

Abstract
Response regulators undergo regulated phosphorylation and dephosphorylation at conserved aspartic acid residues in bacterial signal transduction systems. OmpR is a winged helix-turnhelix DNA-binding protein that functions as a global regulator in bacteria and is also important in pathogenesis. A detailed mechanistic picture of how OmpR binds to DNA and activates transcription is lacking. We used NMR spectroscopy to solve the solution structure of the C-terminal domain of OmpR (OmpRC) and to analyze the chemical shift changes that occur upon DNA binding. There is little overlap in the interaction surface with residues of PhoB that were reportedly involved in protein/protein interactions in its head-to-tail dimer. Multiple factors account for the lack of overlap. One is that the spacing between the OmpR half-sites is shorter than observed with PhoB, requiring the arrangement of the two OmpR molecules to be different from that of the PhoB dimer on DNA. A second is the demonstration herein that OmpR can bind to its high affinity site as a monomer. As a result, OmpRC appears to be capable of adopting alternative orientations depending on the precise base composition of the binding site, which also contributes to the lack of overlap. In the presence of DNA, chemical shift changes occur in OmpR in the recognition α-helix 3, the loop between β-strand 4 and α-helix 1, and the loop between β-strands 5 and 6. DNA contact residues are Val203 (T), Arg207 (G), and Arg209 (phosphate backbone). Our results suggest that OmpR binds to DNA as a monomer and then forms a symmetric or asymmetric dimer, depending on the binding site. We propose that during activation OmpR binds to DNA and undergoes a conformational change that promotes phosphorylation of the N-terminal receiver domain, the receiver domains dimerize, and then the second monomer binds to DNA. The flexible linker of OmpR enables the second monomer to bind in multiple orientations (head-to-tail and head-to-head), depending on the specific DNA contacts.
All cells must sense and respond to changes in their environment to survive. In prokaryotes, signal transduction is largely achieved via two-component regulatory systems (for reviews see Ref. 1). In their simplest form, the first component is a sensor kinase, usually a membrane protein that is phosphorylated on a conserved histidine residue in response to an environmental signal. The phosphoryl group is subsequently transferred onto a conserved aspartic acid of the second component, the response regulator. The modified output of the response regulator is most often a change in gene expression, but some effector domains exhibit altered enzymatic activity or protein/protein interactions. Response regulators are grouped into three subfamilies based on the structural similarity of their effector domains: OmpR/PhoB, NtrC/DctD, and NarL/FixJ.
The EnvZ/OmpR two-component system regulates reciprocal expression of the porin genes ompF and ompC in response to changes in the osmolality of the growth medium (2). At low osmolality, OmpF is the major porin in the outer membrane, whereas at high osmolality OmpC is the predominant porin. The two porins do not differ from one another by the size of their pores (3, 4), although OmpC has a slower flow rate (5) because of electrostatic pore potential (3). In recent years, it has become apparent that OmpR is a global regulator, regulating at least 125 genes, including small RNAs, in response to unknown signals (6, 7). Deletion of ompR/envZ had the second most dramatic phenotype compared with deletion of all the other two-component systems in Escherichia coli (4). OmpR also plays an important role in pathogenesis, particularly in Salmonella enterica, Yersinia, and Shigella, where it turns on type III secretion systems and down-regulates flagellar gene expression (8-17). In S. enterica serovar Typhimurium, regulation of virulence gene expression is EnvZ-dependent but not driven by osmolality (18). OmpR directly activates the SsrA/B two-component regulatory system that switches on type III secretion, enabling S. enterica sv. Typhimurium to replicate in macrophages and cause a systemic infection (13, 18). Thus, in a wide variety of organisms, OmpR plays a central regulatory role in controlling expression of both housekeeping and virulence genes.
OmpR is the archetype example from the largest subfamily of response regulators in E. coli. It is a two domain response regulator, composed of an N-terminal receiver or phosphorylation domain (19) and a C-terminal DNA binding domain (20). The two domains are joined by a long, flexible linker that is susceptible to proteolysis (21). Phosphorylation in the N terminus increases the affinity of the C terminus for DNA (22-25). This communication is bi-directional in that DNA binding also stimulates phosphorylation (26) and requires the domains to be linked (27). The dynamic nature of OmpR has prevented the determination of a crystal structure of the full-length protein, and co-crystallization of the C terminus bound to DNA has not been achieved. Thus, although OmpR is an important regulator, many structural and functional features have not yet been resolved. For example, sedimentation velocity and sedimentation equilibrium experiments indicate that from 0 to 38 μm OmpR is 90% monomeric,4 yet it reportedly binds DNA as a dimer (23, 25, 28, 29). Cellular concentrations of OmpR are ∼1-3 μm (30). It is presumed, based on structural homology to other response regulators, that dimerization occurs upon phosphorylation and promotes interaction along its N-terminal α4-β5-α5 face (31). It was suggested that a common mechanism of dimerization is employed by the OmpR subfamily of response regulators, even though substantial functional differences exist among subfamily members. This view has been recently challenged by a crystal structure of the RegX3 response regulator from Mycobacterium tuberculosis, which demonstrated a domain-swap mode of dimerization (32).
In this study, we present the results from our NMR studies of the DNA binding domain of OmpR (OmpRC). Changes in chemical shifts that occurred upon DNA binding enabled us to identify the susceptible residues. Phenotypic analysis further highlighted a role for these residues in activating transcription. Based on these results, we propose that OmpR binds to its high affinity site as a monomer making contacts with T and G bases via Val203 and Arg207 and phosphate backbone contacts via Arg209 in the recognition helix (α-helix 3). A second OmpR molecule binds subsequently. Weak protein/protein interactions in the dimer facilitate interaction with the DNA of the second, lower affinity half-site. The molecular basis for this reduced affinity is now apparent, the result of deviation from the preferred core sequence of GTXTCA (5′-3′ of the template strand, where X is A or T). Our results also account for the structural basis of the reduced affinity of an OmpR2 mutant V203M for ompC (33).
EXPERIMENTAL PROCEDURES
Expression and Purification of Full-length OmpR and OmpRC—Full-length OmpR protein was purified as described previously (34) with the following modifications: OmpR was subcloned into pFR29* and expressed and isolated from strain JMS6210 (35). One liter of cells was grown to an absorbance of 1.0 at A600 and induced for 4 h with 1 mm IPTG.5 The cell pellet was lysed by sonication in TGED, pH 7.6, with the addition of 1 mm 4-(2-aminoethyl)benzenesulfonyl fluoride and 10 μg lysozyme (Sigma). After centrifugation at 18,000 × g for 30 min, the supernatant was recovered, and solid ammonium sulfate was added to 45% saturation at 4 °C. Precipitates formed in 60 min were recovered by centrifugation, dissolved in 10 ml of TGED, pH 7.6, and dialyzed against the same buffer. The dialyzed sample was applied to a HiPrep DEAE-Sepharose column previously equilibrated with TGED, pH 7.6, and proteins were eluted with a linear NaCl gradient from 0 to 400 mm. Fractions positive for OmpR on SDS-PAGE were pooled and applied to a HiPrep SP-Sepharose column previously equilibrated with TGED, pH 7.6, and proteins were eluted with a linear NaCl gradient from 0 to 700 mm. Positive OmpR fractions were pooled and were 90% pure.
OmpRC was purified in the same manner as OmpR but with the following modifications. The pellet from 1 liter of cells was resuspended in 10 ml of TGED, pH 5.7, with 1 mm 4-(2-aminoethyl)benzenesulfonyl fluoride and 3 μg of DNase I (grade II, Roche Applied Science) and gently agitated for 1 h at room temperature. 10 μg of lysozyme was added prior to sonication. After centrifugation at 18,000 × g for 30 min, the supernatant was filtered through a 0.45-μm filter and applied directly to a HiPrep SP-Sepharose column previously equilibrated with TGED, pH 5.7. Proteins were eluted with a linear gradient 0 to 1 m NaCl and resolved as a doublet on 18% SDS-PAGE.
For NMR analysis, uniformly labeled OmpRC was obtained by growing cells in isotopically enriched minimal media containing 0.6% Na2HPO4, 0.3% KH2PO4, 0.15% NaCl, 1 mm MgCl2, 0.1 mm CaCl2, 0.5% basal medium Eagle vitamin solution (Invitrogen). (15NH4)2SO4 (1 g/liter) and unlabeled glucose (4 g/liter) were used for 15N labeling or 1.5 g/liter (15NH4)2SO4 and 2 g/liter of 13C-enriched glucose for double labeling in H2O or D2O.
NMR Spectroscopy and Data Processing—All NMR measurements were performed in either 10% D2O, 90% H2O or 100% D2O at 25 °C on a Bruker DRX 800 instrument. Standard homonuclear two-dimensional and heteronuclear two- and three-dimensional experiments were acquired for the backbone and side chain and NOE constraint assignments and 3J coupling constants (36). Data were processed and analyzed using commercial software SYBYL (Tripos Inc., St. Louis). All 1H dimensions were referenced to internal 2,2-dimethyl-2-silapentane-5-sulfonate, and 13C and 15N were indirectly referenced to 2,2-dimethyl-2-silapentane-5-sulfonate (37).
The assignment of the sequence-specific backbone resonance assignment of 109 amino acid residues of OmpRC was achieved through a combination of standard two- and three-dimensional double and triple resonance NMR experiments (38). More than 70% of all side chain resonances were assigned for OmpRC.
Structural Restraints and Structural Calculations—NMR distance restraints were collected from three different NOESY spectra as follows: three-dimensional NOESY-15N-HSQC (mixing time 120 ms) for amide protons, three-dimensional NOESY-13C-HSQC in D2O (mixing time 100 ms) for aliphatic protons, and two-dimensional 1H-NOESY in D2O (mixing time 100 ms) for aromatic protons. NOE restraints were grouped into four distance ranges: strong, 1.8-2.8 Å; medium, 1.8-4.0 Å; weak, 1.8-5.0 Å; and very weak, 1.8-6.0 Å for backbone-backbone NOE correlations, whereas an additional 0.5 Å was added to the upper bound of the NOE restraints for backbone-side chain and side chain-side chain NOEs (39). Pseudo-atom corrections were applied for distances involving methyl protons, aromatic ring protons, and nonstereospecifically assigned methylene protons (40). Hydrogen bond restraints were employed in areas of regular secondary structures, displaying characteristic NOE cross-peaks. Each deduced hydrogen bond was represented by two distance constraints: 1.7-2.4 Å for HN-O and 2.7-3.4 Å for N-O. Dihedral angle restraints were derived from 3JHNHα coupling constants obtained from a three-dimensional HNHA experiment and were included in the refinement protocol. The ϕ angles were restrained to -60 ± 30° for 3JHNHα < 6.0 Hz and dNN > dαN, -120 ± 50° for 3JHNHα = 8.0-9.0 Hz, and -120 ± 30° for 3JHNHα > 6.0 Hz. Further restraints for ϕ and ψ were added on the basis of the consensus chemical shift index (41) and NOE patterns characteristic of secondary structure as follows: helical residues, -60 ± 40° (ϕ) and -50 ± 50° (Ψ), β-strand residues, -120 ± 40° (ϕ) and -130 ± 50° (Ψ).
Structure calculations were performed with the program DYANA 1.5 (42) using a 40,000-step energy minimization procedure. For the initial rounds of structure calculations, only sequential, intra-residual, medium range NOEs and unambiguous long range NOEs and coupling constants were used. Subsequently, all other long range NOEs and hydrogen bonds were introduced in consecutive steps. 100 structures were calculated in each round, and of these, the 20 structures with the lowest target functions were used to analyze restraint violations and to assign additional NOE restraints for the following round. This process was repeated until more than 90% of the NOE peaks in the spectra had been assigned, and all violations were eliminated. In the final stage, the 20 structures with the lowest target functions were used for structural analyses. All subsequent analyses of the structure and graphic representations of the three-dimensional structures were performed using MOLMOL (43) and PROCHECK-NMR (44).
Construction of an ompC1 Site Mutation by PCR—To construct an ompC1-lacZ fusion, DW245 was digested with NdeI and PstI, and the ompC1-lacZ fragment was subcloned into pAH125 that had been cleaved with same enzymes (45). The plasmid pJR714 was used as a template to make the ompC1 site G to A mutation. The ompC1 region was amplified using OmpC071 (5′-GTCGACAATCTGGTAACTTTTATC-3′) and LacZ075 (5′-GATATCCTGCACCATCGTCTGC-3′) primers and two complementary oligonucleotides containing the desired mutation and then subcloned into the pGEM-T easy vector. The PCR contained 10 ng of double-stranded DNA template pJR714, 10 μm of each primer, 2.5 mm each deoxynucleoside triphosphate, 15 mm MgCl2, and 2.5 units of TaqDNA polymerase (Invitrogen) in 20 μl. This plasmid, pJR731, was cleaved by using SalI and EcoRV and then subcloned into pJR714 after cleavage with the same enzymes. The mutation was sequenced to confirm the presence of the desired mutation. pJR731 was integrated into the chromosomal attB site by direct transformation of E. coli MC4100 hosts (WT and ompR mutant) carrying a helper plasmid as described previously by Haldimann and Wanner (45).
Construction of ompR Mutations by PCR—To create the OmpR mutants, ompR was amplified using OmpR061 and OmpR062 or EnvZ061 primers and then subcloned into the pGEM-T easy vector. Two complementary oligonucleotides containing the desired mutation were used for the PCR. The PCR contained 10 ng of double-stranded DNA template, 10 μm of each primer, 2.5 mm of each dNTP, 15 mm MgCl2, and 2.5 units of Taq DNA polymerase (Invitrogen) in 20 μl. The reaction was amplified by 25 cycles at 95 °C (30 s), 55 °C (30 s), and 72 °C (1 min). The products were used as a template in a second PCR using OmpR061 and OmpR062 primers. The product of the second PCR was subcloned into the pGEM-T easy vector. The plasmids were sequenced to confirm the presence of the desired mutation.
Overexpression and Purification of OmpR Mutants—To overproduce the mutant OmpR proteins, each plasmid was cleaved with NdeI and BamHI and ligated into the expression vector pET15b (Novagen) that had been cleaved with the same enzymes. The ligation mixture was used to transform E. coli strain DH5α, and individual transformants were screened for the presence of the appropriate recombinant plasmid. The resulting plasmids encoded a mutant OmpR with a His6 tag at the N terminus. Each plasmid was transformed into E. coli strain BL21(DE3). Cells were incubated with moderate shaking at 37 °C until the absorbance at 600 nm reached a value of 0.6. IPTG was added to a final concentration of 1 mm, and incubations were continued for 3 h. The cells were collected by centrifugation and resuspended in lysis buffer (50 mm NaH2PO4, 300 mm NaCl, 10 mm imidazole). Lysozyme and protease inhibitor (4-(2-aminoethyl) benzenesulfonyl fluoride-hydrochloride) were added to a final concentration of 1 mg/ml and 1 mm, respectively. The mixture was incubated on ice for 30 min. Samples were frozen at -70 °C and thawed and sonicated six times, each for 30 s. The cell extract was clarified by centrifugation for 30 min at 12,000 rpm at 4 °C. The proteins were purified by affinity chromatography according to the manufacturer's procedure (Qiagen, Valencia, CA).
β-Galactosidase Assays—All of the OmpR mutants were subcloned into pBluescript KS vector using XbaI and BamHI. The resulting plasmids were cleaved with XbaI and HindIII and ligated into the pFR29* that had been cleaved with the same enzymes. pFR29* is a derivative of pFR29 from which the envZ gene has been excised (46). The plasmids were used to transform strains MH225, MH513, MH225.101, and MH513.101. MH225 contains a chromosomal ompC-lacZ fusion and MH513 contains a chromosomal ompF-lacZ fusion (47). The .101 strains contain a small in-frame deletion in the ompR gene (48). An overnight culture of bacteria was diluted 1:100 and grown to mid-log phase in minimal A medium with 15% sucrose (ompC-lacZ) or without sucrose (ompF-lacZ). IPTG was then added to a final concentration of 1 mm, and cells were inoculated for 2 h. Expression of mutant OmpR proteins was confirmed by a Western blot. Transcription of the ompF-lacZ and ompC-lacZ fusions was monitored by β-galactosidase assays as described previously by Miller (49). All β-galactosidase measurements were performed in triplicate with at least two independent cultures.
OmpR Phosphorylation Assays—5 μm OmpR or mutant OmpR protein was incubated with 50 mm Tris-HCl, pH 7.6, 50 mm KCl, 20 mm MgCl2 in 100 μl. Phosphorylation was initiated by adding 15 mm of ammonium hydrogen phosphoramidate. The reaction mixture was incubated for 1 h at room temperature and stopped by addition of 20 mm EDTA. Phosphorylation of OmpR and OmpR mutant proteins was determined by separation on a C4 column using reversed phase HPLC as described (24).
DNA Binding—Electrophoretic mobility shift assays were employed for measurements of DNA binding. The 219-bp upstream region of ompF, extending from residues +71 to -148 (containing OmpR-binding sites F1, F2, and F3), was amplified by PCR using [γ- 32P]ATP-labeled OmpF051-1 and unlabeled OmpF052-1 as the primers (Table 2). For ompC, the 196-bp upstream region of ompC (+40 to -156 containing OmpR-binding sites C1, C2, and C3) was amplified by PCR using [γ-32P]ATP-labeled OmpC052 and unlabeled OmpC051 as the primers (Table 2). The labeled 219- and 196-bp DNA (∼1 × 103 cpm) fragments were incubated with various concentrations of phosphorylated mutant or wild type OmpR protein for 20 min at 30 °C in a 20-μl reaction mixture containing 1× binding buffer (4 mm Tris-HCl, pH 7.4, 20 mm KCl, 2 mm EDTA, 1 mm dithiothreitol) and 1 μg of poly(dI-dC)). Following the incubation, the samples were separated by electrophoresis on a 5% nondenaturing polyacrylamide gel and exposed to film.
TABLE 2.
Oligonucleotides used in this study
Oligonucleotides used in this study. The name of the oligonucleotide is listed in the left column, in the middle column is the sequence (5′-3′) of the non-template strand. Bases that were added to promote double strand formation are outside of the brackets. The bases of the high affinity C1b site are enclosed in parentheses and reversed in the C1b® oligonucleotide. The core binding sites are underlined. The right column lists the application of the oligonucleotide. The bases that were not complementary to the corresponding genes are in bold.
| Oligonucleotide | Oligonucleotide sequence (5′ → 3′) | Use |
|---|---|---|
| SsrB | CATCGATTAATCTGACTG | NMR |
| C1-half (C1b) | AC (ATTTTGAAACATCTA) T | NMR |
| C1a-C1b | AC (TTTACATTTT (GAAACATCTA)) T | NMR |
| C1a-C1b® | AC (TTTACATTTT (TAGATGTTTC)) T | NMR |
| OmpC051 | GCTTGATGTTAGGTGCTTATTTCG | Gel mobility shift assay |
| OmpC052 | GCCACTGCATACTGATTAACCCTC | Gel mobility shift assay |
| OmpF051-1 | CATCACGTCTCTATGGAAATATG | Gel mobility shift assay |
| OmpF052-1 | CTGCCGTCAATAAGTTCTGTC | Gel mobility shift assay |
| OmpR061 | CATATGCAAGAGAACTACAAGATTC | OmpR mutant construction |
| OmpR062 | GGATCCTCATGCTTTAGAGCCGTC | OmpR mutant construction |
| EnvZ061 | CCATATGGTTAAAGGCACGGG | OmpR mutant construction |
| 162-1 | GATGCCGCTCTGTAGCGGTGAG | OmpR mutant construction |
| 162-2 | CTCACCGCTACAGAGCGGCATC | OmpR mutant construction |
| 163-1 | CCGCTCACCTGTGGTGAGTTTG | OmpR mutant construction |
| 163-2 | CAAACTCACCACAGGTGAGCGG | OmpR mutant construction |
| 164-1 | CTCACCAGCTGTGAGTTTGCG | OmpR mutant construction |
| 164-2 | CGCAAACTCACAGCTGGTGAG | OmpR mutant construction |
| 173-1 | GAAGGCACTGTGTAGCCATCCG | OmpR mutant construction |
| 173-2 | CGGATGGCTACACAGTGCCTTC | OmpR mutant construction |
| 176-1 | GTCAGCCATTGTCGTGAGCC | OmpR mutant construction |
| 176-2 | GGCTCACGACAATGGCTGAC | OmpR mutant construction |
| 183-1 | GCTCTCCCGCTGTAAGCTGATG | OmpR mutant construction |
| 183-2 | CATCAGCTTACAGCGGGAGAGC | OmpR mutant construction |
| 184-1 | CTCCCGCGATTGTCTGATGAAC | OmpR mutant construction |
| 184-2 | GTTCATCAGACAATCGCGGGAG | OmpR mutant construction |
| 191-1 | CTTGCCCGTTGTCGTGAATATTC | OmpR mutant construction |
| 191-2 | GAATATTCACGACAACGGGCAAG | OmpR mutant construction |
| 212-1 | GCCGCATGTGTGAAGAAGATC | OmpR mutant construction |
| 212-2 | GATCTTCTTCACACATGCGGC | OmpR mutant construction |
| 215-1 | GTGGAAGAATGTCCAGCGCATC | OmpR mutant construction |
| 215-2 | GATGCGCTGGACATTCTTCCAC | OmpR mutant construction |
| 224-1 | CGCGTTACATTCAGTGTGTCTGGGGTC | OmpR mutant construction |
| 224-2 | GACCCCAGACACACTGAATGTAACGCG | OmpR mutant construction |
| 225-1 | GTTACATTCAGACCTGCTGGGGTCTG | OmpR mutant construction |
| 225-2 | CAGACCCCAGCAGGTCTGAATGTAAC | OmpR mutant construction |
| 226-1 | CAGACCGTCTGTGGTCTGGGC | OmpR mutant construction |
| 226-2 | GCCCAGACCACAGACGGTCTG | OmpR mutant construction |
| 232-1 | GGCTACGTCTGTGTACCGGACG | OmpR mutant construction |
| 232-2 | CGTCCGGTACACAGACGTAGCC | OmpR mutant construction |
| R207A-F | CAGATTTCGGCTCTGCGCCGC | OmpR mutant construction |
| R207A-R | GCGGCGCAGAGCCGAAATCTG | OmpR mutant construction |
| R209A-F | CACCATGCGAGCCAGACGCG | OmpR mutant construction |
| R209A-R | CGCGTCTGGCTCGCATGGTG | OmpR mutant construction |
RESULTS
Resonance Assignment and the Structure of OmpRC—A 1H-15N HSQC spectrum of 15N-labeled OmpRC with corresponding residue assignments indicated is shown in Fig. 1. In this spectrum, 86 expected correlation peaks for the backbone amide resonances were assigned. The spectrum also shows side chain NH2 resonance peaks from asparagine and glutamine side chains whose resonances were not included in the structural calculation. The pattern of short and medium range NOEs and 3JHNHα coupling constants served to identify the secondary structural elements of OmpRC (Fig. 2). The pattern of dNN(i, i + 2), dαN(i, i + 3) and dαN(i, i + 4) NOE connectivities with 3JHNHα coupling constants smaller than 6 Hz indicated the presence of three α-helices (Ser163-Ser174, α1; Arg182-Ala189, α2; and Asp202-Val212, α3). Characteristic long range backbone NOE connectivities (i-j >4) and large coupling constants (>9.0 Hz) obtained from HNHA experiments were used to define the β-strands. Based on this standard, six β-strands were identified (Ile138-Ala139, β1; Phe143-Leu145, β2; Glu151-Arg154, β3; Glu157-Pro160, β4; Gln223-Thr224, β5; and Tyr230-Val 231, β6). These six β-strands form an N-terminal four-stranded β-sheet (β1 to β4), and a C-terminal anti-parallel β-hairpin (β5 and β6).
FIGURE 1.

Two-dimensional 1H-15N HSQC spectrum at 800 MHz of the OmpR C terminus (OmpRC) obtained at 25 °C and pH 6.5. The backbone amide resonances are labeled with the one-letter amino acid code and the residue number (based on the full-length protein). The peaks connected by a solid line are from the NH2 groups of side chains. W226(S) refers to the side chain of Trp226 (note the different location from the backbone amide resonance).
FIGURE 2.

Solution structure of OmpRC. A, stereo superposition of 20 selected conformers with the lowest target functions from the final DYANA calculations. The secondary structural elements are labeled β1-6 and α1-3. B, ribbon diagram of the minimized mean structure is shown in one orientation (left) and rotated 90o (right); C, contact surface with the presentation of the electrostatic polarization is shown (blue, positive; red, negative), where the orientation in A is the same as the left panels of B and C and the right panel is rotated 180o. The secondary structure elements are indicated in A and B. The figures were created using MolMol.
After resonance assignment, the three-dimensional structure was calculated using the program DYANA. A total of 1346 inter-proton restraints and 45 dihedral angle constraints were used in the final round of the structural calculation. Several N-terminal residues were excluded in the structural calculation because of their disordered nature. At the final stage, 100 conformers were calculated, of these, 20 DYANA conformers with the lowest target function were selected to represent the structure (Fig. 2A). The coordinates have been deposited in the RCSB Protein Data Bank with the accession code 2JBP. For the defined region of OmpRC(Val137-Val233), the r.m.s.d. of the mean structure was 0.87 ± 0.19 Å for backbone atoms and 1.61 ± 0.25 Å for all nonhydrogen atoms. In the calculated structures, 63.3% of the residues were in the most favored region on a Ramachandran plot using the program PROCHECK-NMR (44), 30.6% were in the additionally allowed region; 4.6% were in the generously allowed region, and 1.5% were in the disallowed region. The structural statistics are summarized in Table 1.
TABLE 1.
Data collection and refinement statistics
Structural statistics for the best 20 DYANA OmpRC conformers are shown.
| NMR-derived restraints | |
| Total interproton restraints | 1349 |
| Intraresidue (|i - j| = 0) | 218 |
| Sequential (|i - j| = 1) | 443 |
| Medium range (1 < |i - j| < 5) | 286 |
| Long range (|i - j| > 4) | 402 |
| Hydrogen bonds | 48 |
| Dihedral angles (ϕ, ψ) | 45 |
| Residual violationsa | |
| DYANA target function (Å) | 0.37 ± 0.05 |
| Upper limit | |
| Sum (Å) | 2.2 ± 0.3 |
| Maximum (Å) | 0.19 ± 0.02 |
| van der Waals | |
| Sum (Å) | 1.9 ± 0.2 |
| Maximum (Å) | 0.12 ± 0.02 |
| Average r.m.s.d. to mean structure (Å)b | |
| Backbone atoms N, Cα, C′ (Å) | 0.87 ± 0.19 |
| All heavy atoms (Å) | 1.61 ± 0.25 |
| Ramachandran plot (% residues) | |
| Residues in most favored regions | 63.3 |
| Residues in additional allowed regions | 30.6 |
| Residues in generously allowed regions | 4.6 |
| Residues in disallowed regions | 1.5 |
Under Residual violations, the values are means ± S.D.
Residues are in secondary elements.
Structural Analysis of OmpRC—The structure of OmpRC consists of an N-terminal four-stranded antiparallel β-sheet, a bundle of three α-helices packed against two antiparallel β-sheets, and a C-terminal hairpin (see the ribbon diagram in Fig. 2B). The antiparallel β-hairpin is parallel to α-helix 3, which positions two DNA contact loops on the same side of the DNA contact α-helix 3 (see below). The topology of OmpRC is β1-β2-β3-β4-α1-α2-α3-β5-β6. Side chains contributed by each of the secondary structure elements form the hydrophobic core of OmpRC. An x-ray structure of OmpRC was determined previously (50, 51). The averaged structure from the current NMR study aligns well with the x-ray structure of OmpRC with an r.m.s.d. value of 0.97 Å for the backbone atoms of well structured regions. The major differences between the x-ray and NMR structures reside in the unstructured loop regions. These sequences tend to be flexible and can adjust their conformations according to different environments (see below). The electrostatic potential surface of the protein (Fig. 2C) displays a continuous positively charged surface that is expected to promote DNA binding.
OmpRC/DNA Interaction—Despite the wealth of information regarding the function of OmpR, a detailed picture of how OmpR interacts with DNA is sorely lacking. The structure of the DNA binding domain was determined over 10 years ago, yet no co-crystal structures of OmpR bound to DNA exist. The OmpR homologue PhoB binds to DNA as a head-to-tail dimer (52). One distinct difference between the OmpR and PhoB systems is that PhoB-binding sites are well conserved, whereas OmpR sites are not. A consensus PhoB site contains two GTCG repeats separated by 10 bp (53). OmpR DNA-binding sites previously identified contain a GTXTCA motif (5′-3′ on the template strand). Because this motif is not conserved in both half-sites, we considered the possibility that OmpR might bind to DNA as a monomer at the higher affinity half-site. We tested interactions between OmpRC and two DNA sequences. First we examined whether or not OmpR could interact with a DNA-binding site lacking the GTXTCA motif (listed in Table 2). This site was derived from an AT-rich binding site to which the SsrB response regulator in the NarL subfamily binds (54). An NMR sample was prepared containing 15N-enriched OmpRC and a 17-bp SsrB site in a 1:2 molar ratio of protein to DNA. The presence of this DNA-binding site perturbs the OmpR structure, as shown in Fig. 3A. The perturbed resonances include Thr162, Ser163, and Gly164 (located in the loop between β4 and α1 and the N-terminal end of α-helix 1), Phe166, Lys170, and Val173 in α1, Leu180 in the α1-α2 loop, Gly191 (α2-α3 loop), Gln223 and Thr224 in β5, Val225, the Trp226 side chain, Gly227 and Gly229 in the loop between β-strands 5 and 6, and residues Ser200, Arg207, and Arg210 in α-helix 3. The changes in chemical shift patterns support a DNA contact surface that includes α-helix 3, the loop between β4 and α1, and the loop between β5 and β6 (summarized in Fig. 4A; see also supplemental Table 1). This surface is similar to that of the OmpR homologue PhoBc in the DNA-bound complex (52). Although the SsrB DNA is not an OmpR-binding site, our results indicate that the DNA binding surface of OmpR undergoes chemical shift changes in the presence of AT-rich DNA.
FIGURE 3.

Interactions between the OmpR DNA binding domain and two DNA sites.
A, OmpRC in the presence of an SsrB DNA-binding site
(green, 1:2 ratio of OmpRC to DNA). The label identifying
Gln223 is above the black spectrum; it is
completely shifted in the presence of SsrB DNA. B, OmpRC
in the presence of the OmpR DNA half-site C1b (red, 1:2 ratio of
OmpRC to DNA). The HSQC spectrum in black is the spectrum
of the DNA-free OmpRC. The spectra were acquired at 35 °C, and
in 25 mm phosphate buffer, pH 6.5. The data indicate that the
chemical shift perturbation is greater when the C1 site is used, and that the
monomeric OmpR DNA binding domain can form a stable complex with a strong OmpR
half-site. The perturbed residues that are well resolved in each spectrum are
labeled, and the arrows trace the positions of the perturbed
residues. C, summary of the chemical shift changes observed with
OmpRC in the presence of a C1b site. The vertical axis
corresponds to average chemical variations
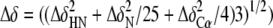 measured on the spectra of the DNA-free OmpRC and the
OmpRC-C1b DNA complex. The amino acid residue number is listed on
the x axis and the secondary structure element is at the top
of the graph, where S signifies a β-strand and α is
α-helix. D, OmpRC in the presence of the DNA
half-site C1a overlaid with the spectrum of OmpRC in the presence
of the DNA half-site C1b at a 1:2 ratio of OmpRC to DNA
(red, C1b site; black, C1a site).
measured on the spectra of the DNA-free OmpRC and the
OmpRC-C1b DNA complex. The amino acid residue number is listed on
the x axis and the secondary structure element is at the top
of the graph, where S signifies a β-strand and α is
α-helix. D, OmpRC in the presence of the DNA
half-site C1a overlaid with the spectrum of OmpRC in the presence
of the DNA half-site C1b at a 1:2 ratio of OmpRC to DNA
(red, C1b site; black, C1a site).
FIGURE 4.

A, surfaces of OmpRC that are affected by DNA binding. A ribbon diagram of the OmpR DNA binding domain is shown. The residues whose resonances are perturbed when the full-length C1 DNA site is added are highlighted in red, and individual amino acids are labeled. These residues define a DNA contact surface for OmpR. See text for further details. B, model of OmpRC bound to C1 DNA. In this model, we assume that the conserved DNA contact residues identified in the PhoB-DNA complex are the same in the OmpR-DNA complex and that the DNA is in the ideal B-form. The complexed structure is calculated from the program DYANA, using the distant constraints between the conserved DNA bases and the conserved DNA contact residues derived from the PhoB-DNA complex, the distant constraints derived from the DNA-free OmpR structure and B-form DNA. A family of 50 structures was calculated, and the one with the lowest DYANA energy is illustrated here. The residues in magenta (Val203, Arg207, and Trp226) are the DNA contact residues from helix 3 of OmpR (Fig. 3). The binding sites are listed below the figure, and DNA contact residues are indicated in red.
The OmpR-binding sites identified previously by DNase I footprinting contain ∼18 bp (23, 25). In the structure of the PhoBc-DNA complex, PhoB forms a dimer on a 20-bp DNA-binding site. It has been proposed that OmpR also binds as a dimer, and each OmpR molecule interacts with a half-site, although previous attempts to measure OmpR binding to a half-site were not successful (25, 26, 29). To address this issue, we examined the interaction between OmpRC and a single OmpR half-site (Fig. 3B). A titration experiment was performed in which C1b DNA was added at ratios of 1:0.5, 1:1, and 1:2 OmpR to DNA. At a ratio of 1:0.5, a mixture of resonances exchange between DNA-bound and DNA-free states. At the OmpR DNA ratio of 1:0.5, two sets of resonances are observed as follows: one representing the resonances from the DNA-free OmpRc and the other representing the resonances from the DNA-bound OmpR. After a molar ratio of 1:1 OmpR to DNA was reached, the characteristic resonances observed in the DNA-free OmpR sample were completely shifted to the DNA-bound positions. The values of the perturbed chemical shifts did not depend on the protein to DNA ratio, and the exchange between the DNA-free OmpRc and the DNA-bound OmpRc under NMR conditions is slow, because no obvious line broadening is observed for the resonance peaks when the OmpR to DNA ratio is 1:0.5. On the basis of the NMR titration experiments, preliminary three-dimensional HNCA and NOESY HSQC experiments, the complex is stably formed and has a half-life of at least several hundred milliseconds as required by our NOESY mixing time (140 ms). A much more extensive perturbation pattern of OmpR resonances was observed for the OmpR C1b half-site DNA interaction than for the OmpR nonspecific DNA interaction, indicating that the two modes of binding are distinct. The residues that were most severely affected include the following: Ile138 (β1), Lys142 in the turn connecting β1 and β2; Thr162, Ser163, and Gly164 in the loop between β4 and α1; Phe153, Glu155, and Phe166 in β3; Lys170, Ala171, and Val173 in α1; Asp183 (α2), Gly191 (α2-α3 loop); Ser200, Arg207, Arg209, Arg210, and Val212 in α3; Asp215 (loop after α3); and Gln223, Thr224, Val225, the Trp226 side chain, Gly227, and Gly229 in the loop between β5 and β6. Thus, the NMR perturbations firmly establish that a surface, including α-helix 3 of OmpRC, is the DNA contact surface (see Fig. 4). This surface is identical to the DNA contact surface identified in the PhoB-DNA complex. When OmpRC binds to DNA, the chemical shift perturbations to residues are caused by changes in chemical environments, such as close proximity to DNA bases, or structural rearrangements, or a combination of both events. In the HSQC spectrum of the OmpR-C1b-DNA complex, each perturbed resonance is in a well separated region and can be traced to an easily identified single position (see for example, residues Gly164 and Thr224 that are perturbed in both complexes; shown in Fig. 3B, highlighted with arrows). A summary of the chemical shift changes is shown in Fig. 3C. Thus, the HSQC spectrum reveals that OmpRC bound to a C1b DNA half-site resides in a chemically homogeneous environment, indicating that it is structurally homogeneous. Because the C1b DNA half-site contains the OmpR DNA-binding motif positioned in the center and lacks symmetry, the data also indicate that OmpR binds to this site as a monomer. This result is in contrast to previous studies that claimed that two OmpR∼P molecules are necessary for binding (29). This experiment also indicates that the OmpRC interaction with its half-site is substantial enough to produce chemical shift changes. Such low affinity binding to a C1b half-site (Kd ≥ 1.5 μm)6 was not apparent in previous studies in which the presence of a C1 half-site did not stimulate phosphorylation (26), or in mobility shift assays (25, 29), indicating the higher sensitivity of NMR to detect protein conformational changes. A similar result was obtained with the C1a half-site (compared with C1b in Fig. 3D), indicating that OmpRC can bind to C1a without first having to bind to C1b. The HSQC spectrum indicates that a similar binding mode is employed, but an uneven signal pattern suggests a more dynamic nature of the complex, such as a faster dissociation of the OmpRC-C1a complex.
OmpRC DNA Contact Residues—The DNA contact residues are shown in Fig. 4. Our NMR results suggest that the DNA contact surface of OmpR is similar to that of PhoB. Thus, we assume that the α-helix 3 of OmpR is the major groove DNA contact surface. α-Helix 3 of OmpR is highly homologous to that of PhoB, except that its N terminus is unwound, increasing the length of the loop between α-helices 2 and 3 compared with PhoB. Based on the structure of the PhoB-DNA complex, we propose that Arg207 contacts a highly conserved guanine base on the template strand in the core sequence GTXTCA. Val203 contacts the conserved thymine adjacent (3′) to the guanine in the template strand and Arg209 makes contacts with the phosphate backbone. The C1b site promotes higher affinity binding, one explanation is that the base adjacent to the thymine (X in the core sequence) is also a thymine and enables Val203 to contact the methyl group through van der Waals contacts (Fig. 4B). In the C1a half-site, this base is an adenine, and clearly the methyl-methyl contacts of Val203 to thymine are preferred. On the opposite strand, the conserved adenine and cytosine bases were found to be important for OmpR/DNA interaction in an in vivo screen (55). In addition, the base present four nucleotides 3′ from the primary guanine contact with Arg207 (in the template strand) is a cytosine in C1b, whereas it is an adenine in C1a. This GC pair may provide a better contact point for the adjacent aspartate 202. These key differences likely account for the reduced affinity of OmpRC for C1a compared with C1b (see “Discussion”).
Mutation of the Core G Eliminates OmpR Binding at ompC1b—To test the prediction that Arg207 contacts the highly conserved guanine base on the template strand in the core sequence GTX- TCA, we mutated it to adenine and measured the β-galactosidase activity of a substituted chromosomal ompC1b-lacZ fusion (shown in Fig. 5). OmpR activated the wild type fusion, exhibiting a high level of β-galactosidase activity (>4,500 units), whereas only 10 units of activity were observed with the G-substituted fusion. The low level of activation by wild type OmpR of the G-substituted fusion was identical to the very low activity observed in an ompR null strain (Fig. 5, 2nd and 4th columns). This result highly suggests that Arg207 makes a direct contact with the G base because mutation of this base completely eliminated activity. Furthermore, the opposite experiment, i.e. substitution of Arg207 with alanine, also eliminated activity (Fig. 6).
FIGURE 5.

Mutation of a key guanine base in the OmpR-binding site eliminates transcription of ompC. A C1b-binding site was placed just upstream of the ompC promoter and fused to the lacZ gene, then integrated on the chromosome at λ att in an ompR null strain (see “Experimental Procedures” for details). Expression of OmpR stimulated activity of the wild type-binding site (1st column), and in the absence of OmpR, there was no activity (2nd column). Substitution of the guanine in the binding site completely eliminated the ability of OmpR to bind to this site and activate transcription (3rd column). The error bars represent the standard deviation, n = 3.
FIGURE 6.

β-Galactosidase assays of alanine-substituted OmpR mutants. Left panel, the results from an ompF-lacZ fusion strain are shown; right panel shows the results from an ompC-lacZ fusion strain. The 1st column is the activity in the presence of wild type OmpR; the 2nd column is an ompR null strain. The individual mutants are listed below the columns.
Substitution of Proposed DNA Contact Residues Eliminates ompF and ompC Activation—Because recognition helix residues Arg207 and Arg209 were predicted to be base contacts, we constructed alanine substitutions by PCR and examined their effect on ompF-lacZ and ompC-lacZ transcription (Fig. 6). Alanine substitution reduced activity essentially to the level of the ompR null strain (Fig. 6, compare 3rd and 4th columns with 2nd column). Because substitution of Arg207 with alanine eliminated activity, this result suggests that Arg207 makes a direct contact with the guanine base, because mutation of this base eliminated activity (Fig. 5).
OmpRC Can Bind to DNA in a Reverse Orientation—We next examined the binding of OmpRC to a full-length binding site (Fig. 7A, red). For these experiments, OmpR-binding sites were organized in tandem along the DNA, and the pattern of chemical shift changes was similar to what we observed with a C1 half-site (Fig. 7A, black). The molecular weight of the dimeric OmpRC-DNA complex increased to about 50 kDa, reducing the resonance intensities of the complexes (even with the partially deuterated OmpR DNA binding domain used for complex formation). The intensities of several residues were influenced specifically by tandem DNA and exhibited a dramatic reduction in intensity, including Lys142, Leu147, Thr149, Gly191, Val212, and Thr224 (Fig. 7A, red). In this experiment, we employed an OmpR to DNA ratio of 2:1. Because only one set of resonance peaks is observed for the OmpRC-DNA complex, OmpRC dimers must be formed on the full-length C1 site. Otherwise, resonance peaks characteristic of the unbound protein would be observed. Furthermore, the resonance patterns of the OmpR dimer are nearly identical to the OmpR monomer bound to the C1b site (Fig. 3B). The data indicate that OmpRC employs similar DNA recognition schemes in the dimeric OmpR-DNA complexes and the monomeric OmpR-C1b or OmpR-C1a complexes. Thus, protein/protein interactions between two OmpRC molecules have little if any effect on the OmpR/DNA interaction.
FIGURE 7.

Detection of potential dimerization surfaces of the OmpR DNA binding domain. A, HSQC spectrum is acquired with the dimeric OmpRC-DNA complex, in which the native C1a-C1b DNA-binding site is in a sequential, tandem arrangement supporting a head-to-tail dimer orientation (red). It is overlaid with the HSQC spectrum (black) acquired from the monomeric OmpRC-C1b complex. See Table 2 for sequences. B, HSQC in red is the same as in A and is overlaid with an HSQC spectrum (black) acquired from the dimeric OmpRC-DNA complex in which the C1a-C1b site is in an inverted tandem arrangement (C1a-C1b®) and supports symmetric or head-to-head orientation of OmpRC on DNA. The resonances from the symmetric OmpR dimer (black) and the anti-symmetric dimer (red) overlap well with each other and with the resonances from the monomeric OmpRC-DNA complex. The average line widths are similar.
In the PhoB-DNA complex, the PhoB/PhoB interaction is limited to charged interactions between two protein molecules (52); the interactions between two OmpR DNA binding domains are also weak. Because we only see one set of resonances from the two OmpR molecules bound to DNA, the data indicate that both OmpR protomers bind to DNA with similar DNA contacts, and there is no further structural rearrangement compared to when OmpR binds to a C1 half-site as a monomer. This result raises the question as to whether the head-to-tail arrangement of OmpR bound to DNA is really necessary.
Because previous studies of cysteine-substituted cross-linked OmpR suggested that OmpR could adopt an alternative, symmetrical orientation (56), we examined whether native OmpRC could bind to DNA in a reverse orientation. An oligonucleotide was engineered to contain OmpR-binding sites in which the orientation of the C1b half-site was reversed. We then examined the chemical shift perturbations that occurred in the presence of this DNA (referred to as C1a-C1b®). The pattern of chemical shift changes was slightly different in the two orientations (see for example V173 in Fig. 7, A and B, and G164 in Fig. 7B). In the experiment shown, the molar ratio of OmpRC to DNA is 2:1; at this ratio, the conformation of OmpRC is completely shifted to the DNA-bound state. It is evident that OmpR is bound to both half-sites, because the chemical shift changes are different from those observed in the presence of either the full-length or the single half-site. These data indicate that the OmpR DNA binding domain can bind to its DNA sites either as a symmetric dimer or an asymmetric dimer, and the orientation of the dimer depends on the orientation of the two single C1 half-sites (see “Discussion”). Our data further show that the resonance patterns of the two dimeric OmpRC-DNA complexes are almost identical. The data suggest that there are no further structural rearrangements when OmpRC binds to either of the long DNA sites, and therefore no strong protein/protein interactions occur between the two OmpRC molecules in either of the dimeric OmpR-DNA complexes.
Substitutions in DNA-sensitive Residues of OmpR Result in Altered Phenotypes—Many OmpR residues that underwent chemical shift changes in the presence of DNA had not been previously isolated as DNA binding mutants in prior mutagenesis screens (see supplemental Table 1). To examine the effect of substitutions at these positions, we replaced the native amino acid with a cysteine residue by PCR. The mutants were expressed in an E. coli host lacking ompR and containing either an ompF-lacZ or an ompC-lacZ transcriptional fusion. The results of β-galactosidase assays with the OmpR mutants compared with the wild type protein are shown in Fig. 8. As can be seen in the figure, many of the cysteine substitutions retained activity (e.g. G164C, V173C, P176C, M186C, V212C, D215C, and V225C). Surprisingly, substitutions T224C and W226C were completely unable to activate transcription, whereas the substituted residue inbetween (V225C) was as active as the wild type OmpR. To examine these extreme phenotypes in more detail, we purified the mutant proteins to compare their phosphorylation and DNA binding properties.
FIGURE 8.

β-Galactosidase assays of cysteine-substituted OmpR mutants. Left panel, the results from an ompF-lacZ fusion strain are shown; right panel shows the results from an ompC-lacZ fusion strain. The 1st column is the activity in the presence of wild type OmpR, the second is an ompR null strain. The individual mutants are listed below the columns.
DNA Binding Assays—To distinguish between a DNA binding mutant and a transcriptional activation (“positive control”) mutant, we measured DNA binding directly using an electrophoretic mobility shift assay (Fig. 9). Addition of the wild type OmpR protein to a labeled ompF or ompC promoter fragment resulted in the appearance of a shifted complex. Neither the T224C nor the W226C mutant was capable of binding to the DNA. In contrast, the V225C mutant was capable of DNA binding, but bound with lower affinity (Fig. 9, compare lanes 3 and 4 of WT and V225C). Thus, the experiment indicates that the cysteine substitutions in the wing were unable to activate the porin genes because they failed to bind DNA, not because they could not interact with RNA polymerase. Substitution of Trp226 with alanine or arginine was equally defective (data not shown). Interestingly, the V225C substitution had a lower affinity for DNA compared with the wild type protein, but it was still able to activate ompF and ompC transcription to levels similar to wild type (Fig. 8). The lower affinity for DNA was most likely the result of its impaired phosphorylation (see below).
FIGURE 9.

Electrophoretic mobility shift assays of OmpR and OmpR mutants binding to the ompF and ompC regulatory region. A 219-bp ompF and a 196-bp ompC promoter fragment of the upstream, untranslated region were radioactively labeled with [γ-32P]ATP and then used as a DNA probe. The radiolabeled fragments were mixed with increasing amounts of phosphorylated OmpR or mutant OmpR∼P. Lanes 1-4 correspond to 0, 50, 100, and 200 nm of phosphorylated wild type OmpR or the mutant OmpR as indicated above the lanes, with 0.7 nm of an ompF or ompC DNA fragment used for each lane in the binding reaction.
Phosphorylation Properties of the Wing Mutants—OmpR is phosphorylated by its cognate kinase EnvZ (57) or by small molecule phosphodonors such as acetyl phosphate and phosphoramidate (21). In either case, phosphorylation occurs at aspartate 55 (24, 58). Recently, several OmpR mutants have been characterized that highlight the inter-domain communication between receiver and effector domains. For example, a substitution in the recognition helix exhibits altered DNA binding behavior and phosphorylation properties, whereas a substitution in the phosphorylation site affects both phosphorylation and DNA binding (33, 59). Phosphorylation of OmpR is easily determined by its altered mobility on a C4 column using reversed phase HPLC (24). It was therefore of interest to compare the phosphorylation properties of the wing mutants with the wild type protein (Table 3). Compared with wild type OmpR, the mutants were severely deficient in phosphorylation. T224C and W226C were phosphorylated to 10 and 18% compared with the wild type protein (wild type = 100%). Although the V225C mutant exhibited wild type levels of ompF and ompC expression (Fig. 8), it was only 40% phosphorylated compared with wild type OmpR. Presumably, this lower level of phosphorylation accounts for its reduced affinity for ompF and ompC DNA observed in the mobility shift assay shown in Fig. 9 but is sufficient to activate transcription.
TABLE 3.
Phosphorylation of OmpR and OmpR mutant proteins
The amino acid substitution is listed in the 1st column. The percent of the protein that was phosphorylated by incubation with acetyl phosphate for 60 min is compared in the 2nd column. In the 3rd column, this value is converted to a percent where the phosphorylation of the wild type protein represents 100%. Wild type and mutant proteins were phosphorylated and analyzed by HPLC as described under “Experimental Procedures.”
| Phosphorylation by acetyl phosphate after 60 min | Phosphorylation compared with wild type | |
|---|---|---|
| % | % | |
| Wild type | 77.3 | 100 |
| T224C | 7.5 | 10 |
| V225C | 31.2 | 40 |
| W226C | 13.9 | 18 |
| Wild type + dithiothreitol | 79.8 | 100 |
| W226C + dithiothreitol | 72.0 | 90 |
Disulfide Bond Formation of Wing Mutant W226C Inhibits Phosphorylation—The HPLC profile of the Trp226 mutant contained multiple peaks. One of them was slightly diminished, and a new peak emerged in the presence of phosphoramidate (data not shown). During protein purification, it became evident that a significant fraction of the protein was forming a disulfide bond. We then examined the phosphorylation of W226C in the presence and absence of the reducing agent dithiothreitol and compared it with the wild type protein (Table 3). The presence of dithiothreitol had no effect on OmpR, and it was 80% phosphorylated in 60 min. The mutant W226C exhibited a slightly reduced level of phosphorylation, ∼90% of the wild type, but significantly higher than in the disulfide-bonded form. In contrast, the disulfide-bonded dimer was only 18% phosphorylated (Table 3). This result indicates that tail-to-tail dimer formation at Trp226 in the wing of the full-length protein inhibits phosphorylation in the N-terminal receiver domain. It is not simply disulfide bond formation that inhibits DNA binding (Fig. 9), because alanine or arginine substitutions at this same position were also incapable of expressing ompF and ompC (data not shown).
The DNA Contact Sites Are Different at Different OmpR-binding Sites—OmpR-binding sites are poorly conserved and difficult to predict. In S. enterica sv. Typhimurium, OmpR activates expression of genes on a pathogenicity island that are required for survival in macrophages. OmpR-binding sites at the ssrA gene were identified by DNase I footprinting (18). This site lacked the G base conserved in the porin gene-binding sites. It was therefore of interest to determine whether the R207A mutant, which was incapable of activating ompF and ompC, was capable of activating ssrA (Fig. 10). We tested an array of OmpR mutants, some were defective in activating ompF and ompC as shown in Fig. 8 and others exhibited wild type behavior. Not surprisingly, R207A was as effective as the wild type OmpR in stimulating β-galactosidase activity of the ssrA-lacZ fusion, suggesting that OmpR employs slightly different DNA contacts at its diverse binding sites. In contrast, the R209A substitution was completely defective at activating ssrA, ompF, and ompC, as were G191C and W226C. OmpR and mutant protein expression levels were comparable, based on Western blots (data not shown).
FIGURE 10.

The effect of OmpR mutants on β-galactosidase activity of ssrA-lacZ. OmpR wild type or mutants (listed below the relevant column) were expressed in an ompR null strain containing ssrA-lacZ in single copy (54). Overnight cultures were diluted 1:100 and grown to mid-log phase in LB media; IPTG was added to a final concentration of 1 mm and incubated for 2 h. Expression of mutant OmpR proteins was confirmed by a Western blot. Transcription of the ssrA-lacZ fusion was monitored by β-galactosidase assay as described previously (49). All β-galactosidase measurements were performed in triplicate with at least two independent cultures. The level of activity of wild type OmpR was set to 100%, and the mutants were expressed relative to it.
DISCUSSION
OmpR plays a critical role as a global regulator in many prokaryotes. In S. enterica, it regulates the expression of the genes on pathogenicity island 2 (SPI-2) and is required for S. enterica sv. Typhimurium to survive and replicate in macrophages (13, 18). Even though OmpR has been studied intensively and the structures of OmpRC and several homologues have been solved, there are still many unanswered questions. One of them is how OmpR binds to DNA. The OmpR homologue PhoB binds to DNA as a head-to-tail dimer. Its binding sites consist of two 7-bp direct repeats separated by an A/T-rich region of 4 bp, situated ∼10 bp upstream of the -10 region (53). An OmpR-binding site contains a GTXTCA motif in its DNA-binding site. If OmpR were bound to DNA in the same manner as PhoB, i.e. as an asymmetric head-to-tail dimer, additional amino acids should undergo chemical shift changes in the presence of the full C1 site compared with the half-site. Our present results indicate that this is not the case (Fig. 3, B and D, and Fig. 7, A and B, and supplemental Table 1). In the presence of a half-site or a full-length site, the perturbed residues were nearly identical, suggesting that either an OmpR dimer is symmetric (tail-to-tail or head-to-head) or that OmpR binds to DNA as a monomer, or only extremely weak dimeric interactions exist. Furthermore, our results indicate that OmpR can bind to the high affinity site C1b in either orientation. This result corroborates previous results in which OmpR cross-linked head-to-head could still bind to DNA (56). Our NMR data of the OmpR-C1 half-site-DNA complex further support the notion that OmpR binds to C1 as a monomer (Fig. 3, B and D) and is in contrast to previous claims that two OmpR molecules are required to bind to DNA (29). The tail-to-tail orientation seems unlikely to occur in the full-length protein in vivo, because the disulfide-bonded W226C mutant inhibited phosphorylation of the receiver domain (Table 3). Furthermore, a cysteine substitution at this position of full-length OmpR did not form cross-links in the presence of the homo-bifunctional reagent bismaleimidohexane (56). Consistent with this view is the observation that a mutant with similar disulfide binding properties as W226C (e.g. G227C) was isolated in a mutagenesis screen that contained only the C terminus of OmpR (60). The structure of the OmpR homologue HP1043 from Helicobacter pylori was recently solved by NMR and binds as a symmetrical dimer to an inverted repeat (61), indicating that additional OmpR subfamily members can bind in a head-to-head orientation as we first described (56). Hp1043 forms a rigid dimer through its receiver domain and binds to DNA as a monomer, as we described for OmpR in this study.
Comparison of the PhoB and OmpR Dimer Interface—OmpR and PhoB DNA binding domains are highly homologous, and their binding sites both contain a GT in their core binding sites that make recognition helix contacts. Our NMR and phenotypic analyses further suggest that the DNA binding schemes used by the two DNA binding domains are also similar. However, a major difference in DNA recognition by OmpR and PhoB affects how the dimer is formed. Comparing the spacing between the two G bases of each half-site, OmpR has 9 bp (GTTTCAAAATGTAA), whereas PhoB has 10 bp (GTCATAAAGTTGTCA) between them. Thus, the distance and the angle between the two OmpR DNA binding domains in the dimer are different from those of the PhoB DNA binding domains in the dimer. Our study demonstrates that the OmpR DNA binding domain can form a dimer on the DNA in either orientation. It also suggests that protein/protein interactions between OmpR DNA binding domains are extremely weak or absent. If true, it raises the possibility that at highly complex promoters (e.g. SPI-2 in S. enterica sv. Typhimurium), OmpR may interact with other regulatory proteins in some transcriptional complexes (54). The low specificity of OmpR for DNA likely contributes to its role as a global regulator and enables it to activate transcription of horizontally acquired genes in pathogenesis.
Although OmpR and PhoB Are Structurally Similar, Key Differences Exist—In the PhoB/DNA co-crystal structure, both protomers establish extensive protein-protein contacts. For example, residues in the 5′-protomer of β-strands 3 and 4 make contacts with residues in the β-hairpin (β-strands 6 and 7). Most of these residues are not conserved in OmpR (52). However, when we compare OmpR and PhoB bound to DNA (Fig. 11), it is evident that important differences exist. In our model, a likely interaction site is the loop between β-strands 5 and 6 on one molecule (as in PhoB), but instead of interacting with β-strands 3 and 4, OmpR interacts with the loop between α-helix 2 and α-helix 3 on the other molecule. Furthermore, because there is one fewer base inbetween the OmpR half-sites, OmpR molecules must rotate 36° on the DNA, reducing the amino acid contacts between protomers.
FIGURE 11.

Comparison of OmpR and PhoB dimers on DNA. In the top panel, OmpR dimers on an ompC1-binding site are shown. In the lower panel, the PhoB dimer from Ref. 52 is shown.
Structural Insight into the V203M (OmpR2) Mutant—V203M was one of the earliest OmpR mutants isolated; it was referred to as an OmpR2 mutant because it conferred an ompF constitutive, ompC minus phenotype (47, 62). In a more recent study, it was found to discriminate between F1- and C1-binding sites, binding with higher affinity to F1 and lower affinity to C1 than the wild type OmpR (33). It is now possible to speculate as to the structural basis for this reduced affinity. The side chain of Val203 contacts the methyl group of thymine in the conserved GT sequence. Valine has a rather short side chain, whereas methionine has a long side chain. The methionine would have to shift its contact, and it is better able to do this when the next base is an adenine, enabling the methionine to contact the thiamine base-paired to this adenine. Consistent with this view, a single base change in the C1-binding site restored high affinity binding by the mutant. This base change substituted an adenine for thymine at the X position of the GTXTCA core (33).
Steps Leading to OmpR DNA Binding and Activation—Phosphorylation of PhoB is required for DNA binding, and structural analysis of DrrB also suggests that phosphorylation would remodel the interdomain interface, relieving inhibitory contacts and exposing the recognition helix. OmpR is different from these other homologues, in that it can bind DNA in the absence of phosphorylation (24). If the PhoB model were appropriate for OmpR, we would expect that many resonances from the DNA binding domain of OmpR would be perturbed in the presence of the full-length protein. To our surprise, the chemical shift perturbation of the DNA binding domain of OmpR was minimal (data not shown). Thus, our data suggest that the activation pathway for OmpR is different. Based on the results presented here, we propose that the pathway that leads to OmpR activation is as follows (Fig. 12). The C terminus of OmpR binds to DNA with high affinity at the 3′ half-site (Fig. 12, C1b) as a monomer (Fig. 12, step 1). As a consequence of binding, a conformational change is transmitted to the N terminus, promoting phosphorylation (Fig. 12, step 2) and possibly domain swapping (32). Separation of the two domains is aided by the longer, more dynamic linker connecting them (27). Thus, phosphorylation does not stimulate the receiver domain to release inhibitory interactions between the DNA binding domain and the receiver domain as it does with PhoB. Rather, it promotes a conformation that enhances dimerization between the receiver domains of two OmpR molecules (Fig. 12, step 3). Dimerization then enables DNA binding to the lower affinity upstream site by the second protomer (Fig. 12, C1a, step 4).
FIGURE 12.

Sequence of events for OmpR activation and DNA binding. Step1, OmpR binds as a monomer to the high affinity C1b site located 3′ to its upstream half-site C1a. Step 2, conformational change occurs in the N-terminal receiver domain as a consequence of the C terminus binding to DNA. This promotes phosphorylation of Asp55 in the receiver domain. Step 3, as a consequence of phosphorylation, the receiver domains form symmetric dimers. Step 4, dimerization brings the second OmpRC domain to its lower affinity site, promoting DNA binding by the second OmpRC. If the orientation of C1b is reversed, the dimer still forms, presumably because the longer, more flexible linker enables OmpRC to bind.
Supplementary Material
Acknowledgments
We are grateful to Peter Gettins (University of Illinois, Chicago) for helpful comments on the manuscript. The Bruker DRX800 was purchased with funds provided by Grant BIR0079604 from the National Science Foundation.
This work was supported in part by National Institutes of Health Grant GM-058746 and National Science Foundation Grant MCB-0613014 (to L. J. K.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Table 1.
Footnotes
L. J. Kenney and K. van Holde, unpublished results.
The abbreviations used are: IPTG, isopropyl thiogalactopyranoside; r.m.s.d., root mean square deviation; NOE, nuclear Overhauser effect; NOESY, nuclear Overhauser effect spectroscopy; HSQC, heteronuclear single quantum coherence; HPLC, high pressure liquid chromatography.
L. J. Kenney, unpublished observations.
References
- 1.Hoch, J. A., and Silhavy, T. J. (eds) (1995) Two-component Signal Transduction, American Society for Microbiology, Washington, DC
- 2.van Alphen, W., and Lugtenberg, B. (1977) J. Bacteriol. 131 623-630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Basle, A., Rummel, G., Storici, P., Rosenbusch, J. P., and Schirmer, T. (2006) J. Mol. Biol. 362 933-942 [DOI] [PubMed] [Google Scholar]
- 4.Cowan, S. W., Schirmer, T., Rummel, G., Steiert, M., Ghosh, R., Pauptit, R. A., Jansonius, J. N., and Rosenbusch, J. P. (1992) Nature 358 727-733 [DOI] [PubMed] [Google Scholar]
- 5.Nikaido, H., Rosenberg, E. Y., and Foulds, J. (1983) J. Bacteriol. 153 232-240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oshima, T., Aiba, H., Masuda, Y., Kanaya, S., Sugiura, M., Wanner, B. L., Mori, H., and Mizuno, T. (2002) Mol. Microbiol. 46 281-291 [DOI] [PubMed] [Google Scholar]
- 7.Guillier, M., and Gottesman, S. (2006) Mol. Microbiol. 59 231-247 [DOI] [PubMed] [Google Scholar]
- 8.Dorman, C. J., Chatfield, S., Higgins, C. F., Hayward, C., and Dougan, G. (1989) Infect. Immun. 57 2136-2140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dorrell, N., Li, S. R., Everest, P. H., Dougan, G., and Wren, B. W. (1998) FEMS Microbiol. Lett. 165 145-151 [DOI] [PubMed] [Google Scholar]
- 10.Bernardini, M. L., Fontaine, A., and Sansonetti, P. J. (1990) J. Bacteriol. 172 6274-6281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bernardini, M. L., Sanna, M. G., Fontaine, A., and Sansonetti, P. J. (1993) Infect. Immun. 61 3625-3635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brzostek, K., Raczkowska, A., and Zasada, A. (2003) FEMS Microbiol. Lett. 228 265-271 [DOI] [PubMed] [Google Scholar]
- 13.Lee, A. K., Detweiler, C. S., and Falkow, S. (2000) J. Bacteriol. 182 771-781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lindgren, S. W., Stojiljkovic, I., and Heffron, F. (1996) Proc. Natl. Acad. Sci. U. S. A. 93 4197-4201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pickard, D., Li, J., Roberts, M., Maskell, D., Hone, D., Levine, M., Dougan, G., and Chatfield, S. (1994) Infect. Immun. 62 3984-3993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raczkowska, A., and Brzostek, K. (2004) Acta Microbiol. Pol. 53 11 [Google Scholar]
- 17.Romling, U., Sierralta, W. D., Eriksson, K., and Normark, S. (1998) Mol. Microbiol. 28 249-264 [DOI] [PubMed] [Google Scholar]
- 18.Feng, X., Oropeza, R., and Kenney, L. J. (2003) Mol. Microbiol. 48 1131-1143 [DOI] [PubMed] [Google Scholar]
- 19.Kato, M., Aiba, H., Tate, S., Nishimura, Y., and Mizuno, T. (1989) FEBS Lett. 249 168-172 [DOI] [PubMed] [Google Scholar]
- 20.Tate, S., Kato, M., Nishimura, Y., Arata, Y., and Mizuno, T. (1988) FEBS Lett. 242 27-30 [DOI] [PubMed] [Google Scholar]
- 21.Kenney, L. J., Bauer, M. D., and Silhavy, T. J. (1995) Proc. Natl. Acad. Sci. U. S. A. 92 8866-8870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aiba, H., Nakasai, F., Mizushima, S., and Mizuno, T. (1989) J. Biochem. (Tokyo) 106 5-7 [DOI] [PubMed] [Google Scholar]
- 23.Harlocker, S. L., Bergstrom, L., and Inouye, M. (1995) J. Biol. Chem. 270 26849-26856 [DOI] [PubMed] [Google Scholar]
- 24.Head, C. G., Tardy, A., and Kenney, L. J. (1998) J. Mol. Biol. 281 857-870 [DOI] [PubMed] [Google Scholar]
- 25.Huang, K. J., and Igo, M. M. (1996) J. Mol. Biol. 262 615-628 [DOI] [PubMed] [Google Scholar]
- 26.Ames, S. K., Frankema, J., and Kenney, L. J. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 11792-11797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mattison, K., Oropeza, R., and Kenney, L. J. (2002) J. Biol. Chem. 277 32714-32721 [DOI] [PubMed] [Google Scholar]
- 28.Rampersaud, A., Harlocker, S. L., and Inouye, M. (1994) J. Biol. Chem. 269 12559-12566 [PubMed] [Google Scholar]
- 29.Yoshida, T., Qin, L., Egger, L. A., and Inouye, M. (2006) J. Biol. Chem. 281 17114-17123 [DOI] [PubMed] [Google Scholar]
- 30.Cai, S. J., and Inouye, M. (2002) J. Biol. Chem. 277 24155-24161 [DOI] [PubMed] [Google Scholar]
- 31.Toro-Roman, A., Mack, T. R., and Stock, A. M. (2005) J. Mol. Biol. 349 11-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.King-Scott, J., Nowak, E., Mylonas, E., Panjikar, S., Roessle, M., Svergun, D. I., and Tucker, P. A. (2007) J. Biol. Chem. 282 37717-37729 [DOI] [PubMed] [Google Scholar]
- 33.Tran, V. K., Oropeza, R., and Kenney, L. J. (2000) J. Mol. Biol. 299 1257-1270 [DOI] [PubMed] [Google Scholar]
- 34.Walthers, D., Tran, V. K., and Kenney, L. J. (2003) J. Bacteriol. 185 317-324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Slauch, J., and Silhavy, T. J. (1989) J. Mol. Biol. 210 291-292 [DOI] [PubMed] [Google Scholar]
- 36.Clore, G. M., and Gronenborn, A. M. (1998) Trends Biotechnol. 16 22-34 [DOI] [PubMed] [Google Scholar]
- 37.Wishart, D. S., Bigam, C. G., Yao, J., Abildgaard, F., Dyson, H. J., Oldfield, E., Markley, J. L., and Sykes, B. D. (1995) J. Biomol. NMR 6 135-140 [DOI] [PubMed] [Google Scholar]
- 38.Kanelis, V., Forman-Kay, J. D., and Kay, L. E. (2001) IUBMB Life 52 291-302 [DOI] [PubMed] [Google Scholar]
- 39.Clore, G. M., Gronenborn, A. M., Nilges, M., and Ryan, C. A. (1987) Biochemistry 26 8012-8023 [DOI] [PubMed] [Google Scholar]
- 40.Wuthrich, K., Billeter, M., and Braun, W. (1983) J. Mol. Biol. 169 949-961 [DOI] [PubMed] [Google Scholar]
- 41.Wishart, D. S., and Case, D. A. (2001) Methods Enzymol. 338 3-34 [DOI] [PubMed] [Google Scholar]
- 42.Guntert, P., Mumenthaler, C., and Wuthrich, K. (1997) J. Mol. Biol. 273 283-298 [DOI] [PubMed] [Google Scholar]
- 43.Koradi, R., Billeter, M., and Wuthrich, K. (1996) J. Mol. Graphics 14 51-55 [DOI] [PubMed] [Google Scholar]
- 44.Doreleijers, J. F., Rullmann, J. A., and Kaptein, R. (1998) J. Mol. Biol. 281 149-164 [DOI] [PubMed] [Google Scholar]
- 45.Haldimann, A., and Wanner, B. L. (2001) J. Bacteriol. 183 6384-6393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Russo, F. D., and Silhavy, T. J. (1991) J. Mol. Biol. 222 567-580 [DOI] [PubMed] [Google Scholar]
- 47.Hall, M., and Silhavy, T. J. (1981) J. Mol. Biol. 151 1-15 [DOI] [PubMed] [Google Scholar]
- 48.Hall, M. N., and Silhavy, T. J. (1981) J. Mol. Biol. 146 23-43 [DOI] [PubMed] [Google Scholar]
- 49.Miller, J. (1972) Experiments in Molecular Genetics, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
- 50.Kondo, H., Nakagawa, A., Nishihira, J., Nishimura, Y., Mizuno, T., and Tanaka, I. (1997) Nat. Struct. Biol. 4 28-31 [DOI] [PubMed] [Google Scholar]
- 51.Martinez-Hackert, E., and Stock, A. M. (1997) Structure (Lond.) 5 109-124 [DOI] [PubMed] [Google Scholar]
- 52.Blanco, A. G., Sola, M., Gomis-Ruth, F. X., and Coll, M. (2003) Structure (Lond.) 10 701-713 [DOI] [PubMed] [Google Scholar]
- 53.Makino, K., Amemura, M., Kawamoto, T., Kimura, S., Shinagawa, H., Nakata, A., and Suzuki, M. (1996) J. Mol. Biol. 259 15-26 [DOI] [PubMed] [Google Scholar]
- 54.Feng, X., Walthers, D., Oropeza, R., and Kenney, L. J. (2004) Mol. Microbiol. 54 823-835 [DOI] [PubMed] [Google Scholar]
- 55.Pratt, L. A., and Silhavy, T. J. (1995) Mol. Microbiol. 17 565-573 [DOI] [PubMed] [Google Scholar]
- 56.Maris, A., Walthers, D., Mattison, K., Byers, N., and Kenney, L. J. (2005) J. Mol. Biol. 350 843-856 [DOI] [PubMed] [Google Scholar]
- 57.Igo, M. M., and Silhavy, T. J. (1988) J. Bacteriol. 170 5971-5973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Delgado, J., Forst, S., Harlocker, S., and Inouye, M. (1993) Mol. Microbiol. 10 1037-1047 [DOI] [PubMed] [Google Scholar]
- 59.Mattison, K., Oropeza, R., Byers, N., and Kenney, L. J. (2002) J. Mol. Biol. 315 497-511 [DOI] [PubMed] [Google Scholar]
- 60.Tsuzuki, M., Aiba, H., and Mizuno, T. (1994) J. Mol. Biol. 242 607-613 [DOI] [PubMed] [Google Scholar]
- 61.Hong, E., Lee, H. M., Ko, H., Kim, D. U., Jeon, B. Y., Jung, J., Shin, J., Lee, S. A., Kim, Y., Jeon, Y. H., Cheong, C., Cho, H. S., and Lee, W. (2007) J. Biol. Chem. 282 20667-20675 [DOI] [PubMed] [Google Scholar]
- 62.Nara, F., Matsuyama, S., Mizuno, T., and Mizushima, S. (1986) Mol. Gen. Genet. 202 194-199 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.