

Summary
As cells undergo oncogenic transformation and as transformed cells arrive at metastatic sites, a complex interplay occurs with the surrounding stroma. This dialogue between tumor and stroma ultimately dictates the success of the tumor cells in the given microenvironment. As a result, understanding the molecular mechanisms at work is important for developing new therapeutic modalities. Proteases are major players in the interaction between tumor and stroma. This review will focus on the role of proteases in modulating tumor-stromal interactions of both primary breast and prostate tumors as well as at bone metastatic sites in a way that favors tumor growth.
Keywords: Proteases, Tumor-stromal Interaction, Tumor Progression, Bone Metastasis
Introduction
According to the American Cancer Society, approximately 560,000 people die every year as the result of cancer [1]. Of these, nearly all have bone metastases. Some cancers, including breast, prostate, kidney, thyroid, and lung have extreme proclivity to metastasize to bone. Breast cancer is the most common cancer in females, while prostate cancer is the most common cancer in males and both often have bone as their first site of metastasis [1]. In fact, in one autopsy study of patients with prostate cancer, 90% of patients with metastases had bone metastases [2]. As these cancers metastasize to bone, it is not without consequence. Consequences of bone metastasis include intractable bone pain, increased fracture risk, hypercalcemia, and leukoerythroblastic anemia, to name a few. As a result, progression to bone metastasis represents an ominous sign in the pathogenesis of both breast and prostate cancer.
While breast and prostate cancer both have bone as their primary site of metastasis, the bone metastatic profile of prostate cancer differs from the breast cancer [3]. As cells undergo oncogenic transformation, they acquire the ability to divide in the absence of growth signals, avoid apoptosis, stimulate angiogenesis, and develop the ability to invade other tissues and establish metastatic colonies [4]. However, cancer cells do not accomplish this in isolation. Rather, they harness their power in the tumor microenvironment only by recruiting the surrounding normal host cells to facilitate their growth and progression. The tumor cells and host cells undertake a complex interplay with bidirectional communication.
Normal bone remodeling involves a balance between osteolysis and bone synthesis with both being necessary for normal bone physiology. As a result of the interaction of tumor cells with the bone stroma, the normal physiologic balance between osteoblasts synthesizing bone and osteoclasts resorbing bone is distorted. In all focal metastatic bone lesions there are components of both new bone synthesis as well as osteolysis [5]. However, while osteoblastic synthesis of bone and osteolysis remain interconnected, certain malignancies tend to favor one end of the spectrum. Prostate cancer-induced bone metastases tend to demonstrate focal osteogenesis predominantly [5–7]. All prostate cancers metastatic to bone are space-occupying lesions, induce bone resorption in addition to bone formation, and this has been documented histologically by bone histomorphometry and by the measurement of urinary markers of bone resorption. [5]. To the contrary, mammary tumor-induced bone metastases are predominantly osteolytic [3; 5; 8].
Because bone metastases represent a common clinical sequela in both prostate and breast cancers, it is important to understand the complex interplay between tumor and host cells in the local microenvironments of both the primary tumors and in the bone metastases. The processes of primary tumor growth, dissemination, and establishment of secondary colonies are driven by a diverse group of factors including cytokines, growth factors, and proteases. Modification of such factors is often a crucial step in furthering tumor growth and metastasis. The proteases are a varied group of proteins that have been shown to be involved in modifying tumor-stromal interaction through activation or inactivation of various cytokines and growth factors and modification of adhesion molecules in a manner that favors tumor growth. Within this group of proteins, two families of proteases have been demonstrated to be particularly important in this context—the matrix metalloproteinases (MMPs) and the cathepsins. This review will focus on the role of proteases in modulating tumor-stromal interactions in the breast and prostate microenvironments in facilitating cancer progression and in altering the bone microenvironment to promote establishment of metastases.
Proteases
Matrix Metalloproteinases (MMPs)
The MMPs are a family of zinc-dependent proteases [9]. These proteases were originally recognized for their ability to degrade a variety of extracellular matrix (ECM) components. The MMPs have two conserved domains common to all MMPs which contain a zinc binding domain and a cysteine-switch. The cysteine-switch is found in the propeptide domain while the zinc-binding motif is found in the catalytic domain. The interaction between the cysteine-switch and zinc maintains the latency of the pro-MMPs [10]. The family has grown to contain over 20 different enzymes. Currently, there are 24 known MMP genes in humans including a duplicated MMP-23 gene [11–14]. On the basis of the ECM components cleaved, the MMPs can be divided into collagenases, gelatinases, and stromelysins/matrilysins (Table 1). Moreover, MMPs are also classified as secreted MMPs or membrane-associated (MT-MMPs) (Table 1).
Table 1.
Matrix Metalloproteinases
| MMP FAMILY | NAME | NUMBERED NAME | SUBSTRATES | EFFECT/PRODUCT |
|---|---|---|---|---|
| Stromelysins | ||||
| Stromelysin-1 | MMP-3 | IL-1β | Activation | |
| MMP-1,3,7,8,9,13 | Activation | |||
| TNF-α | Activation | |||
| α-anticymotrypsin | Inactivation | |||
| E-cadherin | ||||
| RANKL | sRANKL | |||
| Plasminogen | Angiostatin | |||
| Perlecan | Increased FGF, | |||
| Decorin | Increased TGF-β | |||
| Stromelysin-2 | MMP-10 | MMP-1,8,10 | Activation | |
| Stromelysin-3 | MMP-11 | IGFBP-1 | Increased IGF | |
| Matrilysin | MMP-7 | Decorin | Increased TGF-β | |
| Plasminogen | Angiostatin, | |||
| MMP-2,7 | Activation | |||
| TNF-α | Activation | |||
| RANKL | sRankl | |||
| Metalloelastase | MMP-12 | Plasminogen | Angiostatin | |
| Gelatinases | ||||
| Gelatinase A | MMP-2 | Decorin | Increased TGF-β | |
| TGF-β2 | Activation | |||
| MMP-1,2,13, | Activation | |||
| TNF-α | Activation | |||
| IL-1β | Activation | |||
| Gelatinase B | MMP-9 | TGF-β2 | Activation | |
| IL-1β | Activation | |||
| TNF-α | Activation | |||
| Plasminogen | Angiostatin | |||
| Collagenases | ||||
| Collagenase-1 | MMP-1 | Perlecan | Increased FGF | |
| MMP-1,2 | Activation | |||
| TNF-α | Activation | |||
| α1-antichymotrypsin | Inactivation | |||
| Collagenase-2 | MMP-8 | MMP-8 | Activation | |
| Collagenase-3 | MMP-13 | MMP-9,13 | Activation | |
| α1-antichymotrypsin | Inactivation | |||
| Membrane-type | ||||
| MT1-MMP | MMP-14 | MMP-2,13 | Activation | |
| Cell surface CD44 | Release | |||
| Cell surface tTG | Release | |||
| MT2-MMP | MMP-15 | Cell surface tTG | Release | |
| MT3-MMP | MMP-16 | MMP-2 | Activation | |
| Cell surface tTG | Release | |||
| MT4-MMP | MMP-17 | MMP-2 | Activation | |
| MT5-MMP | MMP-21 | MMP-2 | Activation | |
| Unclassified | ||||
| Enamelysin | MMP-20 | |||
| MMP-19 | ||||
| MMP-23 | ||||
| MMP-24 | ||||
| Reviewed in 13, 14, 23 |
A naturally occurring class of proteins that inhibit the MMPs has been recognized and is known as the tissue inhibitors of metalloproteinases (TIMPs). In addition, α2-macroglobulin is capable of inhibiting the MMPs [11]. As their name implies, the TIMPs are primarily responsible for inhibition of MMPs in various tissues while α2-macroglobulin is responsible for inhibition in body fluids. α2-macroglobulin inhibits the MMPs by binding to and trapping the MMP, which triggers their rapid clearance via receptor-mediated endocytosis through the low density lipoproteins receptor-related protein-1 [15]. The balance between naturally occurring inhibitors and MMPs in the tumor microenvironment modulates the malignant phenotype. While a myriad of roles for the various MMPs in both physiologic and pathologic conditions have been elucidated, this review will focus on their role in the progression of cancer, with particular attention to the tumor microenvironment.
Cathepsins
Cathepsins are the other important class of proteases in the complex interplay between tumor and stroma. These proteases are less unified than the MMPs and are divided into the aspartic, cysteine, and serine cathepsins. The majority of cathepsins are lysosomal proteases, but some are secreted or membrane-associated. Of these, the cysteine cathepsins are the best characterized [16]. There are 11 human cysteine cathepsins (Table 2), some of which are constitutively expressed in all cells (e.g. Cathepsin B) and some in only certain cell types (e.g. Cathepsin K). Several of these proteases, including Cathepsin K, are secreted and the substrate of each of these proteases varies greatly depending on the location, particularly lysosomal versus extracellular [16]. Like MMPs, naturally occurring inhibitors of cysteine cathepsins exist [17]. These inhibitors include Cystatins A, B, and C. Akin to the MMPs, a multitude of roles in both physiologic and pathologic conditions has been ascribed to the cathepsins. This review will discuss the role of cathepsins in the tumor microenvironment.
Table 2.
Cysteine Cathepsins
| NAME | ALTERNATE NAME | CELLULAR EXPRESSION |
|---|---|---|
| Cathepsin B | Tumor cells, Tumor-associated macrophages, Fibroblasts, Osteoclasts, Neutrophils, Endothelial Cells, Mast cells | |
| Cathepsin C | Dipeptidyl peptidase | Tumor-associated macrophages, Fibroblasts, Neutrophils, Mast cells, T cells |
| Cathepsin F | Tumor cells, Myoepithelial cells | |
| Cathepsin H | Tumor cells | |
| Cathepsin L | Tumor cells, Tumor-associated macrophages, Fibroblasts, Endothelial cells, Myoepithelial cells | |
| Cathepsin K | Tumor cells, Tumor-associated macrophages, Fibroblasts, Osteoclasts, Myoepithelial cells | |
| Cathepsin O | ||
| Cathepsin S | Tumor cells, Tumor-associated macrophages, Mast cells, Endothelial cells | |
| Cathepsin V | Cathepsin L2, Cathepsin U | Tumor cells, Tumor-associated macrophages |
| Cathepsin W | T cells | |
| Cathepsin X | Cathepsin Y | Tumor cells, Tumor-associated macrophages |
| Reviewed in 16 |
Modulation of Tumor-Stromal Interaction
Microenvironment of Primary Breast and Prostate Cancers
Proteases have been shown to be involved in tumor progression as early as conversion to a premalignant phenotype, for example, MMP-3 has been implicated during this very early step. MMP-3 expression results in changes in the microenvironment of mammary epithelial cells that facilitates conversion to a premalignant phenotype as well as epithelial-to-mesenchymal transformation [18]. MMP-3 mediates these changes in part by cleaving E-cadherin, which results in its loss from cell to cell contacts and subsequent relocalization of β-catenin (Figure 1). This triggers the release of keratinocyte growth factor, which is expressed by mesenchymal cells under normal physiological conditions [18].
Figure 1. Modification of primary tumor microenvironment by proteases.
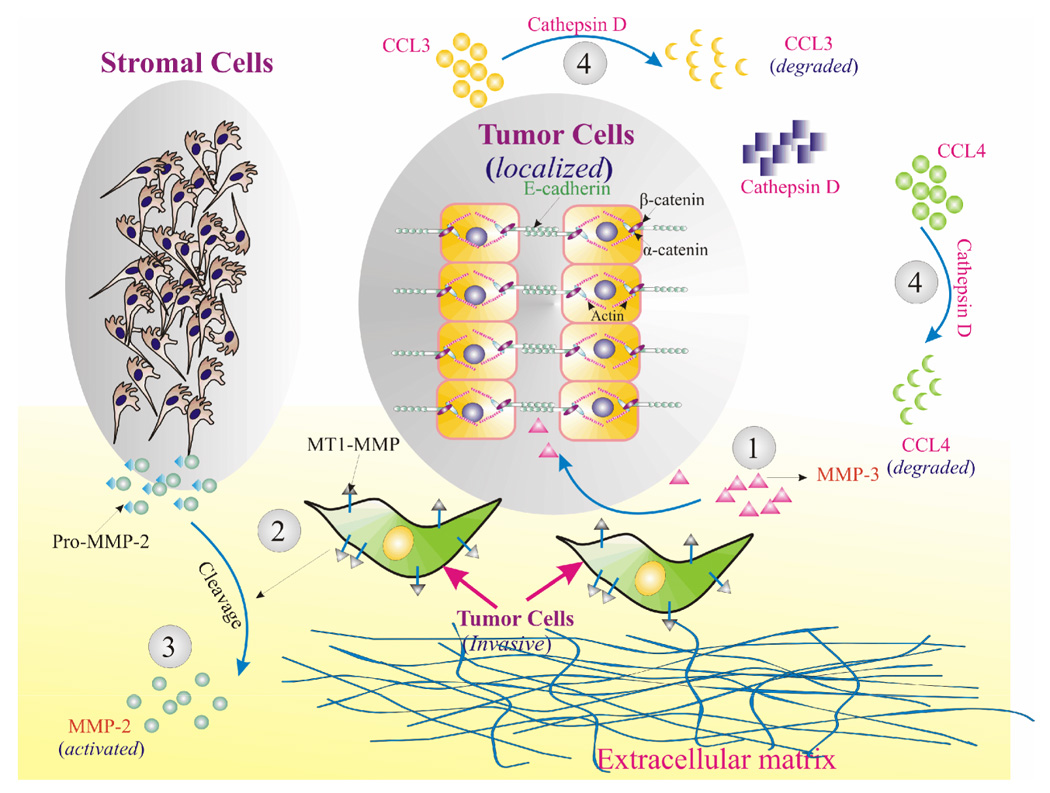
1) MMP-3 production alters the microenvironment to favor conversion to a premalignant phenotype. MMP-3 cleaves E-cadherin which disrupts cell-cell junctions and releases β-catenin to translocate to the nucleus where it can upregulate mesenchymal genes such as keratinocyte growth factor [18]. 2) As tumor cells become invasive and form invadopodia, MT1-MMP accumulates at the surface of the invadopodia. MT1-MMP can then activate pro-MMP-2 produced by stromal cells. 3) Active MMP-2 is concentrated in this way near the invadopodia and can cleave the extracellular matrix, clearing a path for the invading tumor cells [26]. 4) Stromal cells also produce chemokines in response to aberrations such as tumor cell growth. These chemokines include CCL3 and CCL4 and are capable of generating an immune response. Tumor cells produce Cathepsin D which can cleave both CCL3 and CCL4, allowing evasion of immune surveillance [34].
The protease-activated receptors (PARs) have also been implicated in oncogenic transformation and invasion. Activation of these receptors leads to downstream signaling events via intracellular G proteins, ultimately resulting in cell adhesion, cell migration, and mitogenesis. Signaling via these receptors occurs by means of proteolytic cleavage which exposes a moiety capable of binding to the body of the receptor [19] Prostate cancer cells have been shown to express PAR-1at a higher level compared to normal prostate tissue [20]. MMP-1 has been shown to cleave PAR-1 and activate the signaling cascade [21]. This suggests that fibroblasts may produce pro-MMP-1 which is subsequently activated by tumor-derived soluble or membrane-bound MMPs (Figure 2A), however, the production of MMP-1 may not be limited solely to the fibroblasts. Active MMP-1 can then activate PAR-1 promoting mitogenesis as well as serve as a chemoattractant driving the cells towards the source of the MMP-1 [21]. Moreover, MMP-1 may also be involved in degradation of the ECM, and may serve to drive cell division, motility, and invasion [21]. Furthermore, tissue factor (TF) and thrombin are often present in the tumor microenvironment due to leaky vasculature within the tumor [22]. Thrombin, a protease, has been shown to activate PAR-1. As a result thrombin in the extracellular milieu can cleave PAR-1 on the surface of tumor cells to activate downstream signaling pathways mediated by it [20]. These downstream signaling events ultimately result in increased MMP expression as well as cell motility. The end result is that various proteases modify the interaction between tumor and stroma via activation of PARs in a manner that favors tumor growth and subsequent invasion.
Figure 2. Tumor-stromal interaction in proteases-dependent migration of malignant cells.

A) Prostate cancer cells express PAR-1 [20]. Tumor-derived MMPs activate pro-MMP-1 that is produced by osteoblasts and stromal cells. Activated MMP-1 is capable of cleaving and activating PAR-1 on prostate cancer cells which results in mitogenesis and drives migration of cancer cells towards the source of MMP-1 [21]. B) MMP-1 derived from breast and prostate cancer cells enters the vasculature where it can activate PAR-1 expressed by endothelial cells [36–40]. Activation of PAR-1 induces expression and release of von Willebrand Factor (vWF), IL-1, and P-selectin [40]. This transforms the vascular microenvironment into one that is prothrombotic and proinflammatory that is favorable to tumor cells.
Under normal conditions, cell growth in both the breast and the prostate is tightly and effectively regulated by the surrounding extracellular matrix which is primarily composed of fibrin and collagen. This is accomplished both by sequestering growth factors [23] as well as activating receptors [24]. However, cancer cells evade these controls and rapidly divide within the tight confines of the extracellular matrix. This is accomplished as proteases modulate the normal growth restricting conditions. Expression of membrane bound MT1-MMP has been shown to confer this growth advantage in both tumorigenic and non-tumorigenic cell lines by alteration of the microenvironment, specifically through cleavage of type I collagen matrix [25]. Cross-linked fibrin and collagen are both capable of suppressing morphologic changes, cytoskeletal rearrangements, and cell growth [25]. MT1-MMP allows cells to escape these controls imposed by fibrin and collagen. To this point, only MT1-MMP has been shown to confer such an advantage. However, MT2-MMP and MT3-MMP are capable of collagenolysis and fibrinolysis suggesting that the growth advantage may be provided by the entire MT-MMP family [25].
As breast and prostate epithelial cells progress towards a malignant phenotype and become invasive, MMPs continue to play a central role. Invadopodia are key structures for tumor cell invasion. These membranous projections help to localize enzymes important for extracellular matrix degradation and invasion. Expression of MT1-MMP in invadopodia is vital. MT1-MMP is central to the activation of other MMPs, both soluble and ECM-bound found in the microenvironment [26]. Similar to other MMPs, MT1-MMP is synthesized in an inactive form. However, it is constitutively activated by a furin-like pathway [27]. The constitutively active MT1-MMP accumulates at the invadopodia of tumor cells and activates pro-MMP-2 produced by stromal cells in the microenvironment (Figure 1) Active MMP-2 then degrades extracellular matrix components, allowing the cells to breach the ECM [26; 28].
Heparan sulfate proteoglycans (HSPGs) are an important constituent in the tumor microenvironment [29–31]. The heparan sulfate degrading enzymes heparanase, hyalorunon and its receptor CD44 are shown to be up-regulated in many tumor types. CD44 is a glycoprotein capable of binding a myriad of molecules including hyaluronic acid, fibronectin, and laminin. It is involved in a wide variety of physiologic and pathologic processes including migration of metastatic tumor cells [32]. It is thought that CD44 expressed on the surface of tumor cells anchors the cells to the surrounding extracellular matrix through interaction with fibronectin, collagen, and other ECM components. As previously noted, when cells become invasive, they begin to form invadopodia where MT1-MMP is concentrated [26; 28]. MT1-MMP is also capable of cleaving CD44 resulting in the shedding of the extracellular portion. MT1-MMP cleavage of CD44 promotes migration of the tumor cells as they are released from their anchors to the ECM [33]. Thus, as the invadopodia degrade the basement membrane, the shedding of CD44 promotes migration through the newly formed opening.
Emerging data indicate that functional regulation of HSPGs occurs at heparin sulfate synthesis and enzymes that selectively cleave HSPG’s components [30]. Physiological shedding of syndecan-1 by MMP-7 or MT1-MMP facilitates syndecan-1 dependent regulation of the inflammatory response, which may alter the tumor microenvironment. Moreover, shed syndecan-1 is also important in tumor growth and progression [30].
The role of cathepsins in tumor-stromal interaction is limited to cysteine cathepsins (Table 2). Role of specific cysteine cathepsins in cancer has been shown in different animal studies [16]. These studies demonstrate that both tumor cells and tumor associated stromal cells such as macrophages and endothelial cells express these proteases. The functions of these proteases vary depending on their cellular location and levels of expression. The level and patterns of expression of cysteine cathepsins (by tumor cells and stromal cells) changes during the progression. The precise roles of cysteine cathepsins in cancer are not well defined.
Cathepsin D expression in breast cancer signifies a poor prognosis. Cathepsin D potentially modifies the microenvironment and tumor-stromal interactions in a way that affects immune response and migration of cancer cells [34]. Cathepsin D has been shown to cleave and inactivate macrophage inflammatory protein (MIP)-1α (CCL3), MIP-1β (CCL4), and SLC (CCL21) [34]. CCL3 and CCL4 are typically expressed by the stroma of breast cancers as demonstrated by in situ hybridization, while CCL21 is typically expressed by endothelial cells of capillaries and venules within the tumor tissue [34]. These chemokines are potentially capable of generating an immune response to the newly formed tumor. By degrading chemokines, Cathepsin D modifies the relationship of the tumor to the stroma in a manner that allows tumor cells to escape the immune system and thrive unchecked (Figure 1) [34].
As tumor cells interact with the microenvironment and acquire the ability to invade, through ECM degradation and motility acquisition, the next step in breast and prostate cancer metastasis is intravasation into the blood vessels [4]. MMPs also appear to be central to this process. Data suggest that interaction between urokinase plasminogen activator receptors (uPARs) and MMPs is necessary for intravasation. Marimastat, an inhibitor of MMPs, effectively reduced and nearly eliminated intravasation of tumor cells in one model. However, cells expressing high levels of MMPs were unable to intravasate unless they co-expressed uPARs [35]. Specifically, MMP-9 is thought to be involved in this process [35]. Exactly how MMPs and uPARs interact in order to promote intravasation is unknown.
Finally, as breast and prostate cancer cells intravasate into the circulation, they rely on the body’s network of blood and lymphatic vessels to disseminate to distant sites. Subsequent to intravasation, tumor cells interact with endothelial cells to alter the vascular environment in order to evade the immune system and later in order to extravasate at distant sites. This dialogue between tumor and endothelial cells remodels the intravascular compartment to favor thrombosis, inflammation, cell adhesion, and endothelial activation (Figure 2B). PARs are abundantly expressed by endothelial cells and play a central role in this remodeling [36; 37]. Importantly, an environment that favors thrombosis and inflammation facilitates tumor progression [38; 39]. Tumor-produced MMP-1 released into the vasculature has been shown to activate endothelial PAR-1 [40]. PAR-1 activation on endothelial cells triggers the release of proteins such as von Willebrand Factor, Interleukin (IL)-8, and P-selectin, and ultimately generates a prothrombotic and proinflammatory environment that favors tumor growth and progression [40].
Microenvironment of Bone Metastases
Indirect evidence suggests that expression of MMP-11/stromelysin-3 may be involved in creating new niches to permit the growth of malignant cells to form distant secondary colonies. NIH 3T3 fibroblasts which express MMP-11 show a decreased tumor-free period with respect to development of subcutaneous tumors as compared to similar cells expressing anti-sense MMP-11 RNA [41]. Similarly, MCF7 epithelial cells, which do not express endogenous MMP-11, showed a decreased tumor-free period upon overexpression of MMP-11. Interestingly, however, once established, tumors composed of cells expressing MMP-11 did not grow faster or show histologic disparity compared to those not expressing it [41]. Overall, evidence to date points out that MMP-11 may play a role in preparing the microenvironment for tumor establishment. This may be one example of how proteases are involved in the process of creating the “premetastatic niche” [42; 43]. Chemokines, either secreted from primary tumors or released from the stroma following protease-dependent ECM degradation, are likely responsible for dictating the pattern of metastatic spread and proteases have been shown to modify the activity of chemokines. These chemokines appear to drive the influx of bone-marrow derived, VEGFR1+ hematopoietic cells (HPCs) into specific secondary organs where they can establish a premetastatic niche [42; 43]. Abrogating the influx of these cells suppresses metastasis, suggesting preparation of secondary sites is necessary in order for metastasis to occur. This exciting finding opens up new avenues for diagnosis as well as for therapeutic intervention targeting the tumor-derived chemokines that trigger the influx of these cells as well as the HPC-derived factors that create a microenvironment hospitable for metastases.
Once tumors are established at the secondary sites, MMPs have also been shown to confer a growth advantage as well as increase ECM destruction through interaction with the microenvironment. One metastatic niche where this has been clearly shown is the bone, which is a very common site of metastasis particularly in breast and prostate cancers. Indirect evidence that MMPs play an integral role in bone metastasis comes from therapeutic studies. The bisphosphonate class of drugs has become a mainstay in the treatment of bone metastasis as well as in other bone degrading disorders [44–49]. However, when MDA-MB-231, an estrogen independent breast cancer cells, are injected into the left ventricle of mice and allowed to form bone metastases, expression of TIMP-2 in addition to bisphosphonate treatment markedly reduces the number of osteolytic lesions and increases the overall survival compared to treatment with bisphosphonates alone. The additional benefit provided by TIMP-2 suggests that MMPs play a role in bone metastasis, likely in both the establishment of bone metastases and the subsequent osteolysis [50].
Additional indirect evidence for the importance of proteases in modifying the bone microenvironment comes from reports on maspin expression in the bone microenvironment. Maspin is a serine protease inhibitor that has been shown to be down-regulated in breast cancer cells and that in vitro is capable of inhibiting mammary tumor cell invasion and motility [51–54]. Maspin has also been shown to reduce tumor growth, osteolysis, and angiogenesis in a prostate cancer bone metastasis model. This may be due to inhibition of urokinase-type plasminogen activator (uPA) [55]. However, it provides more indirect evidence for the role of proteases in remodeling the bone microenvironment to create a hospitable environment for tumor cells.
More direct evidence has shown that both MMP-9 and MMP-7 contribute to osteolytic metastatic lesions. These studies utilized a prostate cancer cell line, PC3, which establishes predominantly osteolytic bone metastases and thus is more similar to typical mammary tumor-induced bone lesions than typical prostate cancer-induced bone lesions. As PC3 prostate cancer cells colonize the bone, there is an observed increase in MMP-9 enzymatic activity. Peak MMP-9 activity correlated with osteoclast numbers and primary cellular sources of MMP-9 in this model were the host osteoclasts [56]. In addition, MMP activity, likely MMP-9, is required to recruit osteoclasts at metastatic sites, as treatment with MMP inhibitors or antisense oligonucleotides directed at MMP-9 abrogated osteoclast recruitment [57; 58]. PC3 cells can 13 induce in vitro preosteoclast motility and expression of pro-MMP-9, and the motility is diminished when MMP-9 is silenced by small interfering RNA (siRNA) which corroborates MMP-9’s role in osteoclast recruitment [56].
As osteoclasts are recruited to the metastatic sites, a complex network of tumor-stromal interactions is initiated (Figure 3) [3]. Tumor cells produce factors such as parathyroid hormone related peptide (PTHrP), IL-8, and IL-1. This activates osteoblasts to express receptor activator of NF-κB ligand (RANKL) which activates the newly recruited osteoclasts. In addition, IL-8 that is produced can directly activate osteoclasts in a RANKL-independent manner [59; 60]. The activated osteoclasts then degrade the bone matrix which releases sequestered growth factors that further stimulate the tumor cells. RANKL-dependent activation of osteoclasts requires cell to cell contact between RANKL-expressing osteoblasts and RANK-expressing osteoclasts which limits osteoclast activation and bone resorption. However, MMP-7 has been shown to be capable of cleaving RANKL into a soluble form (sRANKL) that retains functionality [61]. The generation of sRANKL frees the osteoclasts from the requirement of cell to cell contact and allows increased generation of activated osteoclasts [61]. The IL-1 produced, which contributes to the initiation of the vicious cycle by triggering osteoblasts to express RANKL, also appears to be important in inducing the release of proteases that can subsequently modify the microenvironment to favor metastatic colonization [62]. Specifically, IL-1 has been shown to induce MMP-13 production in chondrocytes via a mechanism that involves p38 mitogen-activated protein kinase (MAPK), Jun N-terminal kinase (JNK), and NF-κB [62]. High levels of MMP-13 production can contribute to the vicious cycle but may also be important in steps such as the generation of soluble RANKL.
Figure 3. Role of proteases during tumor-stromal interaction in bone metastasis.

Tumor cells entering the bone microenvironment trigger exploits the normal homeostatic process of bone remodeling for their growth and establishment as metastases. Tumor cells produce MMP-9 which promotes osteoclast recruitment [55] and factors such as PTHrP that promote osteoblast activation and RANKL expression [58] sRANKL can be cleaved off the surface of osteoblasts by MMP-7 [58]. RANKL/sRANKL activates osteoclasts to resorb bone which releases sequestered growth factors. Latent TGF-β can be activated by MMP-9 and MMP-2 and can then promote tumor growth, serve as a chemoattractant for tumor cells, and increase PTHrP expression in tumor cells [3; 63; 66]. IGF-I and IGF-II also promote attraction of tumor cells [63; 64]. With further tumor cell influx and growth, the production of factors such as PTHrP and IL-1 are increased, which leads to increased osteoclast activation and osteolytic metastases. On the other hand, growth factors released following bone resorption, such as TGF-β upregulate the production of endothelin-1 by tumor cells. Endothelin-1 enhances tumor growth and proliferation of osteoblasts leading to new bone formation during osteoblastic metastasis.
Some of the factors released include basic fibroblast growth factor (bFGF), insulin-like growth factor (IGF)-I, IGF-II, epidermal growth factor (EGF), transforming growth factor (TGF)-α, and TGF-β[61]. Each of these has been shown to be a mitogen for prostate cancer cells except for TGF-β which shows both growth inhibiting and promoting effects depending on the phenotype of the prostate cancer cells [63]. As a result, proteases release these sequestered factors that allow prostate cancer cells to thrive in the bone microenvironment. In addition, a number of these growth factors including TGF-β, IGF-I, and IGF-II as well as collagen type I peptides serve as chemoattractants to further attract prostate cancer cells to the bone [64]. Similarly, IGF-I has been shown to serve as chemoattractant for breast cancer cells [65]. In fact, MDA-MB-231 breast cancer cells that overexpress the IGF-I receptor have increased prevalence of bone metastases and conversely expression of dominant negative IGF-I receptors reduces bone metastases [66].
While bone resorption allows release of sequestered TGF-β, the bulk is released in an inactive form due to non-covalent association with latency-associated protein (β-LAP) [67]. While TGF-β in normal tissues promotes chemoattraction of leukocytes, decreases epithelial cell proliferation, and initiates the early stages of tissue healing, it induces a different response in tumor cells. Tumor cells have escaped the growth-restricting conditions of TGF-β, so signaling induced by TGF-β increases angiogenesis, increases the desmoplastic response, and decreases immune surveillance, all favoring malignant growth [67]. TGF-β also significantly contributes to the complex tumor-stromal interaction by increasing production of PTHrP [3; 8]. TGF-β may actually be the most important factor in increasing PTHrP because increased production by MDA-MB-231 breast cancer cells treated with bone resorbing culture supernatants was abrogated by TGF-β blocking antibodies but not by antibodies to IGF-IR, FGF-1, or FGF-2. With introduction of a dominant negative TGF-β type II receptor, a similar result was observed [68]. Thus, TGF-β activation represents a critical link between the vicious cycle and the establishment of bone metastases. MMP-9 and MMP-2 have been shown to be capable of cleaving latent TGF-β and activating it [67]. In support of the idea that TGF-β activated by MMPs is key to continuation of the vicious cycle is the finding that MMP inhibition disrupts bone matrix turnover and tumor growth [57]. The release of sequestered TGF-â with subsequent activation thus represents a potentially vital mechanism for enhanced tumor growth.
Cathepsin K is one of the most abundant proteases produced by osteoclasts in breast cancer-induced osteolytic lesions. Cathepsin K is a key player in the bone-resorbing arsenal of osteoclasts [69]. However, it is also important in the differentiation of osteoclasts. Treatment with anti-sense oligonucleotides to Cathepsin K reduced the number of osteoclasts as shown by a reduction in TRAP-positive cells and percentage of multinuclear cells [69]. As a result, Cathepsin K plays both a direct role in bone resorption as well as in modifying the extracellular milieu to favor osteoclast differentiation which aids in metastatic establishment.
The endothelin axis appears to be intimately tied to osteoblastic metastases and is also tied to the activity of proteases. Osteoblasts express both the endothelin-A (ETA) and ETB receptors [70–72]. Stimulation of these receptors by endothelin-1 (ET-1) triggers mitogenesis of osteoblasts. Cancers that typically cause osteoblastic metastases, including most types of prostate cancer and some types of breast cancer, express both ET-1 and high levels of ETA (but often low levels of ETB) [72]. Thus, tumor-produced ET-1 may stimulate osteoblasts to divide yielding osteoblastic metastases but also may provide autocrine effects supporting growth and survival of tumor cells. In addition, growth factors synthesized by the increased numbers of osteoblasts become sequestered in the newly synthesized bone matrix and also become rich in the local microenvironment which promotes further tumor growth and survival. The osteoblastic nature of the metastases is further supported by inhibition of osteoclastic bone resorption by ET-1 [73].
Proteases appear to be important in supporting the endothelin axis. Several tumor-derived factors seem to support the endothelin axis and promote new bone formation including members of the IGF family and TGF-β [72]. However, both TGF-β and the members of the IGF family are often found in a latent form. The IGFs remain latent due to binding with inhibitory binding proteins (IGFBPs). Thus, the release from these binding proteins is necessary for the IGFs to promote new bone synthesis. Cathepsin D is capable of hydrolyzing IGFBPs, allowing the release of IGFs and subsequent downstream signaling [74]. Urokinase-type plasminogen activator (uPA) is similarly capable of hydrolyzing IGFBPs [75; 76]. The production of these proteases by tumor cells thus represents a potential mechanism by which support of the endothelin axis can occur. Latent TGF-β is also capable of being activated by a variety of proteases including both MMP-2 and MMP-9 [67]. Activation of TGF-β supports the endothelin axis and the subsequent osteoblastic response in two ways. First, active TGF-β stimulates increased production of ET-1 by tumor cells [77], and second, it stimulates osteoblastic activity adding to the osteoblastic stimulation by ET-1 [72]. Thus, proteases are intimately linked to the endothelin axis which is likely one of the most important signaling pathways in osteoblastic metastases.
Conclusion
Oncogenic transformation and metastatic establishment are orchestrated by complex interactions between tumor cells and the local stromal microenvironment. The microenvironment is not static, however. Proteases seem to be key players in modifying tumor-stromal interactions in such a way that favors oncogenic transformation and metastatic colonization. The MMPs and cathepsins appear to be two major classes of proteases involved in this alteration. As a result, in addition to the direct effects of proteases such as degradation of extracellular matrix allowing tumor invasiveness, we need to investigate the variety of roles proteases play in modifying the complex interplay between tumor and stroma. There is emerging data to suggest that proteases target a variety of substrates which might be critical for tumor-promoting or tumor-suppressive effects (e.g. anti-tumor immune responses) [78]. Moreover, the identification of proteases that modify host responses to the tumor will allow us to develop broad range or specific protease inhibitors targeted to relevant proteases. Furthermore, the major shortcoming of many of the preclinical studies is the use of immunocompromised mice for xenogenic transplantation of human tumors. Validation of theses studies using syngeneic pre-clinical studies in immuocompetent mice is necessary to confirm the role of proteases in modification of the tumor microenvironment as well as to decipher any role proteases or other mediators may play in aiding tumors in avoiding immune surveillance. Further investigation will likely reveal novel functions of known proteases in the tumor microenvironment worthy of therapeutic consideration.
Acknowledgements
We thank Michelle Varney, Drs. Joseph Vetro, Joyce Solheim and Parmender Mehta for careful review and critique of this manuscript. A special thanks to Dr. Parmender Mehta for his help in creating graphics for the figures. This work was supported in part by grants CA72781 from the national Institutes of Health (RKS), Nebraska Department of Health and Human Services, Nebraska Research initiative (RKS) and the Howard Hughes Medical Institute Research Training Fellowship (TJW).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007 CA. Cancer J.Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Bubendorf L, Schopfer A, Wagner U, Sauter G, Moch H, Willi N, Gasser TC, Mihatsch MJ. Metastatic patterns of prostate cancer: an autopsy study of 1,589 patients. Hum.Pathol. 2000;31:578–583. doi: 10.1053/hp.2000.6698. [DOI] [PubMed] [Google Scholar]
- 3.Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat.Rev.Cancer. 2002;2:584–593. doi: 10.1038/nrc867. [DOI] [PubMed] [Google Scholar]
- 4.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 5.Goltzman D, Karaplis AC, Kremer R, Rabbani SA. Molecular basis of the spectrum of skeletal complications of neoplasia. Cancer. 2000;88:2903–2908. doi: 10.1002/1097-0142(20000615)88:12+<2903::aid-cncr4>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 6.Roudier MP, Corey E, True LD, Hiagno CS, Ott SM, Vessell RL. Histological, immunophenotypic and histomorphometric characterization of prostate cancer bone metastases. Cancer Treat.Res. 2004;118:311–339. doi: 10.1007/978-1-4419-9129-4_13. [DOI] [PubMed] [Google Scholar]
- 7.Shimazaki J, Higa T, Akimoto S, Masai M, Isaka S. Clinical course of bone metastasis from prostatic cancer following endocrine therapy: examination with bone x-ray. Adv.Exp.Med.Biol. 1992;324:269–275. doi: 10.1007/978-1-4615-3398-6_29. [DOI] [PubMed] [Google Scholar]
- 8.Roodman GD. Mechanisms of bone metastasis. N.Engl.J.Med. 2004;350:1655–1664. doi: 10.1056/NEJMra030831. [DOI] [PubMed] [Google Scholar]
- 9.Birkedal-Hansen H. Catabolism and turnover of collagens: collagenases. Methods Enzymol. 1987;144:140–171. doi: 10.1016/0076-6879(87)44177-3. [DOI] [PubMed] [Google Scholar]
- 10.Van Wart HE, Birkedal-Hansen H. The cysteine switch: a principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc.Natl.Acad.Sci.U.S.A. 1990;87:5578–5582. doi: 10.1073/pnas.87.14.5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCawley LJ, Matrisian LM. Matrix metalloproteinases: they're not just for matrix anymore! Curr.Opin.Cell Biol. 2001;13:534–540. doi: 10.1016/s0955-0674(00)00248-9. [DOI] [PubMed] [Google Scholar]
- 12.Nelson AR, Fingleton B, Rothenberg ML, Matrisian LM. Matrix metalloproteinases: biologic activity and clinical implications. J.Clin.Oncol. 2000;18:1135–1149. doi: 10.1200/JCO.2000.18.5.1135. [DOI] [PubMed] [Google Scholar]
- 13.Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs Cardiovasc. Res. 2006;69:562–573. doi: 10.1016/j.cardiores.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 14.Woessner JF, Jr., Nagase H. Anonymous. New York: Oxford University Press; 2000. Matrix Metalloproteinases and TIMPs (ed. 1) [Google Scholar]
- 15.Strickland DK, Ashcom JD, Williams S, Burgess WH, Migliorini M, Argraves WS. Sequence identity between the alpha 2-macroglobulin receptor and low density lipoprotein receptor-related protein suggests that this molecule is a multifunctional receptor. J.Biol.Chem. 1990;265:17401–17404. [PubMed] [Google Scholar]
- 16.Mohamed MM, Sloane BF. Cysteine cathepsins: multifunctional enzymes in cancer. Nat.Rev.Cancer. 2006;6:764–775. doi: 10.1038/nrc1949. [DOI] [PubMed] [Google Scholar]
- 17.Nomura T, Katunuma N. Involvement of cathepsins in the invasion, metastasis and proliferation of cancer cells. J.Med.Invest. 2005;52:1–9. doi: 10.2152/jmi.52.1. [DOI] [PubMed] [Google Scholar]
- 18.Lochter A, Galosy S, Muschler J, Freedman N, Werb Z, Bissell MJ. Matrix metalloproteinase stromelysin-1 triggers a cascade of molecular alterations that leads to stable epithelial-to-mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. J.Cell Biol. 1997;139:1861–1872. doi: 10.1083/jcb.139.7.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seeley S, Covic L, Jacques SL, Sudmeier J, Baleja JD, Kuliopulos A. Structural basis for thrombin activation of a protease-activated receptor: inhibition of intramolecular liganding. Chem.Biol. 2003;10:1033–1041. doi: 10.1016/j.chembiol.2003.10.014. [DOI] [PubMed] [Google Scholar]
- 20.Cooper CR, Chay CH, Gendernalik JD, Lee HL, Bhatia J, Taichman RS, McCauley LK, Keller ET, Pienta KJ. Stromal factors involved in prostate carcinoma metastasis to bone. Cancer. 2003;97:739–747. doi: 10.1002/cncr.11181. [DOI] [PubMed] [Google Scholar]
- 21.Boire A, Covic L, Agarwal A, Jacques S, Sherifi S, Kuliopulos A. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell. 2005;120:303–313. doi: 10.1016/j.cell.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 22.Monsky WL, Mouta CC, Tsuzuki Y, Gohongi T, Fukumura D, Jain RK. Role of host microenvironment in angiogenesis and microvascular functions in human breast cancer xenografts: mammary fat pad versus cranial tumors. Clin.Cancer Res. 2002;8:1008–1013. [PubMed] [Google Scholar]
- 23.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat.Rev.Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 24.Cukierman E, Pankov R, Stevens DR, Yamada KM. Taking cell-matrix adhesions to the third dimension. Science. 2001;294:1708–1712. doi: 10.1126/science.1064829. [DOI] [PubMed] [Google Scholar]
- 25.Hotary KB, Allen ED, Brooks PC, Datta NS, Long MW, Weiss SJ. Membrane type I matrix metalloproteinase usurps tumor growth control imposed by the three-dimensional extracellular matrix. Cell. 2003;114:33–45. doi: 10.1016/s0092-8674(03)00513-0. [DOI] [PubMed] [Google Scholar]
- 26.Nakahara H, Howard L, Thompson EW, Sato H, Seiki M, Yeh Y, Chen WT. Transmembrane/cytoplasmic domain-mediated membrane type 1-matrix metalloprotease docking to invadopodia is required for cell invasion. Proc.Natl.Acad.Sci.U.S.A. 1997;94:7959–7964. doi: 10.1073/pnas.94.15.7959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Remacle AG, Rozanov DV, Fugere M, Day R, Strongin AY. Furin regulates the intracellular activation and the uptake rate of cell surface-associated MT1-MMP. Oncogene. 2006;25:5648–5655. doi: 10.1038/sj.onc.1209572. [DOI] [PubMed] [Google Scholar]
- 28.Artym VV, Zhang Y, Seillier-Moiseiwitsch F, Yamada KM, Mueller SC. Dynamic interactions of cortactin and membrane type 1 matrix metalloproteinase at invadopodia: defining the stages of invadopodia formation and function. Cancer Res. 2006;66:3034–3043. doi: 10.1158/0008-5472.CAN-05-2177. [DOI] [PubMed] [Google Scholar]
- 29.Gotte M, Yip GW. Heparanase, hyaluronan, and CD44 in cancers: a breast carcinoma perspective. Cancer Res. 2006;66:10233–10237. doi: 10.1158/0008-5472.CAN-06-1464. [DOI] [PubMed] [Google Scholar]
- 30.Sanderson RD, Yang Y, Kelly T, MacLeod V, Dai Y, Theus A. Enzymatic remodeling of heparan sulfate proteoglycans within the tumor microenvironment: growth regulation and the prospect of new cancer therapies. J.Cell Biochem. 2005;96:897–905. doi: 10.1002/jcb.20602. [DOI] [PubMed] [Google Scholar]
- 31.Sanderson RD, Yang Y, Suva LJ, Kelly T. Heparan sulfate proteoglycans and heparanase--partners in osteolytic tumor growth and metastasis. Matrix Biol. 2004;23:341–352. doi: 10.1016/j.matbio.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 32.Naor D, Sionov RV, Ish-Shalom D. CD44: structure, function, and association with the malignant process. Adv.Cancer Res. 1997;71:241–319. doi: 10.1016/s0065-230x(08)60101-3. [DOI] [PubMed] [Google Scholar]
- 33.Kajita M, Itoh Y, Chiba T, Mori H, Okada A, Kinoh H, Seiki M. Membrane-type 1 matrix metalloproteinase cleaves CD44 and promotes cell migration. J.Cell Biol. 2001;153:893–904. doi: 10.1083/jcb.153.5.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wolf M, Clark-Lewis I, Buri C, Langen H, Lis M, Mazzucchelli L, Cathepsin D. specifically cleaves the chemokines macrophage inflammatory protein-1 alpha, macrophage inflammatory protein-1 beta, and SLC that are expressed in human breast cancer. Am.J.Pathol. 2003;162:1183–1190. doi: 10.1016/s0002-9440(10)63914-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim J, Yu W, Kovalski K, Ossowski L. Requirement for specific proteases in cancer cell intravasation as revealed by a novel semiquantitative PCR-based assay. Cell. 1998;94:353–362. doi: 10.1016/s0092-8674(00)81478-6. [DOI] [PubMed] [Google Scholar]
- 36.Steinhoff M, Buddenkotte J, Shpacovitch V, Rattenholl A, Moormann C, Vergnolle N, Luger TA, Hollenberg MD. Proteinase-activated receptors: transducers of proteinase-mediated signaling in inflammation and immune response. Endocr.Rev. 2005;26:1–43. doi: 10.1210/er.2003-0025. [DOI] [PubMed] [Google Scholar]
- 37.Even-Ram S, Uziely B, Cohen P, Grisaru-Granovsky S, Maoz M, Ginzburg Y, Reich R, Vlodavsky I, Bar-Shavit R. Thrombin receptor overexpression in malignant and physiological invasion processes. Nat.Med. 1998;4:909–914. doi: 10.1038/nm0898-909. [DOI] [PubMed] [Google Scholar]
- 38.Camerer E, Qazi AA, Duong DN, Cornelissen I, Advincula R, Coughlin SR. Platelets, protease-activated receptors, and fibrinogen in hematogenous metastasis. Blood. 2004;104:397–401. doi: 10.1182/blood-2004-02-0434. [DOI] [PubMed] [Google Scholar]
- 39.Im JH, Fu W, Wang H, Bhatia SK, Hammer DA, Kowalska MA, Muschel RJ. Coagulation facilitates tumor cell spreading in the pulmonary vasculature during early metastatic colony formation. Cancer Res. 2004;64:8613–8619. doi: 10.1158/0008-5472.CAN-04-2078. [DOI] [PubMed] [Google Scholar]
- 40.Goerge T, Barg A, Schnaeker EM, Poppelmann B, Shpacovitch V, Rattenholl A, Maaser C, Luger TA, Steinhoff M, Schneider SW. Tumor-derived matrix metalloproteinase-1 targets endothelial proteinase-activated receptor 1 promoting endothelial cell activation. Cancer Res. 2006;66:7766–7774. doi: 10.1158/0008-5472.CAN-05-3897. [DOI] [PubMed] [Google Scholar]
- 41.Noel AC, Lefebvre O, Maquoi E, VanHoorde L, Chenard MP, Mareel M, Foidart JM, Basset P, Rio MC. Stromelysin-3 expression promotes tumor take in nude mice. J.Clin.Invest. 1996;97:1924–1930. doi: 10.1172/JCI118624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, Zhu Z, Hicklin D, Wu Y, Port JL, Altorki N, Port ER, Ruggero D, Shmelkov SV, Jensen KK, Rafii S, Lyden D. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820–827. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaplan RN, Psaila B, Lyden D. Bone marrow cells in the 'pre-metastatic niche': within bone and beyond Cancer Metastasis Rev. 2006;25:521–529. doi: 10.1007/s10555-006-9036-9. [DOI] [PubMed] [Google Scholar]
- 44.Zheng Y, Zhou H, Brennan K, Blair JM, Modzelewski JR, Seibel MJ, Dunstan CR. Inhibition of bone resorption, rather than direct cytotoxicity, mediates the anti-tumour actions of ibandronate and osteoprotegerin in a murine model of breast cancer bone metastasis. Bone. 2007;40:471–478. doi: 10.1016/j.bone.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 45.Iguchi H. Molecular mechanism and potential targets for bone metastasis. Gan To Kagaku Ryoho. 2007;34:1–10. [PubMed] [Google Scholar]
- 46.Brown JE, Neville-Webbe H, Coleman RE. The role of bisphosphonates in breast and prostate cancers. Endocr.Relat Cancer. 2004;11:207–224. doi: 10.1677/erc.0.0110207. [DOI] [PubMed] [Google Scholar]
- 47.Varghese S. Matrix metalloproteinases and their inhibitors in bone: an overview of regulation and functions. Front Biosci. 2006;11:2949–2966. doi: 10.2741/2024. 2949–2966. [DOI] [PubMed] [Google Scholar]
- 48.Keller ET. The role of osteoclastic activity in prostate cancer skeletal metastases. Drugs Today (Barc.) 2002;38:91–102. doi: 10.1358/dot.2002.38.2.820105. [DOI] [PubMed] [Google Scholar]
- 49.Teronen O, Konttinen YT, Lindqvist C, Salo T, Ingman T, Lauhio A, Ding Y, Santavirta S, Valleala H, Sorsa T. Inhibition of matrix metalloproteinase-1 by dichloromethylene bisphosphonate (clodronate) Calcif.Tissue Int. 1997;61:59–61. doi: 10.1007/s002239900295. [DOI] [PubMed] [Google Scholar]
- 50.Yoneda T, Sasaki A, Dunstan C, Williams PJ, Bauss F, De Clerck YA, Mundy GR. Inhibition of osteolytic bone metastasis of breast cancer by combined treatment with the bisphosphonate ibandronate and tissue inhibitor of the matrix metalloproteinase-2. J.Clin.Invest. 1997;99:2509–2517. doi: 10.1172/JCI119435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zou Z, Anisowicz A, Hendrix MJ, Thor A, Neveu M, Sheng S, Rafidi K, Seftor E, Sager R. Maspin, a serpin with tumor-suppressing activity in human mammary epithelial cells. Science. 1994;263:526–529. doi: 10.1126/science.8290962. [DOI] [PubMed] [Google Scholar]
- 52.Sheng S, Carey J, Seftor EA, Dias L, Hendrix MJ, Sager R. Maspin acts at the cell membrane to inhibit invasion and motility of mammary and prostatic cancer cells. Proc.Natl.Acad.Sci.U.S.A. 1996;93:11669–11674. doi: 10.1073/pnas.93.21.11669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sheng S, Pemberton PA, Sager R. Production, purification, and characterization of recombinant maspin proteins. J.Biol.Chem. 1994;269:30988–30993. [PubMed] [Google Scholar]
- 54.Biliran H, Jr., Sheng S. Pleiotrophic inhibition of pericellular urokinase-type plasminogen activator system by endogenous tumor suppressive maspin. Cancer Res. 2001;61:8676–8682. [PubMed] [Google Scholar]
- 55.Cher ML, Biliran HR, Jr., Bhagat S, Meng Y, Che M, Lockett J, Abrams J, Fridman R, Zachareas M, Sheng S. Maspin expression inhibits osteolysis, tumor growth, and angiogenesis in a model of prostate cancer bone metastasis. Proc.Natl.Acad.Sci.U.S.A. 2003;100:7847–7852. doi: 10.1073/pnas.1331360100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dong Z, Bonfil RD, Chinni S, Deng X, Trindade Filho JC, Bernardo M, Vaishampayan U, Che M, Sloane BF, Sheng S, Fridman R, Cher ML. Matrix metalloproteinase activity and osteoclasts in experimental prostate cancer bone metastasis tissue. Am.J.Pathol. 2005;166:1173–1186. doi: 10.1016/S0002-9440(10)62337-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nemeth JA, Yousif R, Herzog M, Che M, Upadhyay J, Shekarriz B, Bhagat S, Mullins C, Fridman R, Cher ML. Matrix metalloproteinase activity, bone matrix turnover, and tumor cell proliferation in prostate cancer bone metastasis. J.Natl.Cancer Inst. 2002;94:17–25. doi: 10.1093/jnci/94.1.17. [DOI] [PubMed] [Google Scholar]
- 58.Ishibashi O, Niwa S, Kadoyama K, Inui T. MMP-9 antisense oligodeoxynucleotide exerts an inhibitory effect on osteoclastic bone resorption by suppressing cell migration. Life Sci. 2006;79:1657–1660. doi: 10.1016/j.lfs.2006.05.024. [DOI] [PubMed] [Google Scholar]
- 59.Lu Y, Cai Z, Xiao G, Keller ET, Mizokami A, Yao Z, Roodman GD, Zhang J. Monocyte chemotactic protein-1 mediates prostate cancer-induced bone resorption. Cancer Res. 2007;67:3646–3653. doi: 10.1158/0008-5472.CAN-06-1210. [DOI] [PubMed] [Google Scholar]
- 60.Bendre MS, Margulies AG, Walser B, Akel NS, Bhattacharrya S, Skinner RA, Swain F, Ramani V, Mohammad KS, Wessner LL, Martinez A, Guise TA, Chirgwin JM, Gaddy D, Suva LJ. Tumor-Derived Interleukin-8 Stimulates Osteolysis Independent of the Receptor Activator of Nuclear Factor-{kappa} B Ligand Pathway. Cancer Res. 2005;65:11001–11009. doi: 10.1158/0008-5472.CAN-05-2630. [DOI] [PubMed] [Google Scholar]
- 61.Lynch CC, Hikosaka A, Acuff HB, Martin MD, Kawai N, Singh RK, Vargo-Gogola TC, Begtrup JL, Peterson TE, Fingleton B, Shirai T, Matrisian LM, Futakuchi M. MMP-7 promotes prostate cancer-induced osteolysis via the solubilization of RANKL. Cancer Cell. 2005;7:485–496. doi: 10.1016/j.ccr.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 62.Mengshol JA, Vincenti MP, Brinckerhoff CE. IL-1 induces collagenase-3 (MMP-13) promoter activity in stably transfected chondrocytic cells: requirement for Runx-2 and activation by p38 MAPK and JNK pathways. Nucleic Acids Res. 2001;29:4361–4372. doi: 10.1093/nar/29.21.4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Koeneman KS, Yeung F, Chung LW. Osteomimetic properties of prostate cancer cells: a hypothesis supporting the predilection of prostate cancer metastasis and growth in the bone environment. Prostate. 1999;39:246–261. doi: 10.1002/(sici)1097-0045(19990601)39:4<246::aid-pros5>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 64.Cooper CR, Pienta KJ. Cell adhesion and chemotaxis in prostate cancer metastasis to bone: a minireview. Prostate Cancer Prostatic.Dis. 2000;3:6–12. doi: 10.1038/sj.pcan.4500387. [DOI] [PubMed] [Google Scholar]
- 65.Doerr ME, Jones JI. The roles of integrins and extracellular matrix proteins in the insulin-like growth factor I-stimulated chemotaxis of human breast cancer cells. J.Biol.Chem. 1996;271:2443–2447. doi: 10.1074/jbc.271.5.2443. [DOI] [PubMed] [Google Scholar]
- 66.Yoneda T, Hiraga T. Crosstalk between cancer cells and bone microenvironment in bone metastasis Biochem. Biophys.Res.Commun. 2005;328:679–687. doi: 10.1016/j.bbrc.2004.11.070. [DOI] [PubMed] [Google Scholar]
- 67.Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]
- 68.Yin JJ, Selander K, Chirgwin JM, Dallas M, Grubbs BG, Wieser R, Massague J, Mundy GR, Guise TA. TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J.Clin.Invest. 1999;103:197–206. doi: 10.1172/JCI3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ishikawa T, Kamiyama M, Tani-Ishii N, Suzuki H, Ichikawa Y, Hamaguchi Y, Momiyama N, Shimada H. Inhibition of osteoclast differentiation and bone resorption by cathepsin K antisense oligonucleotides. Mol.Carcinog. 2001;32:84–91. doi: 10.1002/mc.1067. [DOI] [PubMed] [Google Scholar]
- 70.Stern PH, Tatrai A, Semler DE, Lee SK, Lakatos P, Strieleman PJ, Tarjan G, Sanders JL. Endothelin receptors, second messengers, and actions in bone. J.Nutr. 1995;125:2028S–2032S. doi: 10.1093/jn/125.suppl_7.2028S. [DOI] [PubMed] [Google Scholar]
- 71.Takuwa Y, Masaki T, Yamashita K. The effects of the endothelin family peptides on cultured osteoblastic cells from rat calvariae Biochem. Biophys.Res.Commun. 1990;170:998–1005. doi: 10.1016/0006-291x(90)90491-5. [DOI] [PubMed] [Google Scholar]
- 72.Guise TA, Yin JJ, Mohammad KS. Role of endothelin-1 in osteoblastic bone metastases. Cancer. 2003;97:779–784. doi: 10.1002/cncr.11129. [DOI] [PubMed] [Google Scholar]
- 73.Chiao JW, Moonga BS, Yang YM, Kancherla R, Mittelman A, Wu-Wong JR, Ahmed T. Endothelin-1 from prostate cancer cells is enhanced by bone contact which blocks osteoclastic bone resorption. Br.J.Cancer. 2000;83:360–365. doi: 10.1054/bjoc.2000.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Conover CA, Perry JE, Tindall DJ. Endogenous cathepsin D-mediated hydrolysis of insulin-like growth factor-binding proteins in cultured human prostatic carcinoma cells. J.Clin.Endocrinol.Metab. 1995;80:987–993. doi: 10.1210/jcem.80.3.7533776. [DOI] [PubMed] [Google Scholar]
- 75.Koutsilieris M, Frenette G, Lazure C, Lehoux JG, Govindan MV, Polychronakos C. Urokinase-type plasminogen activator: a paracrine factor regulating the bioavailability of IGFs in PA-III cell-induced osteoblastic metastases. Anticancer Res. 1993;13:481–486. [PubMed] [Google Scholar]
- 76.Koutsilieris M, Polychronakos C. Proteinolytic activity against IGF-binding proteins involved in the paracrine interactions between prostate adenocarcinoma cells and osteoblasts. Anticancer Res. 1992;12:905–910. [PubMed] [Google Scholar]
- 77.Le BG, Aubin P, Soliman H, Ropiquet F, Villette JM, Berthon P, Creminon C, Cussenot O, Fiet J. Upregulation of endothelin 1 and its precursor by IL-1 beta, TNF-alpha, and TGF-beta in the PC3 human prostate cancer cell line. Cytokine. 1999;11:157–162. doi: 10.1006/cyto.1998.0407. [DOI] [PubMed] [Google Scholar]
- 78.Lopez-Otin C, Matrisian LM. Emerging roles of proteases in tumour suppression. Nat Rev Cancer. 2007;7:800–808. doi: 10.1038/nrc2228. [DOI] [PubMed] [Google Scholar]