

Abstract
We present evidence that the size of an active site side chain may modulate the degree of hydrogen tunneling in an enzyme-catalyzed reaction. Primary and secondary kH/kT and kD/kT kinetic isotope effects have been measured for the oxidation of benzyl alcohol catalyzed by horse liver alcohol dehydrogenase at 25°C. As reported in earlier studies, the relationship between secondary kH/kT and kD/kT isotope effects provides a sensitive probe for deviations from classical behavior. In the present work, catalytic efficiency and the extent of hydrogen tunneling have been correlated for the alcohol dehydrogenase-catalyzed hydride transfer among a group of site-directed mutants at position 203. Val-203 interacts with the opposite face of the cofactor NAD+ from the alcohol substrate. The reduction in size of this residue is correlated with diminished tunneling and a two orders of magnitude decrease in catalytic efficiency. Comparison of the x-ray crystal structures of a ternary complex of a high-tunneling (Phe-93 → Trp) and a low-tunneling (Val-203 → Ala) mutant provides a structural basis for the observed effects, demonstrating an increase in the hydrogen transfer distance for the low-tunneling mutant. The Val-203 → Ala ternary complex crystal structure also shows a hyperclosed interdomain geometry relative to the wild-type and the Phe-93 → Trp mutant ternary complex structures. This demonstrates a flexibility in interdomain movement that could potentially narrow the distance between the donor and acceptor carbons in the native enzyme and may enhance the role of tunneling in the hydride transfer reaction.
Quantum tunneling effects have been found to contribute to a growing number of enzyme-catalyzed hydrogen transfer reactions (1–6). By virtue of its atomic mass, the behavior of hydrogen is poised between the classical and quantum mechanical regimes.¶ Comparison of the reaction rates for H-, D-, and T-labeled substrates can effectively probe quantum effects under conditions of moderate protium tunneling (7, 8). Given the almost ubiquitous presence of hydrogen transfer events in biological processes, it has become important to determine the role of macromolecular structure in facilitating the tunneling process. Subtle changes in protein structure may be sufficient to shift a reaction toward increasing potential energy barrier penetration, with a concomitant increase in biologic efficiency. We now report evidence in support of such an effect in an enzyme-catalyzed hydride transfer reaction, demonstrating that a single amino acid side chain of horse liver alcohol dehydrogenase (LADH) modulates the degree of quantum behavior and catalytic efficiency in a parallel fashion.
LADH is an 80-kDa dimer, each monomer consisting of well-defined substrate and cofactor binding domains. Binding of NAD+ stabilizes a conformational transition from an “open” to a “closed” form, narrowing the interdomain cleft (9). The zinc-bound alcohol substrate is anchored close to NAD+, allowing direct hydride transfer to the C-4 atom of the nicotinamide ring (10). A general feature of dehydrogenase structures is the conservation of a bulky hydrophobic residue that is positioned against the back side of the nicotinamide ring of bound cofactor (NAD+ or NADP+).‖ In LADH, one of the methyl groups of Val-203 is found to be within van der Waals contact of the C-5 and C-6 atoms of the nicotinamide ring of NAD+ (10). Furthermore, Val-203 is a highly conserved residue among a large family of alcohol dehydrogenase sequences (15). We have altered the size of the residue at position 203 to examine its effect on tunneling and catalytic rate. Furthermore, a comparison of the x-ray crystal structures of ternary complexes of a tunneling and nontunneling mutant provides a structural basis for the observed effects.
METHODS
Preparation of LADH Mutants.
Mutants of LADH were prepared as described (3). The phagemid pBPP/LADH contains the LADH cDNA under the control of the tac promoter and contains the replication origin for f1 bacteriophage (16). The phagemid was rescued from Escherichia coli as a single-stranded DNA (antisense) by infection with helper phage VCSM13 (Stratagene). The single-stranded template was used for site-directed mutagenesis (Amersham mutagenesis kit, version 2) and also for dideoxy DNA sequencing (United States Biochemical, Sequenase 2.0) to confirm mutations. All mutants were expressed in E. coli XL1-Blue (Stratagene) using the phagemid pBPP/LADH. Mutant enzymes were purified according to procedures described by Park and Plapp (16).
Steady-State Kinetics.
Kinetics were measured at pH 7.0 in 300 mM potassium phosphate, 300 mM semicarbazide hydrochloride, and 10 mM NAD+, and the temperature was thermostated to 25.0 ± 0.1°C. Kinetic parameters were determined by monitoring the absorbance increase at 340 nm from the production of NADH. Values of kcat and Km and their standard errors were obtained by a nonlinear fit of the expression: velocity = kcat[S]/(Km + [S]). The concentration of LADH active sites of the wild-type and site-directed mutants at position 203 was determined spectrophotometrically using the extinction coefficient ɛ280 = 18,200 M−1⋅cm−1 [A280 = 0.455 for 1 mg/ml (31)]. The extinction coefficient used for the double mutant Phe-93 → Trp: Val-203 → Ala [ɛ280 = 24,600 M−1⋅cm−1] was determined relative to the wild-type enzyme’s extinction coefficient by a comparative Bradford and spectrophotometric assay.
Primary and Secondary Tritium Isotope Effects.
Kinetic isotope effects were measured under the same reaction conditions as steady-state kinetics by competitively using doubly labeled substrates as described (1, 3, 17). Primary isotope effect measurements involved comparison of the rate of tritium transfer from T-labeled benzyl alcohol to the rate of H-labeled (kH/kT) or D-labeled (kD/kT) benzyl alcohol, which was remotely labeled with 14C. Secondary isotope effects report the relative reaction rate of substrate, which has a H-, D-, or T-labeled noncleaved hydrogen at the benzylic carbon, that is converted to product aldehyde with C-1 labeled. The LADH-catalyzed reaction was quenched at 5–30% conversion, and substrate and products were separated by reverse-phase HPLC. Kinetic isotope effects were then determined by 3H/14C liquid scintillation counting (LKB model 1209 Rackbeta) of the substrates and products.
X-Ray Crystallography of Phe-93 → Trp and Val-203 → Ala LADH Complexes.
Protein solutions of Phe-93 → Trp (16 mg/ml) and Val-203 → Ala (22 mg/ml) contained a 10-fold molar excess of NAD+, 5 mM trifluoroethanol, and 5% (vol/vol) polyethylene glycol 400, and were filtered through a 0.45 μm cellulose acetate filter. Crystals of both complexes were grown at 4°C via vapor diffusion from 4 μl drops equilibrated against 50 mM Tris⋅HCl buffer (pH 8.4), 5 mM trifluoroethanol, and PEG 400 [12% (vol/vol) for Val-203 → Ala enzyme, 15–17% (vol/vol) for Phe-93 → Trp enzyme]. Diffraction data for the Phe-93 → Trp mutant were collected at 277 K from four crystals (see Table 2). Data for the Val-203 → Ala mutant were collected from a single crystal that was flash cooled at 100 K in a N2 stream after a brief rinse in the crystallization solution containing an increased PEG 400 concentration (30%) to allow proper vitrification. Data for both complexes were collected on an R-AXIS II detector (Molecular Structure Corp., The Woodlands, TX) with a Cu rotating anode source running at 50 kV and 100 mA, and were processed using the XKL software package (34). Refinement was performed with X-PLOR (18). The Phe-93 → Trp structure was initially phased with the isomorphous 1.8-Å LADH structure (19). This model was subjected to rigid body refinement with the cofactor, substrate analog, and mutated residue omitted. The cofactor NAD+, trifluoroethanol, and Trp-93 were fitted into Fo − Fc and 2Fo − Fc maps obtained at this stage, and the resulting model was subjected to 100 steps of constrained conjugate gradient refinement, 10 steps of overall B-factor refinement, and 20 steps of individual B-factor refinement. Due to the quality of the initial phasing model, simulated annealing refinement did not improve Rfree (18) and did not alter the active site geometry. Thus, the conjugate gradient-refined model was retained. Solvent molecules were added to the model based on a peak at the 2σ level on 2Fo − Fc and Fo − Fc maps, and favorable hydrogen bonding geometry. This was followed by several rounds of manual adjustment of the substrate position and conjugate gradient refinement.
Table 2.
Data collection and refinement statistics for two LADH mutants
| Phe-93 → Trp | Val-203 → Ala | |
|---|---|---|
| Data Collection | ||
| Space Group | P1 | P1 |
| Cell parameters | ||
| a, Å | 51.8 | 44.4 |
| b, Å | 44.6 | 86.7 |
| c, Å | 93.3 | 90.6 |
| α, degrees | 103.1 | 102.7 |
| β, degrees | 87.6 | 99.2 |
| γ, degrees | 70.6 | 101.6 |
| Solvent content, % | 51 | 39 |
| Monomers/asymmetric unit | 2 | 4 |
| Resolution, Å | 2.0 | 2.5 |
| Unique reflections | 43,756 | 40,627 |
| Completeness, % | 67.6 (82.1)* | 85.0 |
| Rmerge† | 0.064 | 0.088 |
| Refinement | ||
| Resolution range, Å | 10.0–2.0 | 10.0–2.5 |
| Final R factor‡ | 0.203 | 0.214 |
| Free R factor‡ | 0.269 | 0.294 |
| rms deviations observed | ||
| Bond lengths, Å | 0.011 | 0.007 |
| Bond angles, degrees | 2.01 | 1.63 |
| Total water molecules | 116 | 315 |
Completeness to 2.5 Å.
Rmerge = Σ|I0 − Ia|/Σ(Ia), where I0 is the observed intensity and Ia is the average intensity, the sums being taken over all symmetry related reflections.
R factor = Σ|F0 − Fc|/Σ(F0), where F0 is the observed amplitude and Fc is the calculated amplitude.
The 1.8-Å LADH structure (19) was also used as an initial search model for the Val-203 → Ala mutant complex. A molecular replacement rotation search and Patterson correlation refinement indicated a 7° angle between the noncrystallographic 2-fold axes in the two independent dimers in the unit cell. The angular orientations of these axes were used to guide a manual examination of packing interactions. The translation search yielded an unambiguous solution. The resultant model, with the cofactor and substrate analog omitted, was subjected to conjugate gradient positional and B-factor refinement as described above. Fo − Fc and 2Fo − Fc maps were then used to fit NAD+ and trifluoroethanol into the model. Conjugate gradient refinement was repeated, with charges omitted on the alcohol and Zn cation. A harmonic constraint was initially placed on the Zn–alcohol oxygen distance to avoid unusually short Zn–oxygen contacts. Again, simulated annealing refinement produced no change in active site geometry or improvement in Rfree. Solvent-fitting and additional conjugate gradient refinement were carried out as described for the Phe-93 → Trp structure. Noncrystallographic symmetry averaging among the four independent monomers did not improve Rfree and was not imposed.
RESULTS AND DISCUSSION
Our ability to characterize hydrogen tunneling in the hydride transfer catalyzed by mutants of LADH relies on the simple fact that tunneling probability decreases with increasing mass,¶ leading to tunneling in the order H > D > T. Detection of hydrogen tunneling depends on the mass relationship for the rate of reaction of H-, D-, and T-labeled substrates, designated kH, kD, and kT, respectively. Using tritium as a frame of reference, classical behavior predicts that the ratio of ln(kH/kT) to ln(kD/kT) will equal 3.3 (7). By contrast, under conditions of moderate tunneling, when protium tunnels more than deuterium, the relationship between ln(kH/kT) to ln(kD/kT) can greatly exceed 3.3 (1, 8).
To observe a kinetic isotope effect in the hydride transfer step in LADH, this step must be at least partially rate-limiting (1). Early studies of the native form of LADH failed to detect tunneling, a consequence of the inherent kinetic complexity of the wild-type reaction, which is partially rate-limited by product release (3). Thus, subsequent modification of several residues in the alcohol binding pocket were designed to increase the rate of product release. The modifications (Leu-57 → Phe and Phe-93 → Trp) have been found to “unmask” tunneling, leading to ratios for ln(kH/kT) to ln(kD/kT) of 8.5 and 6.1, respectively (3). In the present study, changes in the size and hydrophobicity of the amino acid side chain at position 203 have been carried out with the goal of moderating the hydride transfer step directly (Table 1). To rule out any systematic trend in kinetic properties for the position 203 mutants, Km values for NAD+ were determined. These varied from 3 mM (Val-203 → Leu) to 160–190 μM (Val-203 → Ala and Phe-93 → Trp:Val-203 → Ala) to 610 μM (Val-203 → Gly), relative to a value of 4 μM for wild-type enzyme. Although Km increases upon mutation of Val-203, all mutants (with the possible exception of Val-203 → Leu) display Km values much lower than the experimental NAD+ concentration of 10 mM. Thus, the kinetic isotope effect measurements are expected to reflect the reaction of alcohol with the E⋅NAD+ complex in all cases.
Table 1.
Kinetic parameters and isotope effects for the hydride transfer catalyzed by mutants of LADH
| Parameter | Leu-57 → Phe* | Phe-93 → Trp* | Val-203 → Leu | Val-203 → Ala | Val-203 → Ala: Phe-93 → Trp | Val-203 → Gly |
|---|---|---|---|---|---|---|
| kcat/Km, mM−1⋅s−1 | 8.6 | 4.7 | 1.0 | 0.20 | 0.13 | 0.071 |
| Km, mM† | 0.028 | 0.028 | 1.8 | 1.0 | 2.1 | 1.7 |
| kD/kT primary‡ | 1.83 ± 0.01 | 1.86 ± 0.01 | 1.89 ± 0.01 | 1.88 ± 0.02 | 1.91 ± 0.02 | 1.90 ± 0.01 |
| kH/kT primary‡ | 7.31 ± 0.05 | 7.76 ± 0.03 | 7.82 ± 0.08 | 7.24 ± 0.06 | 7.65 ± 0.06 | 7.53 ± 0.03 |
| kD/kT secondary‡ | 1.033 ± 0.004 | 1.048 ± 0.004 | 1.074 ± 0.004 | 1.058 ± 0.004 | 1.075 ± 0.004 | 1.097 ± 0.007 |
| kH/kT secondary‡ | 1.318 ± 0.007 | 1.333 ± 0.004 | 1.380 ± 0.005 | 1.316 ± 0.006 | 1.325 ± 0.004 | 1.358 ± 0.007 |
| ln(kH/kT)/ln(kD/kT) primary§ | 3.30 ± 0.03 | 3.31 ± 0.02 | 3.23 ± 0.03 | 3.14 ± 0.05 | 3.16 ± 0.02 | 3.16 ± 0.03 |
| ln(kH/kT)/ln(kD/kT) secondary§ | 8.5 ± 1.0 | 6.1 ± 0.5 | 4.5 ± 0.2 | 4.9 ± 0.3 | 3.9 ± 0.2 | 3.3 ± 0.2 |
Previously reported (3).
Km is the Michaelis constant for benzyl alcohol. Errors are less than 10% of value.
Values represent >10 measurements ± the standard error of the mean.
As summarized in Table 1, a decrease in the size of the residue at position 203 leads to a regular decline in both catalytic efficiency (kcat/Km) and tunneling (ratio of ln(kH/kT)/ln(kD/kT)).** As has been seen previously, changes in tunneling are much more readily detected in ln(kH/kT)/ln(kD/kT) for secondary than primary isotope effects (1, 17, 20). This does not imply that there is greater tunneling of the uncleaved secondary hydrogen. Rather, this recurring phenomenon reflects a contribution of the motion of the 2° hydrogen to that of the 1° hydrogen at the transition state [referred to as coupled motion (21)], together with the fact that deviations from classical behavior become much easier to detect the closer the measured isotope effect is to unity.
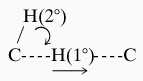 |
Because secondary isotope effects are generally less than 1.4 for kH/kT and 1.1 for kD/kT, they present a highly sensitive probe for deviations from classical behavior. Consistent with this property, Table 1 shows little or no change in the primary ln(kH/kT)/ln(kD/kT), whereas the secondary ln(kH/kT)/ln(kD/kT) decreases from 8.5 to 3.3. The trend in the secondary ln(kH/kT)/ln(kD/kT) is illustrated in Fig. 1, where one sees an almost linear relationship between catalytic efficiency and the signature of tunneling. Once the side chain has been removed completely from position 203 (Gly-203), the enzymatic rate has decreased by more than two orders of magnitude and the evidence for tunneling has been lost.
Figure 1.

Correlation of the log(kcat/Km) and the ratio of ln(kH/kT)/ln(kD/kT) for site-directed mutants of alcohol dehydrogenase. Point A, Val-203 → Gly; point B, Val-203 → Ala:Phe-93 → Trp; point C, Val-203 → Ala; point D, Val-203 → Leu; point E, Phe-93 → Trp; point F, Leu-57 → Phe. Tunneling is indicated when the ratio of ln(kH/kT) to ln(kD/kT) is >3.3 (8).
A feature of the data in Table 1 is the relative insensitivity of the magnitude of the kH/kT isotope effects to the extent of tunneling. From the calculations of Huskey and Schowen (21), it is anticipated that a decrease in tunneling and coupled motion will lead to a decrease in the magnitude of the 1° and 2° isotope effects, respectively. The transition state with the most active enzyme, Val-203, has the charge and bond hybridization properties of the initial alcohol (1, 22). The data reported herein suggest that as semiclassical behavior increases from Val-203 to Gly-203, there is a progression of the transition state toward a more product-like configuration. This would produce compensating increases in the size of the isotope effect as the effects of tunneling diminish.
Inspection of the kD/kT isotope effects provides additional insight into the nature of the hydride transfer step for the series of mutants. We note a small, increasing trend of the primary kD/kT isotope effects. This is consistent with less initial tunneling with deuterium than protium, as well as a progressively later transition state as the series goes from Val-203 to Gly-203.‡‡ It can be seen that the observed decrease in the extent of tunneling correlates almost exclusively with an increase in the secondary kD/kT isotope effect. This result has been predicted from model calculations in which it is shown that the secondary kD/kT isotope effect decreases in a regular fashion as the reaction coordinate frequency, its isotope dependence, and extent of tunneling are increased (J. Rucker and J.P.K., unpublished data). For the mutants with the least degree of tunneling (Phe-93 → Trp:Val-203 → Ala and Val-203 → Gly), the increased magnitude of the secondary kD/kT may also indicate a later transition state. In this scenario, the decrease of hydrogen tunneling could reflect a change of the internal equilibrium constant of the reaction away from an optimal value for tunneling where ΔH° = 0 (23).
The finding that mutations at Val-203 lead to marked reductions in kcat/Km is different from the behavior of alcohol binding pocket mutants (Table 1), which showed relatively little change compared with the wild-type enzyme (3). The reduction in rate for Val-203 mutants is consistent with a chemical step that has become rate-limiting. We note that the double mutant, Phe-93 → Trp:Val-203 → Ala, shows slightly less tunneling than the single mutant, Val-203 → Ala (Table 1 and Fig. 1). This contrasts with the placement of Trp at position 93 in wild-type enzyme, which leads to an increased detection of tunneling due to unmasking of the hydride transfer step (3). We conclude that the hydride transfer step is likely to be fully rate-determining with Val-203 → Ala, making it possible to evaluate the importance of observed differences in tunneling and structure on the hydride transfer step directly.
The effects of these mutations on the ground state conformation of LADH are observed by the determination of the x-ray crystal structures of the Phe-93 → Trp and Val-203 → Ala mutants complexed with NAD+ and the substrate analog trifluoroethanol (Table 2). Comparison of the structures reveals differences in both local active site geometries and in more global interdomain relationships, consistent with the kinetic findings.
In the presence of cofactor and substrate analog, the Phe-93 → Trp mutant crystallizes in a closed conformation. The backbone rms difference between this mutant and the closed wild-type structure (19) is 0.18 Å. Likewise, the relative conformation of the cofactor and substrate in the Phe-93 → Trp mutant structure is very similar to that seen in other high resolution wild-type complexes (10, 19). One methyl group of Val-203 remains in van der Waals contact with the B side of the nicotinamide ring (Fig. 2 Upper). The trifluoroethanol is well positioned for hydride transfer of its pro-R hydrogen to C-4 of the nicotinamide ring. The alcohol methylene carbon is 3.2 Å from the nicotinamide C-4 carbon. This is the point of closest contact between cofactor and substrate, and is similar to the analogous 3.4-Å distance seen in the crystal structure of the ternary complex of wild-type LADH, NAD+, and pentafluorobenzyl alcohol (10).
Figure 2.

Comparison of active site structures from a high tunneling (Phe-93 → Trp, top) and a low tunneling (Val-203 → Ala, bottom) mutant of LADH. Electron density omit maps (Fo − Fc) and resulting models for the residue at position 203 (green), NAD+ (C, purple; O, red; P, white; N, blue), and trifluoroethanol (C, yellow; F, orange; O, red) are illustrated for each structure. Omit maps were generated with trifluoroethanol, NAD+, and residue-203 omitted from the final model and are contoured at σ levels of 2.5 (top) and 2.0 (bottom). (Upper): The nicotinamide ring in Phe-93 → Trp is in van der Waals contact with a methyl group of Val-203. The average donor to acceptor carbon distance among the two independent monomers is 3.2 Å. (Lower): In Val-203 → Ala, van der Waals contact between residue 203 and the nicotinamide ring is removed, causing a shift in ring position (red arrow, see Fig. 3). The average donor to acceptor carbon distance among the four crystallographically independent monomers is 4.0 Å. [Figs. 2 and 4 were rendered using molscript (24), rayscript (E. Fontana, D. Peisach, and E. Peisach, Brandeis University) and rayshade (version 4.0, C. Kolb and R. Bogart, Princeton University).]
The crystal structure of the Val-203 → Ala complex suggests that this mutation compromises the catalytic site geometry and offers an explanation for the reduced catalytic efficiency and diminished tunneling of this mutant. The crystal structure has two dimers per asymmetric unit, in contrast to other closed LADH structures studied at 277 K, which have a single dimer (9, 10, 19), but similar to closed forms studied at 100 K (25, 26). The general position of the cofactor in Val-203 → Ala is similar to that seen in Phe-93 → Trp. However, replacement of Val-203 by Ala removes the steric contact with the B face of the nicotinamide ring (Fig. 2 Lower).
In each of the four crystallographically independent monomers of Val-203 → Ala, the nicotinamide ring is rotated away from the trifluoroethanol, toward the gap left by replacement of the valine at position 203 by the smaller alanine (Fig. 3). The plane of the nicotinamide ring is rotated by an average of 10° relative to the nicotinamide position observed in Phe-93 → Trp or the wild-type enzyme. This rotation is accompanied by the shift of C-4 of the nicotinamide ring by 0.4 Å toward Ala. Although the resolution of the structure limits precise positioning of the substrate, the trifluoroethanol density in each of the four monomers also shows a common shift toward Phe-93, suggesting a less favorable catalytic geometry than in Phe-93 → Trp. As a consequence, the distance between the hydride-donating carbon of trifluoroethanol and the C-4 position of the nicotinamide ring is increased by an average of 0.8 Å relative to that seen in the Phe-93 → Trp structure (Figs. 2 and 3). The diminished tunneling of the Val-203 → Ala mutant is consistent with this increase in distance between the donor and acceptor carbons of the hydride transfer.
Figure 3.

Comparison of the four crystallographically independent nicotinamide rings in Val-203 → Ala (green) with the two independent rings in Phe-93 → Trp (blue). Surfaces are drawn at ≈90% of the van der Waals radii. Overlap of the structures is based on a least-squares alignment of the six independent cofactor binding domains. The nicotinamide rings are viewed edge-on along the glycosidic bond from the ribose, approximately normal to the view shown in Fig. 2. The nicotinamide ring in Phe-93 → Trp is in van der Waals contact with the “upper” methyl group of Val-203. In Val-203 → Ala, the nicotinamide ring rotates (curved arrow) to fill the gap left by replacement to alanine (straight arrow). Further rotation of the ring is prevented by steric contacts with Thr-178 (data not shown). Trifluoroethanol is displayed from the Phe-93 → Trp structure.
Despite an increase in the donor-to-acceptor carbon distance in the Val-203 → Ala mutant, this structure also shows a globally narrower interdomain active site cleft than that observed in either the closed wild-type enzyme or the Phe-93 → Trp mutant. Analysis of the interdomain motion indicates that the narrowing of the cleft in Val-203 → Ala results from an additional ≈0.5-Å rigid body shift of the catalytic domain toward the cofactor domain in each of the four crystallographically independent monomers relative to the conformation typically seen in closed native structures (Fig. 4) (9, 10, 19, 25, 26). The “hyperclosed” conformation observed in the Val-203 → Ala structure may result from the replacement of valine by the smaller alanine at position 203. A similar effect has been observed in the crystal structure of a human β1 alcohol dehydrogenase mutant Arg-47 → Gly (27). However, a hyperclosed structure has also been observed in a native LADH complex with the isosteric NAD+ analog, benzamide adenine dinucleotide, which indicates that an interdomain mutation is not required for this effect (T.D.C., K. Pankiewicz, K. Watanabe, and B.M.G., unpublished result).
Figure 4.

Difference in interdomain conformation between the hyperclosed Val-203 → Ala LADH structure and the closed Phe-93 → Trp structure. Cofactor domains at left were aligned by a least-squares fit of the two crystallographically independent Phe-93 → Trp monomers (red) and the four independent Val-203 → Ala monomers (white). The hyperclosed conformation of the Val-203 → Ala mutant results from an ≈0.5-Å average rigid body translation of its catalytic domain toward its coenzyme domain, when compared with the closed conformation of the Phe-93 → Trp structure. Vectors describing the rigid body translation for each Val-203 → Ala monomer correlate well, all lying within 25° of the average translation vector (arrow). Rotational components of this motion range between 0.5 and 2.5°, but there is little directional correlation between rotation axes for each of the four independent monomers. Analysis of interdomain motion used CCP4 least-squares fitting routine Lsqkab (32, 33). Val-203 (green), NAD+ (purple), Trp-93 (blue), and trifluoroethanol (C, yellow; F, orange; O, red) are displayed from the Phe-93 → Trp structure.
Crystals of the Val-203 → Ala complex are not isomorphous with those of the wild-type closed forms, and have significantly lower solvent content (Table 2). Thus, external forces imposed by crystal packing may contribute to the hyperclosed geometry. Data for the Val-203 → Ala complex were also collected from a flash-frozen crystal at a lower temperature (100 K) than that used for the Phe-93 → Trp mutant (277 K). However, both complexes were grown at the same temperature under similar conditions, and it is unlikely that flash cooling would induce this degree of conformational change (Table 2) without crystal damage. Normal closed geometries are observed in other flash-frozen LADH complexes (25, 26), whereas the mutation-induced hyperclosure described for the human β1 LADH was observed in a nonfrozen crystal (27). Regardless of its origin, the observation of a hyperclosed geometry demonstrates the flexibility in interdomain movement available to a ternary complex of cofactor, substrate, and LADH. In the case of the wild-type enzyme, interdomain mobility of this type may permit narrowing of the distance between the hydride donor and acceptor carbons, accompanied by an optimization of the barrier shape for tunneling (28).
The data presented here provide a link between a protein structure and the kinetic phenomenon of hydrogen tunneling. It is striking that a single residue in LADH can bring about a shift from behavior that is classical (Gly-203) to behavior that incorporates quantum mechanical effects. The gain in catalytic advantage of at least two orders of magnitude in proceeding from Gly-203 → Val (Fig. 1)†† can, in the most straightforward case, be attributed to the observed decrease in bond distance between the reacting carbon centers of alcohol substrate and cofactor (Figs. 2 and 3). However, site-specific mutagenesis experiments may introduce multiple effects influencing catalytic efficiency. At the present time we cannot rule out additional factors [e.g., changes in cofactor geometry (29) or internal equilibrium constants (23)] contributing to a portion of the observed trend in rate among the position 203 mutants. Given the fundamental quantum nature of a particle the size of hydrogen, it is not unexpected that enzymes will have “evolved” to take catalytic advantage of tunneling to enhance their rates. Enzyme systems are likely to vary greatly in the degree to which they utilize quantum behavior to accelerate their rates. Theoretical results (30) suggest that quantum effects for the H-transfer catalyzed by the enzyme carbonic anhydrase are similar to those observed for the noncatalyzed reaction. At the other extreme, recent data with the enzyme lipoxygenase suggest that substrate activation occurs by a mechanism that is almost completely quantum mechanical (5). Examination of the correlation between protein structure and tunneling for an enzyme such as lipoxygenase may reveal more complex changes than those required to transform the LADH system from a classical to a “quantum mechanical” catalyst. In either case, combinations of static and dynamic factors will likely govern differences in hydrogen tunneling observed between these systems.
Acknowledgments
We thank B. Plapp for the plasmid containing the gene for LADH. Discussions with and comments from T. Jonsson, J. Rucker, Y. Kim, A. Kohen, G. Petsko, M. Toney, and S. Tsai are gratefully acknowledged. We also thank B. Plapp, S. Benkovic, and V. Schramm for their critical evaluations of this manuscript. The research in Berkeley was supported by a National Science Foundation grant (J.P.K.), a National Institute of General Medical Sciences postdoctoral fellowship training grant (B.J.B.), and a National Institutes of Health Molecular Biophysics training grant (J.K.C.).
ABBREVIATION
- LADH
horse liver alcohol dehydrogenase
Footnotes
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, Biology Department, Brookhaven National Laboratory, Upton, NY 11973 (entries 1AXE and 1AXG).
The probability of a particle tunneling through a potential energy barrier is proportional to its de Broglie wavelength [λ = h/(2 mE)½]. At a total energy of 20 kJ/mol, the isotopes of hydrogen: protium (H), deuterium (D), and tritium (T) have de Broglie wavelengths of 0.6, 0.5, and 0.4 Å, respectively, which are similar to the typical distance traversed during a hydrogen transfer reaction.
Examples of hydrophobic side chains in contact with the backside of the nicotinamide ring: glyceraldehyde-3-phosphate dehydrogenase-Ile-11, Tyr-317 (11); lactate dehydrogenase-Ile-249, Val-136 (12); dihydrofolate reductase-Phe-103 (13); malate dehydrogenase-Ala-245, Leu-157 (14).
The Val-203 → Leu mutation appears to contradict the correlation of the extent of tunneling and the size of the residue at position 203. However, the β-carbon substitution at this residue is critical for optimal catalysis. Leucine has an isopropyl group and two hydrogens at its β-carbon compared with valine, which has two methyl groups and a hydrogen. In this case, leucine can be thought of as “functionally smaller” than valine. Alternatively, the reduced tunneling with leucine could simply reflect an improper fit of leucine into the pocket behind the nicotinamide ring.
Given the reduced contribution of coupled motion and tunneling to kD/kT measurements, relative to kH/kT, trends due to changes in transition state structure may be easier to discern.
Because the detection of tunneling is largely masked in the wild-type enzyme as a result of kinetic complexity, the actual catalytic advantage due to tunneling may exceed that estimated using the alcohol binding pocket mutant (Leu-57 → Phe) as a frame of reference.
References
- 1.Cha Y, Murray C J, Klinman J P. Science. 1989;243:1325–1330. doi: 10.1126/science.2646716. [DOI] [PubMed] [Google Scholar]
- 2.Grant K L, Klinman J P. Biochemistry. 1989;28:6597–6605. doi: 10.1021/bi00442a010. [DOI] [PubMed] [Google Scholar]
- 3.Bahnson B J, Park D-H, Kim K, Plapp B V, Klinman J P. Biochemistry. 1993;32:5503–5507. doi: 10.1021/bi00072a003. [DOI] [PubMed] [Google Scholar]
- 4.Jonsson T, Edmondson D, Klinman J P. Biochemistry. 1994;33:14871–14878. doi: 10.1021/bi00253a026. [DOI] [PubMed] [Google Scholar]
- 5.Jonsson T, Glickman M H, Sun S, Klinman J P. J Am Chem Soc. 1996;118:10319–10320. [Google Scholar]
- 6.Kohen A, Jonsson T, Klinman J P. Biochemistry. 1997;36:2603–2611. doi: 10.1021/bi962492r. [DOI] [PubMed] [Google Scholar]
- 7.Swain C G, Strivers E C, Reuwer J F, Schaad L J. J Am Chem Soc. 1958;80:5885–5893. [Google Scholar]
- 8.Saunders W H., Jr J Am Chem Soc. 1985;107:164–169. [Google Scholar]
- 9.Eklund H, Samama J-P, Jones T A. Biochemistry. 1984;23:5982–5996. doi: 10.1021/bi00320a014. [DOI] [PubMed] [Google Scholar]
- 10.Ramaswamy S, Eklund H, Plapp B V. Biochemistry. 1994;33:5230–5237. doi: 10.1021/bi00183a028. [DOI] [PubMed] [Google Scholar]
- 11.Skarzynski T, Moody P C E, Wonacott A J. J Mol Biol. 1987;193:171–187. doi: 10.1016/0022-2836(87)90635-8. [DOI] [PubMed] [Google Scholar]
- 12.Abad-Zapatero C, Griffith J P, Sussman J L, Rossmann M G. J Mol Biol. 1987;198:445–467. doi: 10.1016/0022-2836(87)90293-2. [DOI] [PubMed] [Google Scholar]
- 13.Bolin J T, Filman D J, Matthews D A, Hamlin R C, Kraut J. J Biol Chem. 1982;257:13650–13662. [PubMed] [Google Scholar]
- 14.Kelly C A, Nishiyama M, Ohnishi Y, Beppu T, Birktoft J J. Biochemistry. 1993;32:3913–3922. doi: 10.1021/bi00066a010. [DOI] [PubMed] [Google Scholar]
- 15.Sun H-W, Plapp B V. J Mol Evol. 1992;34:522–535. doi: 10.1007/BF00160465. [DOI] [PubMed] [Google Scholar]
- 16.Park D-H, Plapp B V. J Biol Chem. 1991;266:13296–13302. [PubMed] [Google Scholar]
- 17.Bahnson B J, Klinman J P. Methods Enzymol. 1995;249:373–397. doi: 10.1016/0076-6879(95)49042-6. [DOI] [PubMed] [Google Scholar]
- 18.Brünger A T. X-PLOR: A System for X-ray Crystallography and NMR. New Haven, CT: Yale University Press; 1992. , Version 3.1. [Google Scholar]
- 19.Al-Karadaghi S, Cedergren-Zeppezauer E S, Hovmoller S, Petratos K, Terry H, Wilson K. Acta Crystallogr D. 1994;50:793–807. doi: 10.1107/S0907444994005263. [DOI] [PubMed] [Google Scholar]
- 20.Klinman J P. In: Enzyme Mechanism for Isotope Effects. Cook P F, editor. Boca Raton, FL: CRC; 1991. pp. 127–148. [Google Scholar]
- 21.Huskey P, Schowen R. J Am Chem Soc. 1983;105:5704–5706. [Google Scholar]
- 22.Klinman J P. Biochemistry. 1976;15:2018–2026. doi: 10.1021/bi00654a032. [DOI] [PubMed] [Google Scholar]
- 23.Rucker J, Cha Y, Jonsson T, Grant K L, Klinman J P. Biochemistry. 1992;31:11489–11499. doi: 10.1021/bi00161a030. [DOI] [PubMed] [Google Scholar]
- 24.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 25.Cho H, Ramaswamy S, Plapp B V. Biochemistry. 1997;36:382–389. doi: 10.1021/bi9624604. [DOI] [PubMed] [Google Scholar]
- 26.Ramaswamy S, Scholze M, Plapp B V. Biochemistry. 1997;36:3522–3527. doi: 10.1021/bi962491z. [DOI] [PubMed] [Google Scholar]
- 27.Hurley T D, Bosron W F, Stone C L, Amzel L M. J Mol Biol. 1994;239:415–429. doi: 10.1006/jmbi.1994.1382. [DOI] [PubMed] [Google Scholar]
- 28.Antoniou, D. & Schwartz, S. D. (1997) Proc. Natl. Acad. Sci. USA, in press.
- 29.Almarsson Ö, Bruice T C. J Am Chem Soc. 1993;115:2125–2138. [Google Scholar]
- 30.Hwang J-K, Warshel A. J Am Chem Soc. 1996;118:11745–11751. [Google Scholar]
- 31.Shearer G L, Kim K, Lee K M, Wang C K, Plapp B V. Biochemistry. 1993;32:11186–11194. doi: 10.1021/bi00092a031. [DOI] [PubMed] [Google Scholar]
- 32.The CCP4 Suite: Programs for Protein Crystallography. Acta Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 33.Kabsch W. Acta Crystallogr A. 1976;32:922–923. [Google Scholar]
- 34.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]