

Abstract
Plague, one of the most devastating diseases of human history, is caused by Yersinia pestis. In this study, we analyzed the population genetic structure of Y. pestis and the two other pathogenic Yersinia species, Y. pseudotuberculosis and Y. enterocolitica. Fragments of five housekeeping genes and a gene involved in the synthesis of lipopolysaccharide were sequenced from 36 strains representing the global diversity of Y. pestis and from 12–13 strains from each of the other species. No sequence diversity was found in any Y. pestis gene, and these alleles were identical or nearly identical to alleles from Y. pseudotuberculosis. Thus, Y. pestis is a clone that evolved from Y. pseudotuberculosis 1,500–20,000 years ago, shortly before the first known pandemics of human plague. Three biovars (Antiqua, Medievalis, and Orientalis) have been distinguished by microbiologists within the Y. pestis clone. These biovars form distinct branches of a phylogenetic tree based on restriction fragment length polymorphisms of the locations of the IS100 insertion element. These data are consistent with previous inferences that Antiqua caused a plague pandemic in the sixth century, Medievalis caused the Black Death and subsequent epidemics during the second pandemic wave, and Orientalis caused the current plague pandemic.
Keywords: taxonomy, multilocus sequence typing, molecular epidemiology, microevolution
The genus Yersinia contains three pathogenic species, Y. pestis, the causative agent of plague, and the enteric food- and water-borne pathogens Y. pseudotuberculosis and Y. enterocolitica. Y. pestis is primarily a disease of rodents or other wild mammals that usually is transmitted by fleas and often is fatal. Human disease is now rare and usually is associated with contact with rodents and their fleas. However, three former waves of pandemic plague reached Europe by different routes (Fig. 1) and affected very significant portions of the human population: the first pandemic (Justinian’s plague, 541–767 AD) is thought to have been imported from East or Central Africa and spread from Egypt to countries surrounding the Mediterranean (1). The second pandemic (the Black Death and subsequent epidemics from 1346 to the early 19th century) spread from the Caspian Sea to all of Europe and may have been imported from central Asia. The third pandemic began in the mid-19th century in the Yunnan region of China and spread globally via marine shipping from Hong Kong in 1894, the same year that Y. pestis was first described by Yersin (2, 3).
Figure 1.

Routes followed by the three plague pandemic waves, labeled 1, 2, and 3. Circled numbers indicate the regions thought to be the origins of these pandemic waves.
Y. pestis has been subdivided into three biovars (Antiqua, Medievalis, and Orientalis) on the basis of minor phenotypic differences. Epidemiological observations and historical records led to the hypothesis (4) that biovar Antiqua, resident in Africa, is descended from bacteria that caused the first pandemic whereas Medievalis, resident in central Asia, is descended from the bacteria that caused the second pandemic. Bacteria epidemiologically linked to the third pandemic are all Orientalis and are currently widespread.
In addition to the subdivision into three biovars, Y. pestis manifests some degree of restriction fragment length polymorphism according to ribotyping and pulsed-field gel electrophoresis (5), even among strains isolated from a single country (6). However, serotyping and phage typing have not revealed any diversity. Furthermore, the results of DNA–DNA hybridization (7) indicate that Y. pestis is highly related to Y. pseudotuberculosis and the sequences of their 16S rRNAs are identical (8). These results led to the proposal (7) that Y. pestis and Y. pseudotuberculosis should be reclassified as two related subspecies. However, Y. pestis causes fatal bubonic plague and is transmitted by flea bites whereas Y. pseudotuberculosis is transmitted by the fecal–oral route and rarely leads to death. For these reasons and because of the historical importance of Y. pestis for human history, the reclassification of Y. pestis and Y. pseudotuberculosis has been rejected by medical microbiologists. The reasons for the differences in virulence between Y. pestis and Y. pseudotuberculosis are still unknown but may reflect the fact that Y. pestis contains two pestis-specific plasmids (9), one of which encodes murine toxin, a phospholipase D homolog that facilitates colonization of the flea midgut (10),§ thus enhancing host-to-host transmission. The few chromosomally located, virulence-associated properties identified so far in Y. pestis are also found in at least some strains of Y. pseudotuberculosis and are not thought to account for the differences in their disease potential.
DNA sequences of multiple housekeeping genes can be used to deduce the phylogenetic history of bacterial species such as Salmonella enterica (11). Multilocus sequence typing of housekeeping genes revealed the existence of clonal groupings even within bacterial species such as Neisseria meningitidis (12), Streptococcus pneumoniae (13), and Helicobacter pylori (14), which are characterized by high levels of recombination (15, 16). In species in which horizontal genetic exchange is rare, sequence polymorphism reflects the accumulation of mutations at a uniform clock rate and is correlated with the time elapsed since the existence of a last common ancestor. Analyses of sequence variability have revealed that Mycobacterium tuberculosis (17) and Plasmodium falciparum (18) are only several thousand years old. In contrast, the last common ancestor of Escherichia coli and S. enterica existed approximately 140 million years ago (19).
Here, we show that Y. pestis is a highly uniform clone of Y. pseudotuberculosis that arose shortly before the first known pandemics of plague and that the three biovars are phylogenetically distinct, consistent with unique associations with each of the three pandemics.
Materials and Methods
Bacterial Strains.
Seventy-six strains of Y. pestis of all three biovars (Antiqua, Medievalis, and Orientalis) had been isolated between 1942 and 1998 from humans, fleas, and small mammals in diverse countries (Table 1 and Fig. 2). Gene fragments were sequenced from 36 strains, and IS100 typing was done with 49. Thirteen strains of Y. enterocolitica of various bioserotypes and 12 strains of Y. pseudotuberculosis of serotypes I–V also were used for sequencing gene fragments (Table 1).
Table 1.
Alleles of six gene fragments in three Yersinia species
| IP number | Species | Country | Host | Year | Serotype/bioserotype | thrA | trpE | glnA | tmk | dmsA | manB |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 6 strains | pe | Diverse* | Diverse* | 53–84 | Antiqua | 1 | 1 | 1 | 1 | 1 | 1 |
| 7 strains | pe | Diverse† | Diverse† | 47–64 | Medievalis | 1 | 1 | 1 | 1 | 1 | 1 |
| 23 strains | pe | Diverse‡ | Diverse‡ | 42–98 | Orientalis | 1 | 1 | 1 | 1 | 1 | 1 |
| 31830 | ps | U.K. | Human | 1969 | IV | 1 | 3 | 1 | 2 | 5 | 1 |
| 32889 | ps | Spain | Human | 1988 | III | 1 | 3 | 1 | 2 | 5 | 4 |
| 32951 | ps | France | Human | 1990 | II | 1 | 3 | 1 | 2 | 8 | |
| 32934 | ps | France | Monkey | 1989 | II | 3 | 3 | 1 | 5 | 5 | 1 |
| 32949 | ps | France | Human | 1990 | I | 1 | 3 | 1 | 4 | 5 | 2 |
| 32953 | ps | France | Human | 1990 | I | 4 | 3 | 1 | 4 | 6 | 2 |
| 32790 | ps | Italy | Pig | 1986 | I | 4 | 3 | 1 | 4 | 7 | |
| 32817 | ps | Japan | Human | 1986 | V | 5 | 3 | 1 | 6 | 3 | 6 |
| 32821 | ps | France | Human | 1986 | V | 3 | 3 | 1 | 5 | 5 | 5 |
| 31833 | ps | U.K. | Sheep | 1969 | IV | 3 | 3 | 1 | 4 | 4 | 2 |
| 32937 | ps | Argentina | Bovine | 1989 | III | 2 | 2 | 2 | 3 | 2 | 3 |
| 32945 | ps | Argentina | Calf | 1990 | III | 2 | 2 | 2 | 3 | 2 | 3 |
| 383 | ent | Belgium | Human | 1968 | 2 O:9 | 6 | 4 | 7 | 10 | 11 | 10 |
| 21349 | ent | France | Human | 1990 | 2 O:9 | 6 | 4 | 6 | 10 | 11 | 10 |
| 21650 | ent | France | Human | 1990 | 2 O:9 | 6 | 4 | 6 | 10 | 11 | 10 |
| 864 | ent | Belgium | Human | 1970 | 4 O:3 | 6 | 4 | 6 | 10 | 11 | 7 |
| 21699 | ent | France | Human | 1990 | 4 O:3 | 6 | 4 | 6 | 10 | 11 | 7 |
| 134 | ent | Sweden | Human | 1963 | 4 O:3 | 6 | 4 | 6 | 10 | 11 | 7 |
| 885 | ent | U.K. | Dog | 1970 | 2 O:5 | 6 | 4 | 6 | 10 | 11 | 7 |
| 24636 | ent | France | Human | 1995 | 2 O:5 | 6 | 4 | 6 | 12 | 11 | 7 |
| 25963 | ent | France | Human | 1998 | 1A O:5 | 6 | 4 | 5 | 7 | 10 | 9 |
| 21708 | ent | France | Food | 1990 | 1A O:6 | 6 | 4 | 5 | 8 | 9 | 7 |
| 21506 | ent | Spain | Salami | 1990 | 1A O:7,8 | 6 | 4 | 5 | 9 | 9 | |
| Ye8081 | ent | USA | Human | 1B O:8 | 6 | 4 | 4 | 11 | 8 | ||
| WA | ent | USA | Human | 1B O:8 | 6 | 4 | 3 | 13 | 8 |
pe, Y. pestis; ps, Y. pseudotuberculosis; ent, Y. enterocolitica; IP number, Institut Pasteur strain designation. Empty cells indicate the lack of data.
*Kenya (four strains), Congo (one), Nepal (one); human (four), rodent (one), unknown (one).
†Kurdistan (six), Turkey (one); human (one), rodent (five), unknown (one).
‡Vietnam (eight), Madagascar (six), Brazil (five), USA (two), Argentina (one), Turkey (one); human (six), rodent (two), rat (six), flea (five), unknown (four).
Figure 2.

Neighbor-joining phylogenetic tree of band patterns of IS100 in 49 strains of Y. pestis.
Gene Fragments and Sequences.
Oligonucleotide primers were designed based on sequences of dmsA (anaerobic DMSO reductase chain A), glnA (glutamine synthetase, EC 6.3.1.2), manB (phosphomannomutase), thrA (aspartokinase, EC 2.7.2.4), tmk (thymidylate kinase, EC 2.7.4.9), and trpE (anthranilate synthase component I, EC 4.1.3.27) from the Y. pestis Sanger Center Genome Project (http://www.sanger.ac.uk/Projects/Y_pestis/) or from related bacteria in GenBank. The genes were PCR-amplified from chromosomal DNA of Y. pestis or Y. pseudotuberculosis by using the following primer combinations (sequences are 5′–′): dmsA, O1151, GTTCTTGTGGACCGATGCCA, and O1152, AAGATCCGCACCATATCGCC; glnA, O1153, CTGCAGTTATGGACCCGTTC, and O1155, GCGGTAGCCACTTCGTGGTG; manB, O1144, AAAGCCTATGATATTCGTGG, and O1145, TAGTGATGAGCACTCATTTC; thrA, O1141, GGTGACGGTATGCGCACCATGCG, and O1143, GTCAACAATGACCGGGTTCAG; tmk, O1148, TATTGAAGGGCTTGAAGGGG, and O1150, AATGGGTTGGGAGGCATCAAT; trpE, O1146, CCGTATCGAGTTGGAAATGC, and O1147, CACCCGCTTGTACGGTGGCGA. DNA from Y. enterocolitica was amplified by using the following primers: dmsA, O1310, GAGAAATGCGAAATGATTGTGG, and O1152; glnA, O1267, CATTAACGAATCCGACATGG, and O1268, GGTCATACAGGTTTTTGTCC; manB, O1144 and O1145, O1274, GTATTCCAAGTTAAACGTGG, or O1328, TCGTGGATGATCTTCGCGCC; thrA, O1141 and O1290, GGTGTTGGCCTTTTTGTTYGGCG; tmk, O1148 and O1150; trpE, O1331, AACGTCTCACTGCTCGCCTG, and O1308, GGTCTCCTGTGCATGATGCG. Independent PCR products were sequenced from both strands by using dRhodamine-labeled terminators (PE Applied Biosystems 377 sequencer). The sequence reactions were performed by using the amplification primers except for the following genes from Y. enterocolitica: glnA, O1153 and O1312, AATGCGTGTGCCACGTTGTG; thrA, O1315, GTTTGCGCACCTTGCGTGGG, and O1290; trpE, O1331 and O1307, TTACGGGTTTCRTCRGCTTC. Sequences were trimmed to a uniform length for each gene after multiple alignment (pileup, Wisconsin Package, Version 9.1; Genetics Computer Group, Madison, WI). The mean distances between alleles at synonymous (DS) and nonsynonymous (DN) sites were calculated after Jukes–Cantor (20) correction by using dnasp 3.14 (21). Age calculations used the formula:
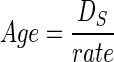 |
where rate is the molecular clock rate. For calculating the age of the pestis sequences, the following formula was used:
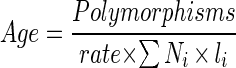 |
where Polymorphisms = 2.996 (95% confidence limit) or 0.693 (50% confidence limit) and Ni × li is the number of sequences times the number of potential synonymous sites in each sequence (18).
Restriction Fragment Length Polymorphism Analysis of the Locations of IS100 in Y. pestis.
A 255-bp IS100 probe was PCR-amplified from DNA of strain 6/69 by using primers IS100-F, AAAACGTTCGAAGAGTATGA, and IS100-R, GATGAGCAGGCGGGGGGCCA, and peroxidase-labeled by using the enhanced chemiluminescence direct nucleic acid-labeling and detection system (Amersham Pharmacia). The probe was used for Southern hybridization with EcoRI-digested genomic DNA from Y. pestis after separation by gel electrophoresis [0.8% agarose, 50 V overnight in TBE buffer (89 mM Tris/89 mM boric acid/2 mM EDTA, pH 8.3)]. Alkaline denaturation, neutralization, and transfer of DNA onto Hybond-N filters (Amersham Pharmacia) with a VacuGene apparatus (Amersham Pharmacia) were performed as described (22). The hybridization patterns were scanned and the computerized data were analyzed by using gelcompar 4.0 (Applied Mathematics, Kortrijk, Belgium). Phylogenetic trees of the hybridization profiles were constructed with gelcompar by using the neighbor-joining method.
Results
Alleles of Six Gene Fragments.
To investigate the population structure of pathogenic Yersinia species, we sequenced ≈400-bp fragments of five housekeeping genes (thrA, trpE, glnA, tmk, and dmsA) and a sixth gene (manB) involved in lipopolysaccharide biosynthesis. Sequences were obtained from 36 diverse strains of Y. pestis, 12 strains of Y. pseudotuberculosis, and 13 strains of Y. enterocolitica, except that we were unable to sequence manB from two strains of Y. pseudotuberculosis and dmsA from three strains of Y. enterocolitica (Table 1). Each unique sequence was assigned a different allele number, resulting in 4–13 alleles for each gene (Table 1). The difference between the various alleles was analyzed by calculating the mean percent difference at both synonymous (% DS) and nonsynonymous (% DN) sites.
All 36 strains of Y. pestis possessed identical alleles for all six gene fragments (Table 1). Furthermore, identical thrA1, glnA1, and manB1 alleles were found in Y. pestis and some strains of Y. pseudotuberculosis. For the three other gene fragments, Y. pseudotuberculosis alleles were found that encoded polypeptides identical to those from Y. pestis (trpE and dmsA) or differed only at nonsynonymous sites (tmk) (Table 2). The distances between the alleles in Y. pestis and Y. pseudotuberculosis were within the range seen in Y. pseudotuberculosis alone, with one minor exception (percent DN for tmk). These observations show that Y. pestis is a highly conserved clone of Y. pseudotuberculosis that differentiated so recently that only few sequence polymorphisms have yet accumulated. If not for tradition, the results also would justify changing the name of Y. pestis to reflect the fact that it is not an independent species.
Table 2.
Mean percent pairwise differences at synonymous (% DS) and nonsynonymous (% DN) sites of six gene fragments
| Gene (size) | Distance | pestis (36)* | pseudotuberculosis (12) | pe → ps | enterocolitica (13)* | pe → ent |
|---|---|---|---|---|---|---|
| thrA (393 bp) | % DS | <0.06 | 1.9 (0–4.4) | 1.6 (0–4.4) | <0.2 | >100 |
| % DN | <0.02 | <0.06 | <0.06 | <0.05 | 12.9 | |
| trpE (351 bp) | % DS | <0.06 | <0.2 | 3.5 | <0.2 | 176 |
| % DN | <0.02 | 0.1 (0–0.4) | 0.06 (0–0.4) | <0.06 | 10.3 | |
| glnA (384 bp) | % DS | <0.06 | 0.3 (0–1.1) | 0.2 (0–1.1) | 3.7 (0–7.9) | 69 (67–70) |
| % DN | <0.02 | <0.06 | <0.06 | 0.05 (0–0.3) | 4.3 (4.2–4.6) | |
| tmk (495 bp) | % DS | <0.04 | 1.4 (0–2.4) | 1.2 (0–2.4) | 6.1 (0–15.5) | 162 (149–206) |
| % DN | <0.02 | 0.05 (0–0.3) | 0.3 (0.3–0.6) | 0.2 (0–0.6) | 4.6 (4.1–4.7) | |
| dmsA (444 bp) | % DS | <0.06 | 2.5 (0–13.0) | 5.5 (3.0–7.2) | 4.4 (0–13.0) | 124 (98–130) |
| % DN | <0.02 | 0.05 (0–0.3) | 0.02 (0–0.3) | 0.3 (0–0.9) | 6.9 (6.9–7.2) | |
| manB (442 bp) | % DS | <0.06 | 4.1 (0–8.4) | 2.3 (0–7.3) | 90 (0–289) | >100 |
| % DN | <0.02 | 0.4 (0–0.9) | 0.2 (0–0.9) | 16 (0–26) | 20 (14–24) |
For datasets lacking sequence variation, the sensitivity of the measurements is indicated by expressing the data as less than the % DS and % DN that would have been observed with a single synonymous or nonsynonymous mutation, respectively.
The number of strains tested are indicated in parentheses after the species designation.
Y. enterocolitica is also relatively uniform. All 13 strains of Y. enterocolitica possessed identical thrA and trpE alleles, and the mean synonymous sequence diversity of glnA, tmk, and dmsA was only 4–6%, comparable to values from E. coli and much lower than those from N. meningitidis (15) and H. pylori (14). manB is exceptionally variable as are other genes involved in lipopolysaccharide biosynthesis in Salmonella (23) and E. coli (24), possibly reflecting at least three distinct instances of import from unrelated bacteria (data not shown).
Neighbor-joining phylogenetic trees (not shown) provided further support for the conclusions described above: all alleles from Y. pestis plus Y. pseudotuberculosis clustered tightly as did all alleles from Y. enterocolitica, other than manB. The most dramatic aspect of the trees was the large distance between Y. pestis/Y. pseudotuberculosis from Y. enterocolitica. Indeed, essentially all the synonymous sites of the six gene fragments and 4–20% of the nonsynonymous sites differed between Y. pestis and Y. enterocolitica (Table 2). These values are comparable to or greater than the level of sequence diversity at housekeeping genes between E. coli and Salmonella (25).
Age Since Descent from a Last Common Ancestor.
If sequence diversity is accumulated at a relatively constant rate (molecular clock hypothesis), the sequence variation at synonymous sites can be used to calculate when a last common ancestor existed. The synonymous clock rate has been calculated by Whittam (26) as 6 × 10−9 synonymous polymorphisms accumulated per year on the basis that E. coli and S. enterica type typhimurium last shared a common ancestor approximately 140 million years ago (19) and that 95% of synonymous sites have been exchanged since then (25). A 5-fold-higher clock rate of 3 × 10−8 was calculated by Guttman and Dykhuizen (27) on the basis that the mutation rate is approximately 10−10 and that E. coli undergo approximately 300 generations per year under natural conditions. The mean percent DS values then directly yield the estimated time elapsed since the existence of the last common ancestor, assuming that the molecular clock rates for Yersinia and E. coli are comparable. The date since separation of Y. pestis and Y. enterocolitica was estimated as 41–186 million years, excluding thrA and manB, for which synonymous polymorphism was saturated. Similarly, the last common ancestor of Y. pestis plus Y. pseudotuberculosis existed 0.4–1.9 million years ago.
The maximal age of Y. pestis can be estimated by a slightly different approach (18) that is necessitated by their lack of sequence variation. According to the Poisson distribution, the upper 95% confidence limit of the number of polymorphisms that could yield zero observed polymorphisms is 2.996 and the 50% confidence limit is 0.693. Substituting these estimates for zero polymorphisms allows calculating the time since the last bottleneck from which all strains of Y. pestis are descended. The extreme values calculated from both clock rate estimates and both confidence limits are 1,056 years [50% confidence limit, Guttman and Dykhuizen (27)] to 20,436 years [95% confidence limit, Whittam (26)]. These calculations also yield the date at which Y. pestis differentiated from Y. pseudotuberculosis, because earlier differentiation (and multiple bottlenecks) should have resulted in the accumulation of polymorphisms whereas all alleles in Y. pestis were identical or nearly identical to alleles still present in Y. pseudotuberculosis. Justinian’s plague was 1,500 years ago, and, therefore, Y. pestis is at least 1,500 years old.
Microevolution Within Y. pestis.
Y. pestis is not totally uniform. Extremely minor sequence variation has been noted in other analyses (28), and restriction analyses revealed considerable variability (5, 29). M. tuberculosis is as uniform as Y. pestis (17), and yet differences between individual strains can be reliably recognized by the restriction fragment length polymorphism caused by variable chromosomal locations of an insertion element (30). Therefore, we determined the restriction fragment length polymorphism pattern of genomic DNA from 49 strains of all three biovars after digestion with EcoRI and hybridization with a probe specific for the IS100 element. Higher-molecular-weight bands that were poorly separated were not evaluated, leaving 17–38 distinct bands per strain. Strains of biovar Antiqua possessed about 30 bands whereas strains of the biovars Medievalis and Orientalis had an average of 20 bands. The number of bands common to each pair of strains was used to construct a neighbor-joining phylogenetic tree (Fig. 2). The three biovars were clustered in independent parts of the tree, without any overlaps, suggesting different origins from a common root for all three. Furthermore, the distances of the branches were shortest for biovar Orientalis and longer for both Medievalis and Antiqua strains. UPGMA (Unweighted Pair Group Mean Average) clustering and splits decomposition yielded comparable results (data not shown). Orientalis is known to have been responsible for most cases of disease in the third pandemic. The longer branches associated with Medievalis and Antiqua suggest that these bacteria have an older evolutionary history and indeed may have survived from former pandemic waves.
Discussion
Y. pestis is a clone of Y. pseudotuberculosis that evolved so recently that it still shares strong sequence homology with that species. The lack of variation at 21,881 synonymous sites within 36 Y. pestis strains shows that Y. pestis probably evolved within the last 1,500–20,000 years. This age estimate assumes a similar clock rate for Yersinia and E. coli. If Y. pestis had a lower mutation rate or fewer generations per year than E. coli, our age estimate would be too low. However, the frequencies of rifampicin-resistant mutants after growth at 28°C for six strains of Y. pestis and three of Y. pseudotuberculosis were comparable to or slightly higher than for E. coli K-12 (data not shown), supporting the use of clock rates that were calibrated for E. coli. A second potential problem is that because of the lack of sequence variation, the calculations estimate the maximal rather than the real age, and the upper limit of 20,000 years may be too large. Because of these considerations, two different clock rates (26, 27) were combined with both 50% and 95% probability limits to yield a large range, 1,500–20,000 years, for the estimate of the age of Y. pestis. This is a realistic estimate of the age of this species and differs markedly from the millions of years that are cited in the popular literature (31).
The selective pressures that led to the recent evolution of Y. pestis are unknown. M. tuberculosis is supposed to have evolved from the bovine pathogen Mycobacterium bovis about 15,000 years ago, a date that is consistent with the domestication of bovines (17, 32). Possible explanations for the evolution of P. falciparum about 5,000 years ago include the lateral transfer to humans from an animal species, changes in human behavior, recent evolution of mosquito vectors, and changes in the host–parasite–vector association (18). None of these explanations necessarily applies to Y. pestis because it infects a variety of small mammals and is not primarily a human pathogen. The apparent correlation between the changed social and economic factors caused by an increase in human population size and the recent evolution of Y. pestis thus may represent pure coincidence. Alternatively, the development of agriculture may have supplied a significantly increased food supply for certain rodents, and their resulting increased population size and behavioral changes may have triggered the evolution of Y. pestis. We note that efficient vectors (the rat flea, Xenopsylla cheopis) and hosts with a history of sylvatic plague (the grass rat, Arvicanthis niloticus) are common in the sources of the first pandemic, East Africa (33) and Egypt (34). Having evolved in one rodent species, Y. pestis then could spread rapidly to a large number of other small mammals that were less affected by human civilization.
What genetic changes were needed for Y. pestis to evolve? Although it has been claimed that mutations in inv (35) and yadA (36) are sufficient to raise the virulence of Y. pseudotuberculosis to that of Y. pestis (37), subsequent work (38) has indicated that these results may reflect problems with nonisogenic strain constructs. Similarly, the Y. pestis hms gene product results in blockage of the flea proventriculus and thus enhances flea-mediated transmission between hosts (39). However, a functional hms locus is not exclusive to Y. pestis and also has been found in certain strains of Y. pseudotuberculosis (40). The only known unique virulence factor is the Pla plasminogen activator that is encoded by the 9.5-kb Y. pestis-specific plasmid pPla and is apparently important for the systemic dissemination of bacteria after s.c. injection (41). Nonetheless, some strains of Y. pestis cured of pPla did not change in virulence for experimental animals, even after s.c. infection (42, 43). Thus, currently, the only unique feature of Y. pestis that is thought to enhance transmission by the flea is the phospholipase D homolog encoded by ymt on the 100-kb pFra plasmid (10).§
The following, highly speculative evolutionary scenario invokes the known features of Y. pestis that distinguish it from Y. pseudotuberculosis and illustrates one possible path for this evolution. Y. pseudotuberculosis occasionally causes fatal septicemia in animals stressed by cold, famine, or capture and can be transmitted occasionally to fleas in nature (44) and in the laboratory (45). The crucial step toward the evolution of pathogenic Y. pestis that can be transmitted by fleas to other mammals may have been the acquisition of the pFra plasmid by a strain of Y. pseudotuberculosis from an unknown donor during cocolonization of the rodent gastrointestinal tract or the flea midgut. The combination of the chromosomally encoded Hms protein (39) and the pFra-encoded phospholipase D homolog (10) might have sufficed for more efficient transfer by ectoparasites to other animals. Subsequent acquisition of the pPla plasmid then might have enhanced the ability to disseminate after inoculation in the skin. IS100 is present in Y. pseudotuberculosis and presumably was inherited by the new strain. IS100 transposition mutations might have coincidentally destroyed the strains’ ability to colonize the gastrointestinal tract and left only transmission via fleas and other vectors as the sole means of survival. If the original hms gene products in Y. pseudotuberculosis were not able to efficiently block the flea proventriculus, the necessity to survive by ectoparasite transmission would have selected for mutations that increase this efficiency. In contrast, proteins necessary for transmission by the fecal–oral route would no longer be needed, leading to the lack of selective pressure against mutations (35) in ure (urease against gastric acid), inv, ail, and yadA (all needed for translocation across the intestinal barrier).
Subsequent microevolution has resulted in the three lineages called biovars Antiqua, Medievalis, and Orientalis. Antiqua reduces nitrate and ferments glycerol, whereas Medievalis does not reduce nitrate and Orientalis does not ferment glycerol (4), as if Antiqua were ancestral to both Medievalis and Orientalis. Devignat combined this observation with historical and epidemiological records and suggested that each of the three pandemic waves was caused by a different biovar (4), namely, the first pandemic wave by Antiqua, the second pandemic wave by Medievalis, and the third and current pandemic by Orientalis. Strains of biovar Antiqua continue to be isolated from East and Central Africa, the supposed source of the first pandemic wave, and Medievalis continues to be isolated in Kurdistan, a region bordering the Caspian Sea through which the second pandemic passed before reaching Europe. The epidemiological associations for the third pandemic are very strong: exclusively biovar Orientalis is isolated from most of the countries contaminated by marine shipping at the end of the last century.
The biovars cluster separately in a phylogenetic tree based on the chromosomal locations of an insertion element (Fig. 2). These data supply molecular evidence supporting the subdivision of Y. pestis into biovars (4). The longer lengths of the Antiqua and Medievalis branches are consistent with the hypothesis that these evolved earlier than Orientalis and with Devignat’s assignment of biovar Antiqua to the first pandemic wave of human plague and biovar Medievalis to the second wave (4).
Acknowledgments
We gratefully acknowledge the helpful comments of Eddie Holmes, Dan Dykhuizen, Guy Baranton, and Sebastian Suerbaum as well as two anonymous reviewers. This work was supported by Grant DFG Ac 36/10-1 from the Deutsche Forschungsgemeinschaft and Grants 97/25200/DCE/CEB (Centre d’Etude du Bouchet) and 93811-77/A000/DRET/DS (Direction de la Recherche et de la Technologie, Ministère de la Défense, France).
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession nos. AJ250236-41 and AJ270406-50).
Hinnebusch, J., Schwan, T., Rudolph, A., Dixon, J., Cherepanov, P. & Forsberg, A., Ninety-Ninth General Meeting of the American Society for Microbiology, May 30–June 3, 1999, Chicago, IL (D/B-236).
References
- 1.Brossollet J, Mollaret H. Pourquoi la peste? Le rat, la puce et le bubon. Paris, France: Gallimard; 1994. [Google Scholar]
- 2.Yersin A. Ann Inst Pasteur. 1894;2:428–430. [Google Scholar]
- 3.Perry R D, Fetherston J D. Clin Microbiol Rev. 1997;10:35–66. doi: 10.1128/cmr.10.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Devignat R. Bull W H O. 1951;4:247–263. [PMC free article] [PubMed] [Google Scholar]
- 5.Guiyoule A, Grimont F, Iteman I, Grimont P A D, Lefèvre M, Carniel E. J Clin Microbiol. 1994;32:634–641. doi: 10.1128/jcm.32.3.634-641.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guiyoule A, Rasoamanana B, Buchrieser C, Michel P, Chanteau S, Carniel E. J Clin Microbiol. 1997;35:2826–2833. doi: 10.1128/jcm.35.11.2826-2833.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bercovier H, Mollaret H H, Alonso J M, Brault J, Fanning G R, Steigerwalt A G, Brenner D J. Curr Microbiol. 1980;4:225–229. [Google Scholar]
- 8.Trebesius K, Harmsen D, Rakin A, Schmelz J, Heesemann J. J Clin Microbiol. 1998;36:2557–2564. doi: 10.1128/jcm.36.9.2557-2564.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brubaker R R. Clin Microbiol Rev. 1991;4:309–324. doi: 10.1128/cmr.4.3.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hinnebusch B J, Fischer E R, Schwan T G. J Infect Dis. 1998;178:1406–1415. doi: 10.1086/314456. [DOI] [PubMed] [Google Scholar]
- 11.Boyd E F, Wang F-S, Whittam T S, Selander R K. Appl Environ Microbiol. 1996;62:804–808. doi: 10.1128/aem.62.3.804-808.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maiden M C J, Bygraves J A, Feil E, Morelli G, Russell J E, Urwin R, Zhang Q, Zhou J, Zurth K, Caugant D A, et al. Proc Natl Acad Sci USA. 1998;95:3140–3145. doi: 10.1073/pnas.95.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Enright M C, Spratt B G. Microbiology. 1998;144:3049–3060. doi: 10.1099/00221287-144-11-3049. [DOI] [PubMed] [Google Scholar]
- 14.Achtman M, Azuma T, Berg D E, Ito Y, Morelli G, Pan Z-J, Suerbaum S, Thompson S, van der Ende A, van Doorn L J. Mol Microbiol. 1999;32:459–470. doi: 10.1046/j.1365-2958.1999.01382.x. [DOI] [PubMed] [Google Scholar]
- 15.Suerbaum S, Maynard Smith J, Bapumia K, Morelli G, Smith N H, Kunstmann E, Dyrek I, Achtman M. Proc Natl Acad Sci USA. 1998;95:12619–12624. doi: 10.1073/pnas.95.21.12619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spratt B G, Maiden M C. Philos Trans R Soc London B. 1999;354:701–710. doi: 10.1098/rstb.1999.0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kapur V, Whittam T S, Musser J M. J Infect Dis. 1994;170:1348–1349. doi: 10.1093/infdis/170.5.1348. [DOI] [PubMed] [Google Scholar]
- 18.Rich S M, Licht M C, Hudson R R, Ayala F J. Proc Natl Acad Sci USA. 1998;95:4425–4430. doi: 10.1073/pnas.95.8.4425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ochman H, Wilson A C. J Mol Evol. 1987;26:74–86. doi: 10.1007/BF02111283. [DOI] [PubMed] [Google Scholar]
- 20.Jukes T H, Cantor C R. In: Mammalian Protein Metabolism. Munro H N, editor. New York: Academic; 1969. pp. 21–132. [Google Scholar]
- 21.Rozas J, Rozas R. Bioinformatics. 1999;15:174–175. doi: 10.1093/bioinformatics/15.2.174. [DOI] [PubMed] [Google Scholar]
- 22.de Almeida A M P, Guiyoule A, Guilvout I, Iteman I, Baranton G, Carniel E. Microb Pathog. 1993;14:9–21. doi: 10.1006/mpat.1993.1002. [DOI] [PubMed] [Google Scholar]
- 23.Reeves P. Trends Genet. 1993;9:17–22. doi: 10.1016/0168-9525(93)90067-R. [DOI] [PubMed] [Google Scholar]
- 24.Stevenson G, Neal B, Liu D, Hobbs M, Packer N H, Batley M, Redmond J W, Lindquist L, Reeves P. J Bacteriol. 1994;176:4144–4156. doi: 10.1128/jb.176.13.4144-4156.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharp P M. J Mol Evol. 1991;33:23–33. doi: 10.1007/BF02100192. [DOI] [PubMed] [Google Scholar]
- 26.Whittam T S. In: Escherichia coli and Salmonella. Curtiss R III, Ingraham J L, Lin E C C, Low K B, Magasanik B, Reznikoff W S, Riley M, Schaechter M, Umbarger H E, editors. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 2708–2720. [Google Scholar]
- 27.Guttman D S, Dykhuizen D E. Science. 1994;266:1380–1383. doi: 10.1126/science.7973728. [DOI] [PubMed] [Google Scholar]
- 28.Buchrieser C, Rusniok C, Franguel L, Couve E, Billault A, Kunst F, Carniel E, Glaser P. Infect Immun. 1999;67:4851–4861. doi: 10.1128/iai.67.9.4851-4861.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lucier T S, Brubaker R R. J Bacteriol. 1992;174:2078–2086. doi: 10.1128/jb.174.7.2078-2086.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Embden J D A, Cave M D, Crawford J T, Dale J W, Eisenach K D, Gicquel B, Hermans P, Martin C, McAdam R, Shinnick T M, et al. J Clin Microbiol. 1993;31:406–409. doi: 10.1128/jcm.31.2.406-409.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karlen A. Man and Microbes: Disease and Plagues in History and Modern Times. New York: Jeremy P. Tarcher; 1995. [Google Scholar]
- 32.Sreevatsan S, Pan X, Stockbauer K, Connell N D, Kreiswirth B N, Whittam T S, Musser J M. Proc Natl Acad Sci USA. 1997;94:9869–9874. doi: 10.1073/pnas.94.18.9869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siongok T K A, Njagi A M, Masaba S. East African Med J. 1977;54:694–699. [PubMed] [Google Scholar]
- 34.Yunker C E, Kaiser M N, Hoogstraal H, Salah A E A. J Egypt Public Health Assoc. 1959;34:43–55. [Google Scholar]
- 35.Simonet M, Riot B, Fortineau N, Berche P. Infect Immun. 1996;64:375–379. doi: 10.1128/iai.64.1.375-379.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Skurnik M, Wolf-Watz H. Mol Microbiol. 1989;3:517–529. doi: 10.1111/j.1365-2958.1989.tb00198.x. [DOI] [PubMed] [Google Scholar]
- 37.Rosqvist R, Skurnik M, Wolf-Watz H. Nature (London) 1988;334:522–525. doi: 10.1038/334522a0. [DOI] [PubMed] [Google Scholar]
- 38.Han Y W, Miller V L. Infect Immun. 1997;65:327–330. doi: 10.1128/iai.65.1.327-330.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hinnebusch B J, Perry R D, Schwan T G. Science. 1996;273:367–370. doi: 10.1126/science.273.5273.367. [DOI] [PubMed] [Google Scholar]
- 40.Buchrieser C, Brosch R, Bach S, Guiyoule A, Carniel E. Mol Microbiol. 1998;30:965–978. doi: 10.1046/j.1365-2958.1998.01124.x. [DOI] [PubMed] [Google Scholar]
- 41.Sodeinde O A, Subrahmanyam Y V B K, Stark K, Quan T, Bao Y, Goguen J D. Science. 1992;258:1004–1007. doi: 10.1126/science.1439793. [DOI] [PubMed] [Google Scholar]
- 42.Samoilova S V, Samoilova L V, Yezhov I N, Drozdov I G, Anisimov A P. J Med Microbiol. 1996;45:440–444. doi: 10.1099/00222615-45-6-440. [DOI] [PubMed] [Google Scholar]
- 43.Welkos S L, Friedlander A M, Davis K J. Microb Pathog. 1997;23:211–223. doi: 10.1006/mpat.1997.0154. [DOI] [PubMed] [Google Scholar]
- 44.Lipaev V M, Antip’eva O A, Al’shevskaya Z T, Chipanina V M, Kozlovskaya O L, Chipanin V I. Zool Zhur. 1970;49:1386–1390. [Google Scholar]
- 45.Blanc G, Balthazard M. C R Soc Biol Paris. 1944;138:811–812. [Google Scholar]