

Abstract
We have investigated mRNA 3′-end-processing signals in each of six eukaryotic species (yeast, rice, arabidopsis, fruitfly, mouse, and human) through the analysis of more than 20,000 3′-expressed sequence tags. The use and conservation of the canonical AAUAAA element vary widely among the six species and are especially weak in plants and yeast. Even in the animal species, the AAUAAA signal does not appear to be as universal as indicated by previous studies. The abundance of single-base variants of AAUAAA correlates with their measured processing efficiencies. As found previously, the plant polyadenylation signals are more similar to those of yeast than to those of animals, with both common content and arrangement of the signal elements. In all species examined, the complete polyadenylation signal appears to consist of an aggregate of multiple elements. In light of these and previous results, we present a broadened concept of 3′-end-processing signals in which no single exact sequence element is universally required for processing. Rather, the total efficiency is a function of all elements and, importantly, an inefficient word in one element can be compensated for by strong words in other elements. These complex patterns indicate that effective tools to identify 3′-end-processing signals will require more than consensus sequence identification.
Knowledge of gene sequences and functions comprises only part of the information necessary to understand biological systems at the molecular level. Equally important are identification and characterization of the regulatory elements that govern the temporal and tissue-specific expression of any individual gene. Discovery of nucleic acid control sequences is difficult, because they are short and often degenerate. Sequence databases, however, can provide means for the statistical identification of prospective signals, given a suitable biological hypothesis for the selection of candidate sequences. We have analyzed polyadenylation control signals in six eukaryotic species by examining 20,842 3′-expressed sequence tags (ESTs). The results should prove useful in designing future experiments to further elucidate the mechanism of mRNA 3′-end-processing.
Control sequences have been commonly investigated through mutagenesis of the untranslated regions in the vicinity of coding sequence on vector constructs or by in vitro footprinting of the binding sites for identified regulatory proteins. Although these methods have successfully characterized many control sites, they are limited in both contextual scope and the number of sequences that can be reasonably investigated. Analysis of control sequences in artificial constructs only partially reproduces conditions in genomic sequence, where signal words may be affected by many factors, such as the use of unrelated but overlapping functional elements (1) and the use of multiple signals of varying strength (2).
The continually expanding sequence databases provide a method to investigate control sequences in silico. Through statistical investigation of multiple sequences known to be associated with a common molecular process, the sequence words most commonly used to regulate that process can be identified in their natural context. Although statistical significance does not necessarily indicate actual biological use, it does provide an informative means to identify potential signals, whose function can subsequently be verified through mutagenesis or other techniques.
Our EST-based analysis of the 3′-end-processing signals in yeast (2) confirmed experimental results and at the same time indicated subtle complexities not previously recognized. We have now extended this analysis to Oryza sativa (rice), Arabidopsis thaliana (arabidopsis), Drosophila melanogaster (fruitfly), Mus musculus (mouse), and Homo sapiens (human). The results not only yield information regarding the control sequences preferred by each species but also provide potential insights into the molecular evolution of the 3′-end-processing mechanism. Studies of protein homology between yeast, mammals, and plants have indicated significant similarities in the subunits of the complexes involved in 3′-end processing (3–6). The present work, however, suggests that the differences also play important roles.
The accurate selection of EST sequences with their 3′-end at the polyadenylate [poly(A)] tail is critical for our analysis. Potential errors or ambiguities include the presence of poly(A) or vector sequence in the database records, mislabeling (inversion of 5′ and 3′) of EST sequence on submission to the databases, ambiguous orientation (sense or antisense) of the archived sequence, hybridization of the oligo(dT) primers to A-rich regions other than the poly(A) tail, and genomic occurrence of the recognition sequences of the restriction endonucleases used for insertion of cDNA into sequencing vectors (2). Poly(A) or vector sequences can be detected relatively easily, and we have done so on the sequences included in this study. As we showed previously, the remaining sources of error can be identified by comparison of EST sequences with complete genomic sequences and associated gene predictions. Because complete sequences are available only for a few organisms, however, we could not use this error correction procedure extensively in the present study. Thus our sets of ESTs likely contain some incorrect sequences. Alignments of the fruitfly ESTs to available contigs (see below) indicate that approximately 25% of those sequences have ambiguous origins.
The 3′-end-processing signal in mammals has been widely investigated [see reviews (7–9)] and has been found to consist of two components: the canonical hexamer AAUAAA, situated 10 to 30 bases upstream of the processing site, and a downstream less sequence-specific U- or UG-rich element. Yeast 3′-end signals [see reviews (8–10)] consist of an upstream efficiency element (optimally UAUAUA), an A-rich positioning element (PE) (optimally AAUAAA), and U-rich regions situated upstream of, downstream of, or flanking a cleavage site (CS). Our study of yeast 3′-end-processing signals showed that the overall efficiency of the signal is a complex function of the individual elements, and specifically that weak control words in one or more elements can be compensated for by stronger words in other elements.
Investigations of plant polyadenylation [see reviews (11, 12)] have indicated that the 3′-end-processing signals resemble those of yeast more than those of mammals. In particular, there appears to be an upstream element (UE) similar to the UAUAUA element of yeast, and the PE has been characterized as A-rich rather than strictly AAUAAA. Also similar to yeast, multiple processing sites for a single transcription unit have been reported in a relatively close-packed arrangement on the order of tens of nucleotides apart. By contrast, studies of alternate polyadenylation in mammals have found competing sites generally separated by hundreds or thousands of nucleotides.
Methods
Data Selection.
3′-ESTs (accession numbers and sequences can be obtained at http://bmerc-www.bu.edu/polyA) were obtained from GenBank. 3′-ESTs are most often generated by reverse transcription initiated by an oligodeoxythymidylate primer that hybridizes to the poly(A) tail found at the 3′-end of mature mRNAs. Sequences are generally processed to remove vector and poly(A) sequence before submission to databases. Therefore, assuming no errors, the 3′-most end of an archived EST sequence corresponds to the site of 3′-end processing (cleavage followed by polyadenylation). We selected sets of ESTs that could be identified with high probability as having originated from the mRNA poly(A) tail. The human, mouse, and fruitfly sequences were inverted, because they had been archived in antisense orientation. All 3′ ends were searched for evidence of either residual poly(A) or vector sequences. Sequences with at least 12 adenines of the final 15 bases were assumed to include poly(A) tails and were truncated to the base preceding the 5′-most adenine. We identified vector sequences by searching for the recognition sites of the restriction enzymes used for insertion of the cDNA into the sequencing vector. If the sequence immediately preceding the recognition site was poly(A), the sequence was truncated and retained for analysis. If the immediately preceding sequence was not poly(A), the sequence was discarded as likely to be a genomic occurrence of the recognition site, which disqualifies it from our analysis.
The sequences were further checked for redundancy (within each organism-specific set) by an intersequence comparison using blast (13), in which every sequence was checked against every other. Any pair of sequences with nucleotide identity greater than 90% of the length of the shorter sequence was assumed to be from the same source, and one was eliminated from the data set. In the human ESTs, 200 additional sequences were eliminated after it was found that they matched an Alu sequence, specifically in such a position as to indicate hybridization of the oligothymidylate primer to a genomic run of 15 adenines.
Sequence Analysis.
Our analysis of the EST sequences consisted of determining the position-dependent occurrence frequencies of single and multiple nucleotides (words), where the position dependence was keyed on the putative 3′-end-processing site. The EST sequences for each organism were initially aligned on the 3′-most end of the sequence. To identify the most common (and presumably strongest) subelements of the putative signals, we aligned the sequences using a 6-nt-wide log-likelihood profile. The profile score at each position was computed as:
 |
1 |
where i is the starting position of the profile in the sequence, qi+j, j is the frequency of the base at i + j occurring at position j in the profile, and pi+j is the frequency of base at position i + j appearing anywhere in the sequence. The ESTs were aligned to the position of maximum profile score, and the profile was recomputed. This process was iterated to approximate convergence on a “best” subelement sequence. We constrained the algorithm not to offset the sequences more than 5 nt in any iteration, thereby restricting the total movement away from the original alignment at the end of the EST sequence. This constraint is especially important in the case of yeast and plants, where the potentially overlapping nature of adjacent signals has been previously discussed (10, 12).
We performed word-based analysis using Z-scores (14).
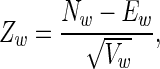 |
2 |
where, for a given word w, Nw is the measured count, and Ew and Vw are estimators of the expected count and variance, respectively. We used Markov h-2 estimators (14) for expected counts and variance, where h is the length of the words examined. Z-scores were computed for the 150 nt upstream of the CS. Examination of the Z-scores revealed approximately normal distributions.
The danger in using Z-scores on the EST data is that, as the total number of words examined decreases, the highest-scoring words can be dominated by extremely unlikely words that appear only a few times. We therefore also investigated the raw counts of each word in each window. Words that score highly both in Z-score and raw count are likely candidates for biological function.
We also measured raw counts and calculated Z-scores in smaller windows within the 3′-end-processing region. The basis of this measurement is our assumption that biologically significant words will occur in a roughly constant position with respect to the processing site. The constant positioning of a specific word is reflected in a correspondingly nonuniform distribution among the counting windows. For windowed Z-scores, the estimators for count and variance from the overall region were scaled for use in the windows, because use of estimates of count and variance from the larger region implicitly assumes a uniform distribution of any specific word across the entire range. The windowed Z-score is then a measure of departure from uniform distribution for any specific word with respect to the CS.
We primarily investigated the occurrence frequencies of 6-nt words, because many previously reported 3′-processing elements have been hexamers. Additionally, the significance of statistical results typically decreases for increasing word sizes in a constant data set.
We searched 48 × 106 nt of fruitfly genomic sequences for alignments of the 3,236 fruitfly EST sequences as follows: to avoid the complication of identification of spliced exons, only the final 60 nt of each EST was aligned to the genomic sequence. We first used a finite-state automaton to search for an alignment with at most two mismatches of the 24 bases that started 60 nt upstream of the EST 3′-end. These alignments were then further checked by using a Smith–Waterman (15) alignment of the final 60 nt of the EST with the indicated genomic sequence. Alignments with no more than six mismatches of any type were accepted. The subsequent identification of ambiguous 3′-ends was performed as described previously (2). We performed the identical search for the 4,069 arabidopsis ESTs, using 38 × 106 nt of genomic sequence.
Results
Nomenclature and Conventions.
The position in the pre-mRNA where 3′-end-processing (cleavage and subsequent addition of poly(A)) occurs is referred to as the CS. Upstream (downstream) relative positioning refers to the 5′ (3′) direction with respect to the CS, and corresponding positions are given as negative (positive) numbers. The A-rich element, frequently AAUAAA, centered approximately on position −20, found in all organisms investigated to date, is referred to as the PE. Any common elements located 5′ to the PE are referred to as UEs. Elements located 3′ to the CS are referred to as downstream elements (DE).
We have examined the nucleotide and word distributions in large sets of ESTs (1,352 yeast, 1,246 rice, 4,068 arabidopsis, 3,236 fruitfly, 6,029 mouse, and 4,337 human) that contain putative 3′-end-processing sites.
PE: AAUAAA Appears to Be Common and Presumed Functional, Although Not Essential, in All Organisms Investigated.
The single nucleotide frequencies at each position relative to the 3′-end of the EST are shown in Fig. 1A; all organisms investigated display a marked peak in adenine residues centered on a position approximately 20 nt upstream from the CS (Fig. 1A). The average abundance of each nucleotide varies significantly between plants and animals, reflecting differences in the sequence composition of the 3′ untranslated regions.
Figure 1.

Single-nucleotide frequencies preceding the 3′-end-processing sites as determined by alignment of 3′-ESTs generated from yeast (1,352), rice (1,246), arabidopsis (4,069), fruitfly (3,236), mouse (6,029), and human (4,427) cDNAs. Positions are given relative to the putative 3′-end-processing site. (A) Sequences aligned on the 3′-most end of the ESTs. (B) Sequences aligned to a 6-nt profile, via an iterative procedure as described in Methods.
Using the iterative profiling technique, described in Methods, we determined the best representation of specific elements located near the CS. As shown in Fig. 1B, the animal sequences are dominated by a signal indicative of the canonical AAUAAA. The plant and yeast signals, however, do not show such a dominant feature. Because the procedure is insensitive to minor differences in relative position of a common feature, such differences cannot explain the reduced specificity of AAUAAA in yeast and plants. Sequence Logo (16) representations of the alignments shown in Fig. 1B are available as supplementary material on the PNAS web site (see www.pnas.org).
Analysis of hexamer usage (Z-scores and appearance frequencies) in a 10-nt-wide window centered 20 nt upstream of the CS revealed that all species show a disproportionate usage of AAUAAA. The percentages of EST sequences containing AAUAAA in the final 50 nt are listed in Table 1. Although the plants and yeast all have high Z-scores of this word, the total number of sequences in which it appears is below 15%; these organisms instead show a statistical overabundance of a variety of A-rich hexamers, without any specific sequence dominating. [Similar results have been reported previously based on small-scale mutagenesis studies (11, 17).] By contrast, 65% of the human and 60% of the fruitfly EST sequences examined contain AAUAAA or AUUAAA (the most common variant) in the final 50 nt. In mouse, the percentage drops to 34%; however, we believe that the mouse ESTs contain more ambiguously identified sequences than the others, primarily because the plots shown in Fig. 1A display more random sequences (base frequencies at or near 25%), especially when compared with the human ESTs. In addition, the mouse ESTs contain sequences from at least eight different cell types. Recent experiments (18) indicate that different mouse cell types are polyadenylated by alternate versions of the processing proteins that could have an altered affinity for specific RNA binding sequences. If separated by cell type, however, the mouse data sets become too small for the analysis we describe here.
Table 1.
Frequency of occurrence of AAUAAA in Positioning Element context as determined by analysis of 3′-EST sequences
| Organism | No. of ESTs | ESTs with AAUAAA
|
|
|---|---|---|---|
| No. | % | ||
| Yeast | 1,352 | 178 | 13.2 |
| Rice | 1,248 | 77 | 6.2 |
| Arabidopsis | 4,069 | 349 | 8.6 |
| Arabidopsis* | 625 | 86 | 13.8 |
| Fruitfly | 3,236 | 1,530 | 47.3 |
| Fruitfly* | 882 | 509 | 57.7 |
| Mouse | 6,210 | 1,618 | 26.1 |
| Human | 4,427 | 2,353 | 53.2 |
Only ESTs matched with genomic sequence.
UEs Appear to Be Used in Plants and Yeast, but Only Rarely in Animals.
The plants and yeast display clear evidence of signal elements upstream from the A-rich PE. In yeast, the upstream efficiency element has been demonstrated to be more important than the PE in selection of the processing site (10), and the most efficient version of the UE was UAUAUA (17). In both rice and arabidopsis, U-rich sequences dominate the last 100 nt (other than the PE region) before the CS. Fig. 1A clearly shows this predominance of uracil residues. Through examination of the pentamer and hexamer usage upstream of the PE, we have determined that sequences containing elements UUGUAU and UUGUAA (or similar words) are significant both in raw counts and in Z-scores (defined in Methods), with a peak usage approximately 60 nt upstream of the CS. Other U-rich words (e.g., UUUUUU) often appear in positions typical for the UE; however, the increased counts of these other words occur throughout the 3′-untranslated region (3-′UTR), therefore their high counts are likely because of the overall U-richness of the 3′-UTR. Interestingly, the optimal yeast UE (UAUAUA) occurs both in rice and A. thaliana, with a positional distribution that peaks between 20 and 30 nt upstream of the CS, indicating that, in these cases, this word may act as a positioning rather than an efficiency element. In general, the apparent position of the UE both in rice and arabidopsis seems to extend further upstream than in yeast.
By contrast, human, mouse, and fruitfly ESTs show no statistical evidence of broad use of signal elements upstream of the AAUAAA PE. Yet, although we found no words with statistical overabundance, inspection of the most abundant words in the near upstream region (approximately 50 nt upstream of the CS) revealed words that resemble the UEs identified for yeast and plants. This could indicate small numbers of genes with upstream control sequences. However, words containing repeated UA or UG have also previously been identified as signals with other functionality, e.g., moderation of mRNA stability (19), suggesting that they may play no role in 3′-end processing in the animals.
The 3′-End-Processing Site Is Often Preceded by U-Rich Segments in Yeast and Plants That Seemingly Have No Counterparts in Animals.
As shown in Fig. 1, the 3′-end-processing sites for plants and yeast commonly occur following U-rich regions; we previously found that yeast polyadenylation sites are also commonly followed by a downstream U-rich region, though the specificity of these two “U-rich” signals is not high in yeast. The most common words found in the sequence immediately preceding the CS in rice, arabidopsis, and yeast is a single cytosine in a longer run of uracils (UUUUCU, UUUCUU, etc.). Yet, whereas this group of words is the most significant in the region surrounding the CS, it appears in less than 25% of the sequences that we examined.
Matching EST Sequences to Genomic Sequence Improves Data Analysis.
In our previous study of yeast, we demonstrated that comparison of EST sequences with genomic sequence greatly improves the quality of our data, because we were able to eliminate ambiguous sequences caused by such effects as internal priming or genomic occurrence of restriction enzyme CSs. We obtained 48 × 106 bases of genomic fruitfly sequence and searched for EST alignments as described in Methods. We successfully aligned 1,190 of the 3,236 EST sequences to genomic sequence. We were able to identify 25 EST sequences followed immediately (in genomic sequence) by probable restriction enzyme recognition sequences and 283 EST sequences followed by A-rich regions (at least 7 of the next 10 nt) that make primer hybridization ambiguous. The remaining 882 fruitfly 3′-EST sequences were analyzed by using both the profile and Z-scores, as described for ESTs without genomic information. A similar search for the 4,069 arabidopsis ESTs in 38 × 106 bases of genomic sequence produced 662 matches, of which 624 were retained as unambiguous 3′-ends.
The PE signal words found for the genome-matched fruitfly ESTs agreed with those found through analysis of the nongenomic ESTs. Fig. 2A shows the result of iterative profile alignments for the PE (centered on position −20), as well as the most common signal words. Fig. 2B shows the same features for the fruitfly DE. As can be seen, the DE is not quite as specific as the UE. The most significant hexamers determined for the fruitfly DE are UGUUUU, UGUGUU, and UUUUUU, all of which have their maximum usage approximately 10 to 20 bases downstream from the CS. These words agree well with the optimum RNA-binding sequence determined for the 64-kDa subunit of mammalian cleavage stimulation factor by selex measurements (20).
Figure 2.

Single-nucleotide frequencies that describe the two principal signal elements of the fruitfly 3′-end-processing signal, as determined by analysis of 882 ESTs that were successfully aligned to contig sequences. By using the iterative profiling procedure defined in Methods, the sequences were successively aligned on (A) the PE (approximate position −20) and (B) DE (approximate position +14).
The 3′-end processing efficiencies of the 18 sequences that differ by a single substitution from the canonical AAUAAA were measured in human (HeLa) cell extracts by Sheets et al. (21). For comparison, we measured the abundance of these words in the fruitfly EST-genome sequences that did not contain the canonical AAUAAA (327 of 882 sequences). The genomic sequences were searched for hexamers that differed from AAUAAA by only a single-base mutation. Under the assumption that the most efficient sequence present is most likely to be used, we ranked the words on the basis of their previously measured efficiencies and counted only the highest-ranked word in each EST sequence. Fig. 3 compares these counts with the measured efficiencies. Although the agreement is not exact, the similarities, especially in the use of words with a single pyrimidine substitution (AUUAAA, CAUAAA, UAUAAA, AAUACA, and AAUAUA) are striking. This result suggests that the occurrences of variants of the signal element correlate with their respective processing efficiencies. A comparison of the same analysis for human and mouse ESTs is available as supplementary material (see www.pnas.org).
Figure 3.

A comparison of the occurrences (in 327 contig-aligned fruitfly EST sequences that did not contain AAUAAA) with in vitro measured [in human HeLa cells (21)] processing efficiencies of sequences similar to the canonical PE AAUAAA. The processing efficiencies are plotted as a percentage of the efficiency of AAUAAA, whereas the occurrences are plotted as a fraction of the total number of sequences examined.
Discussion
Fig. 4 displays a schematic picture of the signal elements that appear to determine the 3′-end-processing sites, along with the words that most frequently occur in each position. Although only the most common words for each element are shown, it is important to reiterate that similar words (differing by at most one or two substitutions) also appear to be functional. In yeast, both mutagenesis experiments and our previous EST-based analysis demonstrated that weak signal words, although reduced in processing efficiency, play an important role in 3′-end-processing site selection.
Figure 4.

Graphical representations of the proposed alignment of 3′-end-processing signals for (A) yeast, (B) plants, and (C) animals, including the most common words found for each subelement.
Plant 3′-Polyadenylation Signals Appear to Be Closer to Those of Yeast than to Those of Animals.
Upstream efficiency elements have been shown to be equally as important as or more important than the PE for yeast polyadenylation site selection (10). There have been reports that plants show similar behavior (11, 12), though the plant polyadenylation signals have not been as widely investigated. The words that we found to be likely UE in rice and arabidopsis are similar to those in yeast, though the most significant words detected for plants correspond to some of the less efficient UEs for yeast (e.g., UUGUAA and UUGUAU). The PEs identified for rice and arabidopsis are also similar to yeast, in that the dominant signal appears to be better characterized as “A-rich” than by any single word. Finally, both rice and arabidopsis show an increased uracil incidence as well as common significant words between the PE and the CS when compared with yeast.
The UEs in Plants and Yeast Show Similarity to the DE in Animals.
In a recent comparison of 3′-end processing among various organisms, the idea was proposed that the UE of yeast and the DE of mammalian sequences are related (22). Our study lends support to this model, in that the DEs found in fruitfly sequences contain words similar to the UEs of both rice and arabidopsis. Taken also in comparison with the UE of yeast, we find a frequent pattern of uracils alternating with purines, with occasional replacement of uracil by cytosine (e.g., UAUAUA, UAUGUA, UUGUAU, UACAUA, UGUGUU, etc.).
Protein–mRNA Interactions Appear to Be Inherently Dynamic Processes That Display Statistical Variability.
If we assume that 3′-end processing has a regulatory function, we should not expect that it is either necessary or desirable for all mRNA species to be polyadenylated with the same efficiency. Even if the same molecular components are responsible for all pre-mRNA polyadenylation [and this is not certain (18)], variation in polyadenylation rates could be mediated through variation of control sequences. Several recent reports provide evidence that differential polyadenylation rates are used in regulation of gene expression. In particular, studies of the Su(f) gene in D. melanogaster (23) revealed 3′-end processing in at least three separate positions, one of which produces a severely truncated transcript that is not translated. Production of the nonfunctional transcript correlated with levels of functional protein, implying a negative self-regulation. Selection among alternate polyadenylation sites in several yeast transcripts has been shown to correlate with carbon source regulation (24).
Our previous investigation demonstrated that the complete yeast 3′-end-processing signal is comprised of as many as four separate elements, all of which contribute to the determination of processing location. The current study further emphasizes the statistical nature of 3′-end-processing, both in the lack of strong consensus sequences for many elements investigated here, as well as in the similarity of EST-measured abundance and in vitro-measured processing efficiencies of AAUAAA-like signals.
Our informatic analysis of 3′-end-processing signals generally agrees with published experimental findings, though it requires a conceptual broadening of the idea of a control sequence to include a set of interacting elements, no one of which is required to have a unique identity. Previous studies, both experimental (20) and analytical (25, 26), seem to suggest a similar conclusion. Such a system seems counterintuitively imprecise [and is further complicated by the involvement of remarkably elaborate protein machinery (20, 27)]. Nevertheless, a moment’s consideration of alternatives helps to rationalize it. For example, a single precisely defined polyadenylation signal (e.g., an octamer) recognized by a single cleavage protein might arise (or be obliterated) fairly frequently through mutation. The choice of a flexible set of elements, no one of which is very strongly bound by the protein machinery, but the binding of which is cooperative, minimizes the chance of either premature or missed 3′-end processing. It also affords another way to modulate the dynamic range of gene expression.
It is clear that the dynamic statistical nature of gene regulatory processes must be considered as more complete models are developed. Recent investigations of transcription promoter sites, for example, have indicated that many lack the consensus “TATA”-box sequence (28). The techniques that we have used here to the study 3′-end-processing signals offer a powerful approach to the statistical investigation of control sequences for other regulatory processes. They require only a suitable biological hypothesis for selection of specific data from the sequence databases. The most powerful argument for such bioinformatic analysis is that it complements classical approaches and has the potential to focus experiments on likely candidate sequences, thereby reducing the necessary range of data collection required for complete characterization of control signals.
Supplementary Material
Acknowledgments
We thank Geoffrey Cooper, Chip Celenza, Tom Gilmore, and Dean Tolan for critical reading of the manuscript. We thank the various EST sequencing efforts for making their data available. J.H.G and C.R.C are supported by Sequenom, Inc. S.C.M. is supported by Genetics Institute, Cambridge, MA. T.F.S. is supported by Department of Energy grant DE-FG02–98ER62558.
Abbreviations
- EST
expressed sequence tag
- CS
cleavage site
- PE
positioning element
- UE
upstream element
- DE
downstream element
- poly(A)
polyadenylate
References
- 1.Trifonov E N. Comput Appl Biosci. 1996;12:423–429. doi: 10.1093/bioinformatics/12.5.423. [DOI] [PubMed] [Google Scholar]
- 2.Graber J H, Cantor C R, Mohr S C, Smith T F. Nucleic Acids Res. 1999;27:888–894. doi: 10.1093/nar/27.3.888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao J, Kessler M M, Moore C L. J Biol Chem. 1997;272:10831–10838. doi: 10.1074/jbc.272.16.10831. [DOI] [PubMed] [Google Scholar]
- 4.Stumpf G, Domdey H. Science. 1996;274:1517–1520. doi: 10.1126/science.274.5292.1517. [DOI] [PubMed] [Google Scholar]
- 5.Preker P J, Ohnacker M, Minvielle-Sebastia L, Keller W. EMBO J. 1997;16:4727–4737. doi: 10.1093/emboj/16.15.4727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chanfreau G, Noble S M, Guthrie C. Science. 1996;274:1511–1514. doi: 10.1126/science.274.5292.1511. [DOI] [PubMed] [Google Scholar]
- 7.Colgan D F, Manley J L. Genes Dev. 1997;11:2755–2766. doi: 10.1101/gad.11.21.2755. [DOI] [PubMed] [Google Scholar]
- 8.Keller W, Minvielle-Sebastia L. Curr Opin Cell Biol. 1997;9:329–336. doi: 10.1016/s0955-0674(97)80004-x. [DOI] [PubMed] [Google Scholar]
- 9.Zhao J, Hyman L, Moore C. Microbiol Mol Biol Rev. 1999;63:405–445. doi: 10.1128/mmbr.63.2.405-445.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo Z, Sherman F. Trends Biochem Sci. 1996;21:477–481. doi: 10.1016/s0968-0004(96)10057-8. [DOI] [PubMed] [Google Scholar]
- 11.Rothnie H M. Plant Mol Biol. 1996;32:43–61. doi: 10.1007/BF00039376. [DOI] [PubMed] [Google Scholar]
- 12.Li Q, Hunt A G. Plant Physiol. 1997;115:321–325. doi: 10.1104/pp.115.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Altschul S F, Madden T L, Schaffer A A, Zhang J, Zhang Z, Miller W, Lipman D J. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schbath S. J Comput Biol. 1997;4:189–192. doi: 10.1089/cmb.1997.4.189. [DOI] [PubMed] [Google Scholar]
- 15.Smith T F, Waterman M S. J Mol Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- 16.Schneider T D, Stephens R M. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo Z, Sherman F. Mol Cell Biol. 1995;15:5983–5990. doi: 10.1128/mcb.15.11.5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wallace A M, Dass B, Ravnik S E, Tonk V, Jenkins N A, Gilbert D J, Copeland N G, MacDonald C C. Proc Natl Acad Sci USA. 1999;96:6763–6768. doi: 10.1073/pnas.96.12.6763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zubiaga A M, Belasco J G, Greenberg M E. Mol Cell Biol. 1995;15:2219–2230. doi: 10.1128/mcb.15.4.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takagaki Y, Manley J L. Mol Cell Biol. 1997;17:3907–3914. doi: 10.1128/mcb.17.7.3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sheets M D, Ogg S C, Wickens M P. Nucleic Acids Res. 1990;18:5799–5805. doi: 10.1093/nar/18.19.5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moreira A, Takagaki Y, Brackenridge S, Wollerton M, Manley J L, Proudfoot N J. Genes Dev. 1998;12:2522–2534. doi: 10.1101/gad.12.16.2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Audibert A, Simonelig M. Proc Natl Acad Sci USA. 1998;95:14302–14307. doi: 10.1073/pnas.95.24.14302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sparks K A, Dieckmann C L. Nucleic Acids Res. 1998;26:4676–4687. doi: 10.1093/nar/26.20.4676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schneider T D. J Theor Biol. 1997;189:427–441. doi: 10.1006/jtbi.1997.0540. [DOI] [PubMed] [Google Scholar]
- 26.Berg O G, von Hippel P H. J Mol Biol. 1987;193:723–750. doi: 10.1016/0022-2836(87)90354-8. [DOI] [PubMed] [Google Scholar]
- 27.Minvielle-Sebastia L, Keller W. Curr Opin Cell Biol. 1999;11:352–357. doi: 10.1016/S0955-0674(99)80049-0. [DOI] [PubMed] [Google Scholar]
- 28.Audic S, Claverie J M. Trends Genet. 1998;14:10–11. doi: 10.1016/S0168-9525(97)01323-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}