | 3.19 | 0.34 | 0.32 | 0.47 |
| Adefovir | 4.44 | 3.22 | 2.28 | 4.38 | 3.18 | 2.11 | 0.06 | 0.04 | 0.17 |
| Tenofovir | 4.09 | 3.42 | 1.93 | 4.38 | 3.18 | 2.11 | −0.29 | 0.24 | −0.18 |
| Cidofovir | 4.6 | 2.49 | 4.57 | 2.66 | 0.03 | −0.16 | |||
| Acyclovir | 3.67 | 3.13 | 3.76 | 3.38 | −0.09 | −0.25 | |||
Experimental pKi values were calculated as the -log(IC50) from values in Table 2.
Calculated pKi values were determined by correlating experimental pKi values to different antiviral physicochemical descriptors using various software programs (see “Experimental Procedures”).
Whereas using wild-type WEKs may provide the more comprehensive view of OAT interaction with organic anions, singling out individual transport properties can provide important information. To isolate Oat1-mediated uptake, WEKs of Oat3-/- mice were tracked with 6-CF. As predicted from Table 3, adefovir and cidofovir (Fig. 5, B and C) had significant inhibition of 6-CF uptake by lowering WEK fluorescence to 20% of control levels. The effects of ddC and ddI were less potent but inhibited presumed Oat1 accumulation of 6-CF to nearly 60% of controls (Fig. 5, D and E). As suggested earlier, mOat1 has higher affinity for cidofovir and adefovir in comparison to its affinity for ddC and ddI (Table 3), data that compares well with the oocyte data presented here.
Oat3-specific uptake was studied in isolation by using WEKs from Oat1-/- mice using 5-CF. Adefovir and cidofovir, each tested at 1 mm (Fig. 6, B and C), showed poor inhibition of presumed Oat3-mediated uptake, with only adefovir causing any statistically significant effects (70% of control). The antivirals ddC and ddI had a major impact on Oat3-mediated uptake of 6-CF (reducing it to 20% of control), in contrast to those found by Oat1-mediated uptake (Fig. 5). Data points obtained from this image analysis reflected values obtained from oocytes fairly closely (Table 2 and supplemental Fig. S1). The combined observations suggest that mOat3 is the major route of ddC elimination. It seems likely that data obtained through this methodology more closely resemble the compound interactions in vivo.
QSAR of Antiviral Affinities—To explain the observed differences in antiviral affinity between OAT isoforms, predictive QSAR models were developed based on antiviral structure. Individual models for OAT ligand binding were built by fitting the data to general Equation 1 using multiple linear regression analysis based on different physicochemical properties of the various substrates. Predictions of pKi based on the models compared with the obtained experimental values are shown in Table 3 and supplemental Fig. S3.
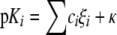 |
(Eq.1) |
The left and right hand side of Equation 1 represent dependent (pKi =-log Ki, in m) and independent variables, respectively. Here, ξi represents the individual physicochemical descriptors (e.g. the number of hydrogen bond donors, acceptors, and rotatable bonds: each scaled by a factor ci). All the parameters and constants (κ) are freely optimizable. The physicochemical significance of coefficients and descriptors is explained in the following section.
mOat1—The major antiviral characteristic determining inhibition is the PSA, which is strongly influenced by the phosphate groups (Fig. 7). The constant κ in Equation 1 forms a base value of 1.495 log units for mOat1 inhibition that is then modulated by the polar surface area terms. Both the coefficient and PSA values are positive implying that an increase in polar surface area from inhibitors increases activity. It should be noted that PSA calculations used in this model reflect topological values and are, therefore, independent of molecular conformations. Poor inhibitors such as ddC, d4T, and ddI have lower PSA (88.15, 78.87, and 88.74 Å2) compared with more active inhibitors such as adefovir, cidofovir, and AZT (146.19, 155.49, and 115.08 Å2). The model was able to explain ∼84% (r2 = 0.841) of variation in experimental activity of all nine mOat1 inhibitors, ranging from 36 to 1479 μm.
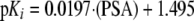 |
(Eq.2) |
FIGURE 7.

Antiviral structural variation and OAT specificity. The chemical structures of the antivirals are shown grouped by OAT preference. The phosphate groups in the mOat1 column demonstrate the need for high polar surface area. Whereas ketone groups (=O) and amine groups (-NH2) (which are hydrogen bond acceptors) are clearly seen among mOat3 preferring antivirals. Compounds that diminish Oat6 binding have a high number of amides (=NH) and alcohols (-OH), which are hydrogen bond donors. Note that ddC and 3TC differ only by a sulfur group, which is effective in increasing affinity for mOat1 but not affecting mOat3 affinity (Table 2). These characteristics can be used to modify for OAT affinity.
mOat3—Two descriptors, i.e. the total number of hydrogen bond acceptors (e.g. amines and ketones) and RB, are responsible for modulating the activity of mOat3 inhibitors. An increase in the number of rotatable bonds is detrimental to mOat3 inhibition, whereas increases in the number of hydrogen bond acceptors is beneficial for mOat3 inhibition. However, a distinct balance is required between RB and hydrogen bond acceptors for optimal inhibition of mOat3. Cidofovir and AZT are the least and most active compounds, respectively, the former containing nine RB, whereas the latter contains six RB. The constant κ in Equation 2 contributes positively (0.51 log units) to the overall equation. The model was able to explain ∼79% (r2 = 0.793) of variation in activity of all nine mOat3 inhibitors.
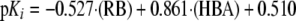 |
(Eq.3) |
mOat6—Antivirals with a large number of hydrogen bond donors (HBA) (e.g. amides and alcohols) are not good substrates for mOat6. The optimizable parameter κ contributes 5.37 log units in Equation 4, the base value for mOat6, inhibition that is then modulated by the hydrogen bond donor term. The coefficient associated with the number of hydrogen bond donors is negative, whereas in this case, the final number is positive indicating that increases in the number of hydrogen bond donors is detrimental to mOat6 inhibition (Equation 4). Less active compounds such as adefovir and tenofovir contain a larger number of hydrogen bond donating groups than the more active compounds d4T, ddC, and AZT. This simple model, with only one descriptor, was able to explain ∼76% (r2 = 0.758) of variation in the inhibition constants of all six mOat6 inhibitors.
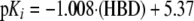 |
(Eq.4) |
DISCUSSION
We have analyzed the interaction of antivirals with organic anion transporters of the SLC22 family using novel methods for visualizing OAT-mediated uptake in wild-type and knock-out whole organ culture systems combined with Xenopus ooctyes. Drugs, toxins, and endogenous compounds that interact with OATs do not appear to act independently of each other. Clinically, many drugs that are OAT substrates are used together, HIV combination therapy being one example. Therefore, a thorough understanding of drug affinities for OATs is required. The present study examines OAT-mediated transport in multiple systems and levels (oocytes, whole embryonic kidneys, and physicochemical characteristics). Computational analyses of the data suggest physicochemical characteristics of antivirals that dictate binding preference for mOat1, mOat3, or mOat6. The WEK system, which likely recapitulates the native OAT intracellular and paracellular environment with greater fidelity than Xenopus oocytes, proved to be directly quantifiable and provides insight into the mechanism(s) by which drugs and/or toxins are able to access intrarenal sites. The approaches described here can tease out these interactions within the whole organ in the context of other drugs and toxins, whereas the analysis of structural characteristics of substrates could be exploited to modify OAT affinity and drug elimination.
The use of QSAR analysis provides mechanistic insight into the differential interaction of antivirals with OATs. We determined that different descriptors are important for explaining the biological activity of different OATs. The balance of hydrogen bond acceptors, such as amine and ketone groups, to the number of rotatable bonds was found to be important for mOat3 inhibition. On the other hand, the polar surface area (phosphate groups) and the number of hydrogen bond donating moieties (e.g. amides and alcohols) were found to be important for mOat1 and mOat6 inhibition, respectively. These data emphasize the distinctiveness of the binding sites of the three OATs, despite their high degree of homology (7) and suggests binding pocket characteristics that can be exploited for specific drug design. The identification of these characteristics may make it possible to differentially circumvent certain intrarenal anion transport processes, whereas modulating others. This concept would presumably apply to other OAT-expressing tissues like choroid plexus, olfactory mucosa, and placenta as well. For example, a viable treatment for prolonging drug action might utilize targeted and differential inhibition of the different OAT isoforms using structure-based drug design, a concept recently highlighted between mOat1 and mOat6 (23).
To analyze antiviral transport at multiple levels, novel WEK assays with wild-type and knock-out tissue were developed and utilized in conjunction with Xenopus oocyte assays. Comparisons clarified the different roles of OAT family members in antiviral uptake. For example, mOat3 was implicated as the major contributor to ddC and ddI transport, and consequently, ddC is shown to be a novel mOat3 substrate. Both ddC and ddI have been shown to be cytotoxic (27-30). Although Oat1 and Oat3 have significant overlap in the proximal tubule segments (S1-S3), rOat3 has also been shown to be expressed in the thick ascending loop, distal tubule, connecting tubule, and the collecting duct of the outer medulla (31, 32). Different parts of the nephron may thus accumulate substrates or toxins at different rates due to the different localization of transporters, thereby affecting cellular toxicity.
In addition, although adefovir, cidofovir, and tenofovir have been shown clinically to cause renal insufficiency (11), the particular local impact on the kidney may be more discrete than ddC or ddI due to more specific proximal tubule localization of Oat1 (31, 32). In fact, the proximal tubule appears to be the most vulnerable during renal ischemia due to its low glycolytic capacity (reviewed in Refs. 33 and 34-37). Plasma levels of adefovir in patients undergoing treatment (10 mg/day) rarely approach the maximal capacity of OATs. Nevertheless, such levels appear to be associated with nephrotoxicity, further highlighting the need to study the exact limits of OAT-mediated clearance of antivirals.
In general, the affinity of antivirals for mOat1 agreed with those found in previous work using cultured cell lines expressing hOat1 (13, 14, 17), and were consistent at multiple levels of analysis in this work. Furthermore, the similarities in affinity of mouse Oat1 to human Oat1, with respect to these antivirals, suggest mouse models of antiviral excretion may mirror those in humans.
Oat3 and Oat6 had little affinity for this group of (acyclic nucleotide analog) antivirals. Furthermore, a major role for Oat1 in the uptake of ddC and ddI seems less likely. For many compounds, Oat1 and Oat3 had complementary affinities for the antivirals tested (i.e. one with high affinity, one with low affinity). Finally, the recently discovered olfactory mucosa transporter (Oat6) was found to have little ability to interact with this group of antiviral drugs, despite high overall sequence homology. This further isolates mOat6 as a functionally separate homolog, possibly as an odorant transporter as postulated earlier (1).
By utilizing OAT knock-out WEKs for isolating the inhibitory profile of individual OATs in whole organ systems, we were able to dissect the relative importance of Oat1 and Oat3 interactions with antivirals in intact tissue. Whereas it is possible that loss of one OAT may impact interactions between the OATs (e.g. by uncoupling required protein regulation mechanisms), the data are largely consistent with published data in oocytes and cell lines, as well as in in vivo experiments (8, 22, 24). The flexibility to modulate OAT regulation in real time (i.e. increase in transcription or protein-protein regulation) can have a significant impact on how drug efflux is studied. Furthermore, it is worth noting that the fluorescent trackers provide new avenues for investigating the role of OATs in embryonic physiology and cell biology, maturation of the OAT system, as well as differences in the interplay of OATs early in fetal development. For instance, it has been suggested that OATs could require a scaffolding complex for particular functions (38), the function of which could be explored visually using this assay. Furthermore, the fetal capacity for handling toxins and drugs can be studied. It was previously shown that OATs are expressed and up-regulated as early as the onset of kidney development (i.e. rat embryonic day 13) (18), whereas it has been suggested that an OAT system (Oat4) regulates the mother-fetal barrier for certain drugs and other compounds in the placenta (39). In light of the prophylactic use of antiviral drugs in pregnant women (40, 41), this information may prove to be of particular clinical importance.
Ultimately, the data has important implications for systems pharmacology, particularly for antivirals in the kidney. There are multiple polyspecific ion transporters in the kidney, including organic cation transporter, organ anion polypeptide transporters, multi-drug resistance proteins (MDR and MRP), etc. (38, 42), which due to the large number of family members will likely have variation in specificity and affinity for substrates (including fluorescent tracking molecules). For instance, NBD-TMA and ASP+ have been used to track organic cation transport (43-45), whereas rhodamine-123 has been used as an MDR1 and MRP1 substrate (46, 47). Transporter-specific fluorescent isoforms could differentially mark the respective transport phenomenon. Thus, the different emission spectra utilized by confocal or two-photon microscopes could be exploited to understand the interplay of different ion transporters with each other and with different substrates in real-time. As systems pharmacology evolve, it may be possible to clarify the effects of transient disease on separate ion transport systems, their reactions to drug pharmacodynamics, and their interactions at the protein level.
Supplementary Material
Acknowledgments
We gratefully acknowledge the excellent technical assistance of Shammy Closson, as well the helpful discussions with Sun-Young Ahn, Megha Nagle, Satish A. Eraly, Hiro Sakurai, Kevin T. Bush, and Igor Tsigelny.
This work was supported by National Institutes of Health Grants AI057695 and HD40011 (to S. K. N.) and DK61425 (to P. W. S.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1-S3.
Footnotes
The abbreviations used are: OAT, organic anion transporter; PAH, p-aminohippurate; ES, estrone-3-sulfate; WEK, whole embryonic kidney culture; 5-CF, 5-carboxyfluorescein; 6-CF, 6-carboxyfluorescein; 4-CFDM, 4-carboxyfluorescein-dimethylester; CF, carboxyfluorescein; NRTI, nucleotide reverse transcriptase inhibitor; QSAR, quantitative structure activity relationship; HIV, human immunodeficiency virus; AZT, azidothymidine; PSA, polar surface area; RB, rotatable bonds; WT, wild type; TRITC, tetramethyl-rhodamine isothiocyanate.
References
- 1.Kaler, G., Truong, D. M., Sweeney, D. E., Logan, D. W., Nagle, M., Wu, W., Eraly, S. A., and Nigam, S. K. (2006) Biochem. Biophys. Res. Commun. 351 872-876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eraly, S. A., Bush, K. T., Sampogna, R. V., Bhatnagar, V., and Nigam, S. K. (2004) Mol. Pharmacol. 65 479-487 [DOI] [PubMed] [Google Scholar]
- 3.Lopez-Nieto, C. E., You, G., Barros, E. J. G., Beier, D. R., and Nigam, S. K. (1996) J. Am. Soc. Nephrol. 7 1301 [Google Scholar]
- 4.Lopez-Nieto, C. E., You, G., Bush, K. T., Barros, E. J., Beier, D. R., and Nigam, S. K. (1997) J. Biol. Chem. 272 6471-6478 [DOI] [PubMed] [Google Scholar]
- 5.Sweet, D. H., Wolff, N. A., and Pritchard, J. B. (1997) J. Biol. Chem. 272 30088-30095 [DOI] [PubMed] [Google Scholar]
- 6.Brady, K. P., Dushkin, H., Fornzler, D., Koike, T., Magner, F., Her, H., Gullans, S., Segre, G. V., Green, R. M., and Beier, D. R. (1999) Genomics 56 254-261 [DOI] [PubMed] [Google Scholar]
- 7.Monte, J. C., Nagle, M. A., Eraly, S. A., and Nigam, S. K. (2004) Biochem. Biophys. Res. Commun. 323 429-436 [DOI] [PubMed] [Google Scholar]
- 8.Burckhardt, B. C., and Burckhardt, G. (2003) Rev. Physiol. Biochem. Pharmacol. 146 95-158 [DOI] [PubMed] [Google Scholar]
- 9.Sweet, D. H. (2005) Toxicol. Appl. Pharmacol. 204 198-215 [DOI] [PubMed] [Google Scholar]
- 10.Young, B. (2005) AIDS Patient Care STDS 19 286-297 [DOI] [PubMed] [Google Scholar]
- 11.Lewis, W. (2003) Prog. Cardiovasc. Dis. 45 305-318 [DOI] [PubMed] [Google Scholar]
- 12.Anderson, P. L., Kakuda, T. N., and Lichtenstein, K. A. (2004) Clin. Infect. Dis. 38 743-753 [DOI] [PubMed] [Google Scholar]
- 13.Wada, S., Tsuda, M., Sekine, T., Cha, S. H., Kimura, M., Kanai, Y., and Endou, H. (2000) J. Pharmacol. Exp. Ther. 294 844-849 [PubMed] [Google Scholar]
- 14.Cihlar, T., Lin, D. C., Pritchard, J. B., Fuller, M. D., Mendel, D. B., and Sweet, D. H. (1999) Mol. Pharmacol. 56 570-580 [DOI] [PubMed] [Google Scholar]
- 15.Takeda, M., Khamdang, S., Narikawa, S., Kimura, H., Kobayashi, Y., Yamamoto, T., Cha, S. H., Sekine, T., and Endou, H. (2002) J. Pharmacol. Exp. Ther. 300 918-924 [DOI] [PubMed] [Google Scholar]
- 16.Cha, S. H., Sekine, T., Fukushima, J. I., Kanai, Y., Kobayashi, Y., Goya, T., and Endou, H. (2001) Mol. Pharmacol. 59 1277-1286 [DOI] [PubMed] [Google Scholar]
- 17.Uwai, Y., Ida, H., Tsuji, Y., Katsura, T., and Inui, K. (2007) Pharm. Res. (N. Y.) 24 811-815 [DOI] [PubMed] [Google Scholar]
- 18.Sweet, D. H., Eraly, S. A., Vaughn, D. A., Bush, K. T., and Nigam, S. K. (2006) Kidney Int. 69 837-845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cihlar, T., and Ho, E. S. (2000) Anal. Biochem. 283 49-55 [DOI] [PubMed] [Google Scholar]
- 20.De Clerck, L. S., Bridts, C. H., Mertens, A. M., Moens, M. M., and Stevens, W. J. (1994) J. Immunol. Methods 172 115-124 [DOI] [PubMed] [Google Scholar]
- 21.De Clercq, E. (2004) J. Clin. Virol. 30 115-133 [DOI] [PubMed] [Google Scholar]
- 22.Eraly, S. A., Vallon, V., Vaughn, D. A., Gangoiti, J. A., Richter, K., Nagle, M., Monte, J. C., Rieg, T., Truong, D. M., Long, J. M., Barshop, B. A., Kaler, G., and Nigam, S. K. (2006) J. Biol. Chem. 281 5072-5083 [DOI] [PubMed] [Google Scholar]
- 23.Kaler, G., Truong, D. M., Khandelwal, A., Nagle, M., Eraly, S. A., Swaan, P. W., and Nigam, S. K. (2007) J. Biol. Chem. 282 23841-23853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sweet, D. H., Miller, D. S., Pritchard, J. B., Fujiwara, Y., Beier, D. R., and Nigam, S. K. (2002) J. Biol. Chem. 277 26934-26943 [DOI] [PubMed] [Google Scholar]
- 25.Laitinen, L., Virtanen, I., and Saxen, L. (1987) J. Histochem. Cytochem. 35 55-65 [DOI] [PubMed] [Google Scholar]
- 26.Cihlar, T., Ho, E. S., Lin, D. C., and Mulato, A. S. (2001) Nucleosides Nucleotides Nucleic Acids 20 641-648 [DOI] [PubMed] [Google Scholar]
- 27.Vidal, F., Domingo, J. C., Guallar, J., Saumoy, M., Cordobilla, B., Sanchez de la Rosa, R., Giralt, M., Alvarez, M. L., Lopez-Dupla, M., Torres, F., Villarroya, F., Cihlar, T., and Domingo, P. (2006) Antimicrob. Agents Chemother. 50 3824-3832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bongiovanni, M., and Tordato, F. (2006) Curr. Med. Chem. 13 2789-2793 [DOI] [PubMed] [Google Scholar]
- 29.Hitchcock, M. J. (1993) Clin. Infect. Dis. 16 Suppl. 1, S16-S21 [Google Scholar]
- 30.Antonelli, A., Brandi, G., Casabianca, A., and Magnani, M. (1997) Biochim. Biophys. Acta 1358 39-45 [DOI] [PubMed] [Google Scholar]
- 31.Kojima, R., Sekine, T., Kawachi, M., Cha, S. H., Suzuki, Y., and Endou, H. (2002) J. Am. Soc. Nephrol. 13 848-857 [DOI] [PubMed] [Google Scholar]
- 32.Ljubojevic, M., Herak-Kramberger, C. M., Hagos, Y., Bahn, A., Endou, H., Burckhardt, G., and Sabolic, I. (2004) Am. J. Physiol. 287 F124-F138 [DOI] [PubMed] [Google Scholar]
- 33.Eraly, S. A., Blantz, R. C., Bhatnagar, V., and Nigam, S. K. (2003) Curr. Opin. Nephrol. Hypertension 12 551-558 [DOI] [PubMed] [Google Scholar]
- 34.Ross, B. D., Espinal, J., and Silva, P. (1986) Kidney Int. 29 54-67 [DOI] [PubMed] [Google Scholar]
- 35.Bonventre, J. V. (1988) Annu. Rev. Med. 39 531-544 [DOI] [PubMed] [Google Scholar]
- 36.Epstein, F. H., Agmon, Y., and Brezis, M. (1994) Ann. N. Y. Acad. Sci. 718 72-81 [PubMed] [Google Scholar]
- 37.Epstein, F. H. (1997) Kidney Int. 51 381-385 [DOI] [PubMed] [Google Scholar]
- 38.Anzai, N., Jutabha, P., Kanai, Y., and Endou, H. (2005) Curr. Opin. Nephrol. Hypertens. 14 472-479 [DOI] [PubMed] [Google Scholar]
- 39.Cha, S. H., Sekine, T., Kusuhara, H., Yu, E., Kim, J. Y., Kim, D. K., Sugiyama, Y., Kanai, Y., and Endou, H. (2000) J. Biol. Chem. 275 4507-4512 [DOI] [PubMed] [Google Scholar]
- 40.Jacqz-Aigrain, E., and Koren, G. (2005) Semin. Fetal Neonatal. Med. 10 139-147 [DOI] [PubMed] [Google Scholar]
- 41.Pacifici, G. M. (2005) Early Hum. Dev. 81 647-654 [DOI] [PubMed] [Google Scholar]
- 42.Dresser, M. J., Leabman, M. K., and Giacomini, K. M. (2001) J. Pharmacol. Sci. 90 397-421 [DOI] [PubMed] [Google Scholar]
- 43.Miller, D. S. (2004) Adv. Drug Deliv. Rev. 56 1811-1824 [DOI] [PubMed] [Google Scholar]
- 44.Pietruck, F., Horbelt, M., Feldkamp, T., Engeln, K., Herget-Rosenthal, S., Philipp, T., and Kribben, A. (2006) Drug Metab. Dispos. 34 339-342 [DOI] [PubMed] [Google Scholar]
- 45.Bednarczyk, D., Mash, E. A., Aavula, B. R., and Wright, S. H. (2000) Pflugers Arch. 440 184-192 [DOI] [PubMed] [Google Scholar]
- 46.Zubercova, O., and Babusikova, O. (1998) Neoplasma 45 53-59 [PubMed] [Google Scholar]
- 47.Twentyman, P. R., Rhodes, T., and Rayner, S. (1994) Eur. J. Cancer 30A 1360-1369 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.