

Abstract
Sphingolipids are important components of eukaryotic cells, many of which function as bioactive signaling molecules. Of these, ceramide is a central metabolite and plays key roles in a variety of cellular responses, including regulation of cell growth, viability, differentiation, and senescence. Ceramide is composed of the long-chain sphingoid base, sphingosine, in N-linkage to a variety of acyl groups. Sphingosine serves as the product of sphingolipid catabolism, and it is mostly salvaged through re-acylation, resulting in the generation of ceramide or its derivatives. This recycling of sphingosine is termed the “salvage pathway”, and recent evidence points to important roles for this pathway in ceramide metabolism and function. A number of enzymes are involved in the salvage pathway, and these include sphingomyelinases, cerebrosidases, ceramidases, and ceramide synthases. Recent studies suggest that the salvage pathway is not only subject to regulation, but it also modulates the formation of ceramide and subsequent ceramide-dependent cellular signals. This review focuses on the salvage pathway in ceramide metabolism, its regulation, its experimental analysis, and emerging biological functions.
Keywords: ceramide, ceramide signal, fumonisin B1, protein kinase C, salvage pathway, sphingosine, sphingolipid
1. Introduction
Ceramide is a bioactive sphingolipids that has been implicated in mediating or regulating many cellular processes, including cell cycle arrest, apoptosis, senescence, and stress responses [1]. Formation of ceramide can be induced by different stimuli such as tumor necrosis factor-α (TNF-α) [2], Fas ligand [3], phorbol ester [4], heat stress [5], oxidative stress [6], ionizing radiation [7], and chemotherapeutics [8].
Multiple metabolic pathways converge upon ceramide, and thus far the de novo pathway and the sphingomyelinase (SMase) pathway (Fig. 1) have been the subject of intense study. The de novo synthesis of ceramide commences with the condensation of serine and fatty acyl-CoA via action of serine palmitoyltransferase, followed by the consecutive action of 3-ketodihydrosphingosine reductase, dihydroceramide synthases, and dihydroceramide desaturase [9, 10]. This pathway can be metabolically induced in response to metabolic loading with either serine or palmitate [11]. It is also activated by chemotherapeutic agents [8], heat stress [5], oxidized LDL [12], and cannabinoids [13]. Studies have implicated the ceramide formed de novo in mediating some of the effects of these inducers on stress responses, and apoptosis [5, 8, 13].
Figure 1. Ceramide synthesis.
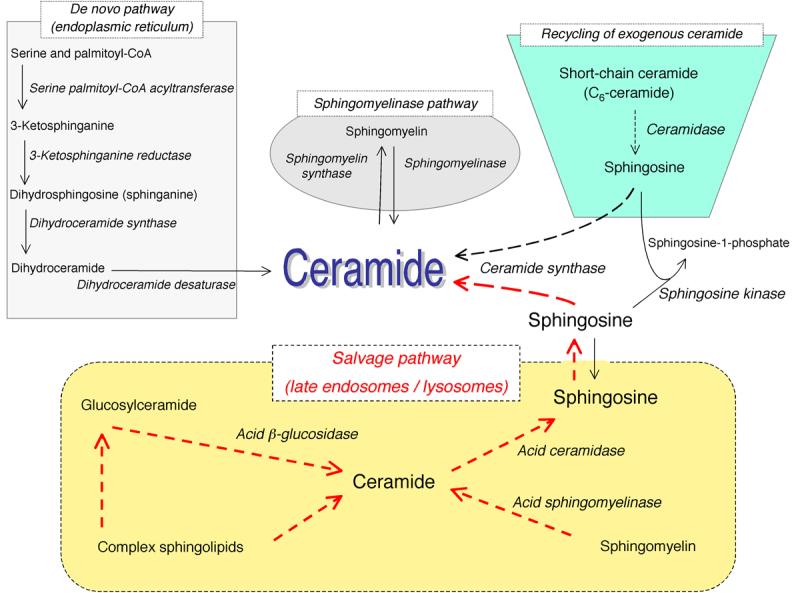
The scheme shows metabolic pathways for ceramide synthesis composed of the sphingomyelinase pathway, the de novo pathway, the exogenous ceramide-recycling pathway, and the salvage pathway. Dotted lines indicate the pathway of ceramide synthesis resulting from recycling/salvaging sphingosine.
Ceramide can be also generated from hydrolysis of sphingomyelin through the action of either acid or neutral SMases [14]. These enzymes break down sphingomyelin to produce ceramide and phosphocholine, and they are stimulated in response to cell treatment with TNF-α [15], Fas ligand [3], or oxidative stress [6].
Recent studies have revealed a more complex mechanism of ceramide accumulation from the catabolism of complex sphingolipids that are broken down eventually into sphingosine, which is then reused through reacylation to produce ceramide. This latter pathway has been referred to as either sphingolipid recycling or the salvage pathway [16-21]. This pathway involves a number of key enzymes that include SMases, possibly glucocerebrosidase (acid-β-glucosidase), ceramidases, and (dihydro)ceramide synthases (Fig. 1).
A growing body of evidence is starting to point toward roles for ceramide generated through the salvage pathway in many biological responses, such as growth arrest [18], apoptosis [22], cellular signaling [23] and trafficking [17]. Indeed, this may emerge as a commonly employed pathway in cell regulation that may even reconcile some of the previous seemingly contradictory results on activation of acid SMase vs the de novo pathway.
Differential activation of these distinct pathways of ceramide formation is possible due to spatial separation of enzymes that contribute to ceramide formation. For example, serine palmitoyltransferase, the limiting enzyme of the de novo pathway is localized in the endoplasmic reticulum [24], and this endoplasmic reticulum-derived dihydrosphingosine serves as a substrate for dihydroceramide biosynthesis through the de novo pathway. By contrast sphingosine is generated from breakdown of complex sphingolipids in lysosomes, and then it could serve as a substrate for ceramide re-generation (the salvage pathway), possibly in endoplasmic reticulum or in endoplasmic reticulum-associated membranes. In addition, it is possible, if not likely, that distinct (dihydro)ceramide synthase isoforms with differential cellular localization may be involved in the de novo vs. the salvage pathway. Cellular homeostasis in response to different stimuli will depend on which pathway of ceramide formation is activated, as well as when and where in the cell this occurs.
The metabolism and role of ceramide and sphingolipids in general have been recently addressed in several reviews [25-29]. This review will focus on the emerging ceramide salvage pathway and its potential roles in physiological and pathological processes.
2. The salvage pathway
2-1 Sphingolipid biosynthesis
Long chain sphingoid bases (dihydrosphingosine and sphingosine) serve as the backbones of mammalian sphingolipids, and are considered the chemically-defining feature of sphingolipids. The de novo biosynthesis of ceramide occurs in the endoplasmic reticulum and commences with the action of serine palmitoyltransferase which condenses preferentially serine and palmitoyl CoA to form 3-ketodihydrosphingosine. This is then reduced to form dihydrosphingosine (sphinganine), which is then acylated by dihydroceramide synthases. The activities for acylation of long chain bases (both dihydrosphingosine and sphingosine) have been found to reside in microsomes. In mammals, six genes that encode (dihydro)ceramide synthase have been recently cloned and termed longevity-assurance homologues (LASSs: LASS1-6)/ceramide synthases (CerSs; CerS1-6) [30-33]. Biochemically, individual ceramide synthase isoforms show substrate preference for specific chain length fatty acyl CoAs, thus generating distinct ceramides with distinct acyl-chain lengths. For example, LASS1 shows significant preference for C18:0 fatty acid CoA, whereas LASS5 and LASS6 preferentially catalyze the acylation of (dihydro)sphingosine with myristoyl-, palmitoyl- and stearoyl-CoA rather than very long-chain fatty acid CoAs [31, 34]. Initial studies regarding the subcellular localization of LASS family members demonstrate that, in over-expression models of genes, tagged-LASS 1/4/5/6 members were commonly observed to reside at the endoplasmic reticulum [31, 32, 35]. Dihydroceramides with distinct fatty acyl moieties are then desaturated by dihydroceramide desaturase to form the corresponding ceramides [36].
Biosynthesis of complex sphingolipids follows the formation of ceramide on the membranes of the endoplasmic reticulum and continues in the Golgi apparatus. This requires either vesicular transport of ceramide to the Golgi or protein-facilitated transport by the action of the ceramide transfer protein CERT [37]. The addition of a glucose residue in a glycosidic linkage to ceramide occurs at the Golgi apparatus, forming glucosylceramide, a predominant precursor of complex glycosphingolipids [21]. In addition to glycosylation, the primary alcoholic moiety of ceramide serves as the attachment site for a phosphate or for a phosphocholine, producing ceramide-1-phosphate or sphingomyelin, respectively [38-40]. These sphingolipids and glycosphingolipids are then distributed to plasma membranes and subcellular organelles and undergo the turnover with degradation and re-generation.
2-2 The sphingosine-ceramide salvage pathway in sphingolipid turnover
Constitutive degradation of sphingolipids and glycosphingolipids [19, 41] takes place in the acidic subcellular compartments, the late endosomes and the lysosomes (Fig. 1). In case of glycosphingolipids, exohydrolases, acting at acidic pH optima, cause the stepwise release of monosaccharide units from the end of the oligosaccharide chains one after the other, leading to the generation of ceramide. Sphingomyelin is converted to ceramide by acid SMase [42]. The common metabolic product, ceramide, can be further hydrolyzed by acid ceramidase to form sphingosine and free fatty acid, both of which are able to leave the lysosome [43], in contrast to ceramide which does not appear to leave the lysosome [44]. The long chain sphingoid bases released from the lysosome may then re-enter pathways for synthesis of ceramide and/or sphingosine-1-phosphate (Fig. 1). The latter lipid is generated by sphingosine kinases (the reader is referred to many excellent reviews on sphingosine kinases) [45-47]. In this review, we will focus on the regeneration of ceramide from breakdown of complex sphingolipids (the salvage pathway), and mostly focus on the lysosomal arm of breakdown of complex sphingolipids.
It should be noted that whereas free dihydrosphingosine is mostly generated by de novo sphingolipid biosynthesis, free sphingosine appears to derive exclusively from the turnover of glycosphingolipids, sphingomyelin or ceramide. Indeed, none of dihydrosphingosine desaturases have been identified so far. Thus all sphingosine should be considered a product of hydrolysis of complex sphingolipids and then ceramide. It follows then that sphingosine-1-phosphate in turn is also strictly a product of sphingolipid breakdown.
The salvage pathway of long chain sphingoid bases, leading to the re-generation of sphingolipids, has been estimated to contribute from 50% to 90% of sphingolipid biosynthesis [16, 48]. These metabolic considerations suggest a crucial role for sphingolipid breakdown in sphingolipid biosynthesis/turnover as well as in cellular signal transduction.
The salvage pathway re-utilizes long chain sphingoid bases to form ceramide through the action of ceramide synthase [20]. Thus, LASS family members probably trap free sphingosine released from lysosome at the surface of the endoplasmic reticulum or in endoplasmic reticulum-associated membranes. Interesting insight into selective fates of free sphingosine has been provided by Sonnino and co-workers [49]. They showed that, in a cell culture model, exogenous sphingolipids in serum that are taken up by the cells directly reach the lysosomes largely to be catabolized to small fragments and water (presumably by being converted first to sphingosine-1-phosphate and then ethanolamine phosphate and palmitaldehyde). In contrast, the majority of endogenous sphingosine is recycled. Importantly, the study suggests that intrinsic turnover ofsphingolipids and glycosphingolipids mostly occurs independently of extracellular lipid-protein complexes such as lipoproteins that must be trapped by cells.
2-3 Deacylation/reacylation of exogenous short-chain ceramide
Short-chain ceramides (C2-ceramide and C6-ceramide) are water-soluble and cell-membrane permeable, and thus, unlike natural long and very long chain ceramides, can be easily delivered to cells [50]. Extensive studies using these short-chain ceramides have been performed to examine the biological aspects of ceramide, revealing intracellular ceramide targets as well as various ceramide signaling pathways [51-55]. Importantly, recent studies also shed light on the metabolic fate of short-chain ceramides [18, 22, 56] (Fig. 1). Exogenous C6-ceramide was subject to deacylation whereas C2-ceramide was defective in deacylation [18, 22], which might arise from the selectivity (substrate preference) of ceramidases. The deacylation process thus becomes a rate-limiting step in determining the extent of availability of sphingosine backbone of exogenous ceramide for further metabolism.
The specific fates of exogenous C6-ceramide were evaluated by employing radio-labeled sphingosine backbones or LC/MS using C17-sphingosine which does not naturally exist [56]. Following C6-ceramide treatment, endogenous long-chain ceramide (mostly C16:0-ceramide) was generated (containing the sphingoid base of the C6-ceramide). Pharmacological approaches using a ceramide synthase inhibitor (fumonisin B1) [57] and metabolic labeling of the C6 fatty acid [18] revealed that this process was attributed to recycling of the sphingosine backbone of C6-ceramide via deacylation/reacylation, but not to the elongation of its fatty acid moiety. Thus, action of ceramidases (or other N-deacylases) results in releasing free sphingosine which in turn undergoes re-acylation in a ceramide synthase-dependent manner. At this point, the specific ceramidases or ceramide synthases (LASSs/CerSs) involved in this recycling are not well defined.
Interestingly, the salvage of the sphingosine backbone of exogenous short-chain ceramide to form endogenous long-chain ceramide was attenuated by brefeldin A, a mycotoxin that causes disassembly of the Golgi apparatus [56]. This mycotoxin is likely to act mainly at the deacylation step. Thus, deacylation of short-chain ceramide is suggested to occur at the intact Golgi apparatus. Mao's group has recently cloned a novel human alkaline ceramidase (termed alkaline ceramidase 2) which selectively localizes to the Golgi apparatus [58]. In light of the Golgi-dependent deacylation of short-chain ceramide, alkaline ceramidase 2 might play a role in deacylation of short-chain ceramide and resultant release of free sphingosine. Accordingly, a unique cross-talk between Golgi alkaline ceramidase and ceramide synthases is presumed to account for the recycling of sphingosine during the turnover of exogenous short-chain ceramides.
3. Experimental analysis of the salvage pathway
There are many reasons for the delayed appreciation and understanding of the salvage pathway. Some have to do with the delayed appreciation of the ‘metabolic connectivity’ of sphingolipid metabolism. At a practical level, the absence of metabolic methodology and absence of selective inhibitors for the key enzymes of the salvage pathway (LASS family members, acid SMase and acid ceramidase) have also contributed to this paucity of studies. The following approaches are recommended in order to ascertain the operation of the salvage pathway and its contribution to ceramide synthesis:
-
i) Fumonisin B1-sensitive/ myriocin-insensitive formation of ceramide
The ceramide salvage pathway differs from the ceramide de novo pathway in metabolic features (catabolism vs anabolism), but these pathways share a singular step where long-chain sphingoid bases are acylated to ceramide in a (dihydro)ceramide synthase-dependent manner. A number of metabolic inhibitors have been discovered to block particular catalytic steps in these pathways [59], and two of the best-studied inhibitors of ceramide synthesis are myriocin/ISP-1 [60] and fumonisin B1 [57]. The former is a selective inhibitor of serine palmitoyltransferase, being selectively capable of blocking the de novo pathway for ceramide synthesis. In contrast, fumonisin B1, a potent inhibitor of (dihydro)ceramide synthases, is able to significantly inhibit both the de novo and the salvage pathways, leading to inhibition of ceramide synthesis as well as reciprocal accumulation of free long chain sphingoid bases. Accordingly, the salvage pathway-derived synthesis of ceramide is sensitive to fumonisin B1 but not to myriocin/ISP-1, and differential sensitivity of ceramide synthesis to the two inhibitors can distinguish the salvage pathway from the de novo biosynthesis. However, caution should be employed in the use of myriocin, since this inhibitor will, if employed over several hours, inhibit all sphingolipid synthesis, including the synthesis of complex sphingolipids. As such, it would also inhibit ceramide formed from the salvage pathway by depleting the precursor sphingomyelin and/or glucosylceramide. Thus, these two pharmacological inhibitors provide powerful tools for determining the pathway responsible for ceramide synthesis.
-
ii) Selective palmitate incorporation into ceramide in chase versus pulse labeling
The metabolic origins of ceramide can be evaluated by metabolic labeling methods using radioactive ceramide precursors (palmitate or serine), in either “pulse labeling” or “steady state labeling” approaches. Activation of the de novo pathway can be detected selectively by pulse labeling with precursors, whereas the salvage pathway can be detected selectively by steady state labeling and analysis of turnover since the label has to be incorporated into complex sphingolipids before further hydrolysis and reincorporation into ceramide. Accordingly, the salvage pathway can be ‘diagnosed’ by metabolic labeling approaches.
-
iii) An increase in free sphingosine
In the steady state, the cellular level of free sphingosine is determined by the net result of metabolic flux between catabolism and anabolism of sphingolipids. An increase in sphingosine has been shown to occur in response to TNF-α [61], serum starvation [62], interleukin-1β [63], bradykinin [64], phorbol ester [17], and Fas ligands [65]. The release of sphingosine is presumed to arise from enhanced degradation of complex sphingolipids, sphingomyelin or ceramide. Subsequently, sphingosine kinases or ceramide synthases can trap free sphingosine, resulting in formation of sphingosine-1-phosphate or ceramide, respectively. Sphingosine formation is not always associated with ceramide synthesis (the salvage pathway). Importantly, the detection of an increase in free sphingosine not only indicates enhancement of sphingolipid catabolism, but also raises the possibility of facilitated sphingosine salvage for ceramide synthesis.
-
iv) Use of RNA interference (RNAi)-based studies to dissect the enzymes involved in the salvage pathway
As shown in Fig. 1, a number of enzymes participate in the salvage pathway, and these include members of the LASS family, acid SMase, and acid ceramidase. The relatively recent development of RNAi [66] has allowed the selective knock-down of these genes individually [23, 67-70]. The implication of several of the above genes along the pathway of ceramide formation provides strong evidence for the operation of this pathway.
4. Regulation of the salvage pathway
4-1, The salvage pathway and protein kinase C (PKC)
Recent studies have begun to implicate members of the PKC of Ser/Thr protein kinases as upstream regulators of the sphingoid base salvage pathway resulting in ceramide synthesis following PKC activation in human breast cancer cell line (MCF-7 cells) [17, 23].Initially, it was shown that PKC activation by phorbol esters stimulated the formation of ceramide, and it was concluded that phorbol esters activated the de novo pathway [4], based on the sensitivity of ceramide formation to fumonisin B1. However, in a more recent study, it was shown that phorbol ester-induced ceramide was not myriocin-sensitive. Labeling studies demonstrated that phorbol esters stimulated turnover of ‘steady state’ labeled sphingolipids but not pulse labeled ceramide, ruling out an involvement of the de novo pathway in ceramide accumulation and indicating the contribution of the salvage pathway. Moreover, mass spectrometric analysis demonstrated an elevation of free sphingosine following PKC activation, further suggesting enhanced degradation of sphingolipids or glycosphingolipids. In light of those results, it was suggested that PKC stimulates the sphingosine-salvage pathway for ceramide synthesis [17].
Interestingly, PKC activation of the salvage pathway induced a selective increase in certain ceramide species (predominantly C16:0-ceramide) following the generation of free sphingosine [17]. Further studies using RNAi revealed an involvement of specific LASS in the phorbol-ester induced ceramide synthesis [23]. In agreement with in vitro substrate preference of LASS5 [34], C16:0-ceramide synthesis was partially attributed to action of LASS5 [23]. Therefore, LASS5 appears to be primarily involved in the re-acylation of salvaged sphingosine.
Cell fractionation studies suggested that the biosynthesis of ceramide in response to phorbol ester led to partial accumulation of ceramide in (or close to) mitochondria (e.g. in mitochondria-associated membranes) [23]. Thus, fumonisin B1-sensitive elevation of C16:0-ceramide was observed in the mitochondria-enriched heavy membrane fraction. Ceramide synthase activity [71] has been observed in mitochondria-associated membranes where free sphingoid bases are converted to (dihydro)ceramide by (dihydro)ceramide synthases [23]. Moreover, ceramide-enriched mitochondria upon treatment with phorbol esters were seen to localize around the nuclear envelope where LASS5 as well as other LASS family members are present [31, 32, 35]. These spatial changes in subcellular compartments (mitochondria, endoplasmic reticulum, and mitochondria-associated membranes) may play a key role in allowing the accumulation of C16:0-ceramide in mitochondria and/or mitochondria-associated membranes.
Additional studies have implicated acid SMase in the generation of ceramide following PKC activation in MCF-7 cells, such that RNAi directed against acid SMase prevented phorbol ester-induced sphingomyelin hydrolysis and caused a significant reduction in the accumulating ceramide [72]. In light of previous studies demonstrating that lysosomal sphingomyelin degradation results in generation of sphingosine which is salvaged for ceramide synthesis [42], acid SMase appears to be a key component in PKC-dependent ceramide synthesis through the salvage pathway.
Studies conducted to determine which PKC isoform is involved in ceramide accumulation showed that specific pharmacologic inhibition or knock-down of the PKCδ isoform completely attenuated ceramide formation following phorbol ester treatment [72]. Additional studies showed that PKCδ activates acid SMase, in a process involving phosphorylation of acid SMase. Thus, PKCδ is likely to elicit ceramide formation through the salvage pathway by controlling sphingolipid turnover at the level of acid SMase (Fig. 2).
Figure 2. Regulation of the salvage pathway and function of ceramide signals.
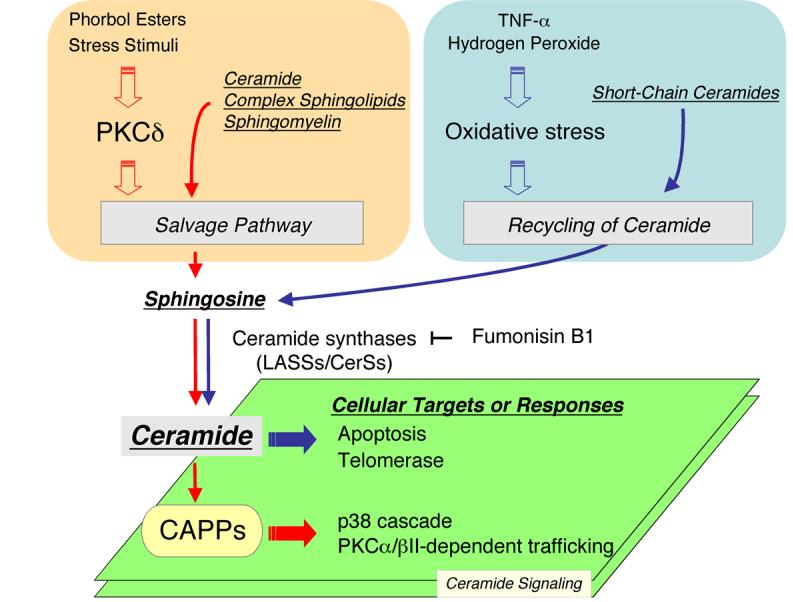
PKC, protein kinase C; CAPP, ceramide-activated protein phosphatase
4-2, Cellular redox state and turnover of short-chain ceramides
Cellular redox state plays an important role in regulating various signal transduction processes [73, 74]. Exposure of cells to hydrogen peroxide induces oxidative stress, which is known to evoke ceramide generation concomitant with sphingomyelin degradation [6]. Sultan et al. demonstrated that hydrogen peroxide accelerated the rate of recycling of sphingosine (derived from short-chain ceramide) for regeneration of endogenous long-chain ceramide [56]. In addition, TNF-α, a known inducer of oxidative stress, also enhanced the generation of long-chain ceramide through the sphingosine recycling pathway. It still remains to be determined whether the effects are on the deacylation or re-acylation steps following TNF-α stimulation. A number of studies have reported on the ability of TNF-α to induce the formation of ceramide through either the SMase or the de novo pathways [2, 15, 75, 76]. Therefore the above studies also suggest activation of the salvage pathway, and perhaps this could encompass the activation of acid SMase and the fumonisin B1-inhibitable accumulation of ceramide. Interestingly, TNF-α also increases sphingosine kinase activity and the accumulation of sphingosine-1-phosphate [77]. In light of the salvage pathway, those findings might imply that TNF-α increases free sphingosine which could then be made available for further metabolism (ceramide and/or sphingosine-1-phosphate).
5. Functions of the salvage pathway
Three pathways (the SMase pathway, the de novo pathway, and the salvage pathway) individually or coordinately contribute to ceramide synthesis, leading to the generation of ceramide signaling and subsequent modulation of downstream cell responses. Little is known on the ceramide signaling governed by the salvage pathway, whereas the other pathways have been better investigated. In this section, results from recent studies that begin to demonstrate roles for the salvage pathway in cellular responses are summarized (Fig. 2).
5-1, Inactivation of the p38 MAPK and role of ceramide-activated protein phosphatases (CAPPs)
Ceramide regulates several signal transduction pathways, and the molecular mechanisms have been investigated extensively. Successfully, some ceramide targets have been identified, including PKCζ [52], kinase suppressor of Ras [78], cRaf [51], cathepsin D [79], and CAPPs composed of protein phosphatase 1 (PP1) and protein phosphate 2A (PP2A) [80-82]. CAPPs belong to the family of Ser/Thr protein phosphatases, and ceramide has been shown to activate CAPPs leading to the dephosphorylation of various proteins including, Bax, Bcl-2, PKCα, Rb protein, SR protein, and Akt, ultimately modulating/mediating various responses of cells [29, 83]. In PKC signaling, ceramide derived from the salvage pathway leads to attenuation/termination of activation of the p38 MAPK through dephosphorylation. Pharmacologic and siRNA-based studies have implicated PP1 catalytic isoforms (PP1c-α, PP1c-β, and PP1c-γ) in mediating the effects of ceramide on p38 MAPK [23]. Moreover, the PP1-dependent dephosphorylation of p38 likely depends on LASS5, since knock-down of LASS5 attenuated PP1 dephosphorylation of p38 concomitant with suppressing ceramide synthesis following stimulation with phorbol ester. Therefore, the link of the salvage pathway to PP1 appears crucial in control of p38 dephosphorylation.
Interestingly, PP1 was found to be recruited to the mitochondrial fraction, enriched in ceramide, following activation of the salvage pathway [23].
5-2, Inhibition of PKC-dependent trafficking by ceramide
Ongoing studies implicate ceramide deriving from the salvage pathway in feedback inhibition of PKCα/βII [17, 84]. Recent studies have shown that these two classical PKCs are sequestered into a subset of recycling endosomes, termed the “pericentrion” which is associated with the Rab11-positive compartment [85, 86]. The formation of the pericentrion regulates endocytosis and results in sequestration of several recycling components (e.g. GM1 ganglioside and CD59) [84]. This sequestration requires dual phosphorylation of PKCα/βII at the carboxyl-terminal sites (Thr638/641 and Ser657/660). Ceramide formed from the salvage pathway was shown to induce the dephosphorylation of PKCα/βII at Thr638/641 in a manner dependent on PP1c-α and PP1c/-β, thus inhibiting translocation of PKCα/βII to the pericentrion. Accordingly, these findings suggest that the salvage pathway-generated ceramide counteracts PKC-dependent trafficking.
5-3, Telomerase activity and recycling of short-chain ceramide
Multiple lines of evidence have demonstrated the ability of endogenous ceramide to inhibit telomerase activity in A549 cells [87, 88]. Moreover, exogenous C6-ceramide was also shown to inhibit telomerase, but interestingly, it was found that these actions are due to generation of endogenous ceramide in a process that requires recycling of the sphingosine backbone of C6-ceramide through deacylation followed by reacylation to mainly C16:0 ceramide [18]. Subsequently, Ogretmen and co-workers showed that the ceramide/sphingosine recycling pathway is regulated by reactive oxygen species at the deacylation and/or reacylation steps [56] . It was also shown that ceramide generated from the salvage pathway is involved in inhibition of the binding of the c-Myc transcription factor to the promoter of hTERT (human telomerase reverse transcriptase) and consequently, inhibition of telomerase activity.
5-4, Recycling/salvage pathways and apoptosis
Takeda et al. reported that, in HaCaT cells, the sphingosine backbone of the short-chain (C6-ceramide) is reacylated to form long-chain ceramide [22]. Interestingly, in these cells, treatment with either C6-ceramide or sphingosine induced apoptosis, which was significantly attenuated by fumonisin B1 pretreatment, thus suggesting a crucial role for the salvage pathway in mediating these apoptotic responses.
Moreover, in some apoptosis models, inhibition of ceramide synthases by fumonisin B1 is known to decrease the susceptibility of cells to apoptosis following treatment with TNF-α [75], phorbol ester [4], or ultraviolet radiation [89]. However, in those models, the contribution of the salvage pathway, as opposed to the de novo pathway, to apoptosis remains unclear, especially since fumonisin B1 blocks both the de novo pathway as well as the salvage pathway.
6. Sphingolipidoses
The break-down of sphingolipids for salvaging sphingosine requires their systematic catabolism via the action of a number of enzymes in acidic compartments (late endosomes/lysosomes) [21, 41, 48]. Genetic defects in several of these hydrolytic enzymes cause various disorders with lysosomal accumulation of the substrate lipids, a group of disorders termed the sphingolipidoses [41, 90-93]. Particularly, some of the known sphingolipidoses might closely associate with aberrant metabolisms of ceramide because of defective activities of ceramide generating/degrading enzymes: Farber's disease, Gaucher disease, and Niemann-Pick type A and B diseases are caused by a deficiency of acid ceramidase [94, 95], glucocerebrosidase (acid-β-glucosidase) [96-98], acid SMase [99, 100], respectively. The salvage pathway is one of the routes for controlling cellular levels of ceramide. Thus, the defects in these enzymes may lead to dysfunction in the salvage pathway, presumably impairing cellular homeostasis mediated by ceramide signaling. However, it remains largely hypothetical at this point.
7. Conclusions
Three pathways contribute to the generation of ceramide, which is emerging as a key bioactive lipid that mediates/regulates several cellular responses. Recent studies have identified the salvage pathway as a novel pathway involved in regulating and subsequent ceramide-mediated signaling. Interestingly, this pathway may already explain and reconcile previous results on the relative roles of the contribution of acid SMase and the ‘de novo’ pathway. The latter has often been implicated based on inhibition by fumonisin B1 which does not distinguish the de novo synthesis from the salvage/recycling pathway. Clearly, at this point, many of the key players and mechanisms of regulation of this pathway remain to be defined. It is expected, however, that much more research will focus on the salvage pathway, and the resultant information will continue to reveal the complexity and importance of this metabolic pathway in cellular responses and in various disorders.
Acknowledgements
This work was supported in part by NIH grants CA 97132. We would like to extend a special thanks to Drs. L. Ashley Cowart and Stefka Spassieva for critical review.
The abbreviations used are
- CAPP
ceramide-activated protein phosphatase
- CerS
ceramide synthase
- LASS
longevity-assurance homologue
- PKC
protein kinase C
- PP1
protein phosphatase 1
- PP2A
protein phosphatase 2A
- RNAi
RNA interference
- SMase
sphingomyelinase
- TNF-α
tumor necrosis factor-α
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hannun YA. Science. 1996;274:1855–1859. doi: 10.1126/science.274.5294.1855. [DOI] [PubMed] [Google Scholar]
- 2.Schutze S, Potthoff K, Machleidt T, Berkovic D, Wiegmann K, Kronke M. Cell. 1992;71:765–776. doi: 10.1016/0092-8674(92)90553-o. [DOI] [PubMed] [Google Scholar]
- 3.Brenner B, Ferlinz K, Grassme H, Weller M, Koppenhoefer U, Dichgans J, Sandhoff K, Lang F, Gulbins E. Cell Death Differ. 1998;5:29–37. doi: 10.1038/sj.cdd.4400307. [DOI] [PubMed] [Google Scholar]
- 4.Garzotto M, White-Jones M, Jiang Y, Ehleiter D, Liao WC, Haimovitz-Friedman A, Fuks Z, Kolesnick R. Cancer Res. 1998;58:2260–2264. [PubMed] [Google Scholar]
- 5.Jenkins GM, Cowart LA, Signorelli P, Pettus BJ, Chalfant CE, Hannun YA. J. Biol. Chem. 2002;277:42572–42578. doi: 10.1074/jbc.M207346200. [DOI] [PubMed] [Google Scholar]
- 6.Goldkorn T, Balaban N, Shannon M, Chea V, Matsukuma K, Gilchrist D, Wang H, Chan C. J. Cell Sci. 1998;111:3209–3220. doi: 10.1242/jcs.111.21.3209. [DOI] [PubMed] [Google Scholar]
- 7.Haimovitz-Friedman A, Kan CC, Ehleiter D, Persaud RS, McLoughlin M, Fuks Z, Kolesnick RN. J. Exp. Med. 1994;180:525–535. doi: 10.1084/jem.180.2.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bose R, Verheij M, Haimovitz-Friedman A, Scotto K, Fuks Z, Kolesnick R. Cell. 1995;82:405–414. doi: 10.1016/0092-8674(95)90429-8. [DOI] [PubMed] [Google Scholar]
- 9.Perry DK. Biochim. Biophys. Acta. 2002;1585:146–152. doi: 10.1016/s1388-1981(02)00335-9. [DOI] [PubMed] [Google Scholar]
- 10.Menaldino DS, Bushnev A, Sun A, Liotta DC, Symolon H, Desai K, Dillehay DL, Peng Q, Wang E, Allegood J, Trotman-Pruett S, Sullards MC, Merrill AH., Jr. Pharmacol. Res. 2003;47:373–381. doi: 10.1016/s1043-6618(03)00054-9. [DOI] [PubMed] [Google Scholar]
- 11.Shimabukuro M, Zhou YT, Levi M, Unger RH. Proc. Natl. Acad. Sci. U. S. A. 1998;95:2498–2502. doi: 10.1073/pnas.95.5.2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kitatani K, Nemoto M, Akiba S, Sato T. Cell. Signal. 2002;14:695–701. doi: 10.1016/s0898-6568(02)00014-1. [DOI] [PubMed] [Google Scholar]
- 13.Gomez del Pulgar T, Velasco G, Sanchez C, Haro A, Guzman M. Biochem. J. 2002;363:183–188. doi: 10.1042/0264-6021:3630183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marchesini N, Hannun YA. Biochem. Cell Biol. 2004;82:27–44. doi: 10.1139/o03-091. [DOI] [PubMed] [Google Scholar]
- 15.Kim MY, Linardic C, Obeid L, Hannun Y. J. Biol. Chem. 1991;266:484–489. [PubMed] [Google Scholar]
- 16.Gillard BK, Clement RG, Marcus DM. Glycobiology. 1998;8:885–890. doi: 10.1093/glycob/8.9.885. [DOI] [PubMed] [Google Scholar]
- 17.Becker KP, Kitatani K, Idkowiak-Baldys J, Bielawski J, Hannun YA. J. Biol. Chem. 2005;280:2606–2612. doi: 10.1074/jbc.M409066200. [DOI] [PubMed] [Google Scholar]
- 18.Ogretmen B, Pettus BJ, Rossi MJ, Wood R, Usta J, Szulc Z, Bielawska A, Obeid LM, Hannun YA. J. Biol. Chem. 2002;277:12960–12969. doi: 10.1074/jbc.M110699200. [DOI] [PubMed] [Google Scholar]
- 19.Riboni L, Viani P, Bassi R, Prinetti A, Tettamanti G. Prog. Lipid Res. 1997;36:153–195. doi: 10.1016/s0163-7827(97)00008-8. [DOI] [PubMed] [Google Scholar]
- 20.Smith ER, Merrill AH., Jr. J. Biol. Chem. 1995;270:18749–18758. doi: 10.1074/jbc.270.32.18749. [DOI] [PubMed] [Google Scholar]
- 21.Tettamanti G. Glycoconj. J. 2004;20:301–317. doi: 10.1023/B:GLYC.0000033627.02765.cc. [DOI] [PubMed] [Google Scholar]
- 22.Takeda S, Mitsutake S, Tsuji K, Igarashi Y. J. Biochem. (Tokyo) 2006;139:255–262. doi: 10.1093/jb/mvj026. [DOI] [PubMed] [Google Scholar]
- 23.Kitatani K, Idkowiak-Baldys J, Bielawski J, Taha TA, Jenkins RW, Senkal CE, Ogretmen B, Obeid LM, Hannun YA. J. Biol. Chem. 2006;281:36793–36802. doi: 10.1074/jbc.M608137200. [DOI] [PubMed] [Google Scholar]
- 24.Mandon EC, Ehses I, Rother J, van Echten G, Sandhoff K. J. Biol. Chem. 1992;267:11144–11148. [PubMed] [Google Scholar]
- 25.Kolesnick RN, Kronke M. Annu. Rev. Physiol. 1998;60:643–665. doi: 10.1146/annurev.physiol.60.1.643. [DOI] [PubMed] [Google Scholar]
- 26.Zheng W, Kollmeyer J, Symolon H, Momin A, Munter E, Wang E, Kelly S, Allegood JC, Liu Y, Peng Q, Ramaraju H, Sullards MC, Cabot M, Merrill AH., Jr. Biochim. Biophys. Acta. 2006;1758:1864–1884. doi: 10.1016/j.bbamem.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 27.Levade T, Auge N, Veldman RJ, Cuvillier O, Negre-Salvayre A, Salvayre R. Circ. Res. 2001;89:957–968. doi: 10.1161/hh2301.100350. [DOI] [PubMed] [Google Scholar]
- 28.Futerman AH, Riezman H. Trends Cell Biol. 2005;15:312–318. doi: 10.1016/j.tcb.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 29.Ogretmen B, Hannun YA. Nat. Rev. Cancer. 2004;4:604–616. doi: 10.1038/nrc1411. [DOI] [PubMed] [Google Scholar]
- 30.Mizutani Y, Kihara A, Igarashi Y. Biochem. J. 2006:531–538. doi: 10.1042/BJ20060379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mizutani Y, Kihara A, Igarashi Y. Biochem. J. 2005;390:263–271. doi: 10.1042/BJ20050291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Riebeling C, Allegood JC, Wang E, Merrill AH, Jr., Futerman AH. J. Biol. Chem. 2003;278:43452–43459. doi: 10.1074/jbc.M307104200. [DOI] [PubMed] [Google Scholar]
- 33.Pewzner-Jung Y, Ben-Dor S, Futerman AH. J. Biol. Chem. 2006;281:25001–25005. doi: 10.1074/jbc.R600010200. [DOI] [PubMed] [Google Scholar]
- 34.Lahiri S, Futerman AH. J. Biol. Chem. 2005;280:33735–33738. doi: 10.1074/jbc.M506485200. [DOI] [PubMed] [Google Scholar]
- 35.Venkataraman K, Riebeling C, Bodennec J, Riezman H, Allegood JC, Sullards MC, Merrill AH, Jr., Futerman AH. J. Biol. Chem. 2002;277:35642–35649. doi: 10.1074/jbc.M205211200. [DOI] [PubMed] [Google Scholar]
- 36.Michel C, van Echten-Deckert G, Rother J, Sandhoff K, Wang E, Merrill AH., Jr. J. Biol. Chem. 1997;272:22432–22437. doi: 10.1074/jbc.272.36.22432. [DOI] [PubMed] [Google Scholar]
- 37.Hanada K, Kumagai K, Yasuda S, Miura Y, Kawano M, Fukasawa M, Nishijima M. Nature. 2003;426:803–809. doi: 10.1038/nature02188. [DOI] [PubMed] [Google Scholar]
- 38.Tafesse FG, Ternes P, Holthuis JC. J. Biol. Chem. 2006;281:29421–29425. doi: 10.1074/jbc.R600021200. [DOI] [PubMed] [Google Scholar]
- 39.Sugiura M, Kono K, Liu H, Shimizugawa T, Minekura H, Spiegel S, Kohama T. J. Biol. Chem. 2002;277:23294–23300. doi: 10.1074/jbc.M201535200. [DOI] [PubMed] [Google Scholar]
- 40.Chalfant CE, Spiegel S. J. Cell Sci. 2005;118:4605–4612. doi: 10.1242/jcs.02637. [DOI] [PubMed] [Google Scholar]
- 41.Kolter T, Sandhoff K. Annu. Rev. Cell Dev. Biol. 2005;21:81–103. doi: 10.1146/annurev.cellbio.21.122303.120013. [DOI] [PubMed] [Google Scholar]
- 42.Riboni L, Prinetti A, Bassi R, Tettamanti G. FEBS Lett. 1994;352:323–326. doi: 10.1016/0014-5793(94)00984-8. [DOI] [PubMed] [Google Scholar]
- 43.Riboni L, Bassi R, Caminiti A, Prinetti A, Viani P, Tettamanti G. Ann. N. Y. Acad. Sci. 1998;845:46–56. doi: 10.1111/j.1749-6632.1998.tb09661.x. [DOI] [PubMed] [Google Scholar]
- 44.Chatelut M, Leruth M, Harzer K, Dagan A, Marchesini S, Gatt S, Salvayre R, Courtoy P, Levade T. FEBS Lett. 1998;426:102–106. doi: 10.1016/s0014-5793(98)00325-1. [DOI] [PubMed] [Google Scholar]
- 45.Olivera A, Spiegel S. Prostaglandins Other Lipid Mediat. 2001;64:123–134. doi: 10.1016/s0090-6980(01)00108-3. [DOI] [PubMed] [Google Scholar]
- 46.Taha TA, Hannun YA, Obeid LM. J. Biochem. Mol. Biol. 2006;39:113–111. doi: 10.5483/bmbrep.2006.39.2.113. [DOI] [PubMed] [Google Scholar]
- 47.Baumruker T, Bornancin F, Billich A. Immunol. Lett. 2005;96:175–185. doi: 10.1016/j.imlet.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 48.Tettamanti G, Bassi R, Viani P, Riboni L. Biochimie. 2003;85:423–437. doi: 10.1016/s0300-9084(03)00047-6. [DOI] [PubMed] [Google Scholar]
- 49.Chigorno V, Giannotta C, Ottico E, Sciannamblo M, Mikulak J, Prinetti A, Sonnino S. J. Biol. Chem. 2005;280:2668–2675. doi: 10.1074/jbc.M407749200. [DOI] [PubMed] [Google Scholar]
- 50.Ghidoni R, Sala G, Giuliani A. Biochim. Biophys. Acta. 1999;1439:17–39. doi: 10.1016/s1388-1981(99)00074-8. [DOI] [PubMed] [Google Scholar]
- 51.Huwiler A, Brunner J, Hummel R, Vervoordeldonk M, Stabel S, van den Bosch H, Pfeilschifter J. Proc. Natl. Acad. Sci. U. S. A. 1996;93:6959–6963. doi: 10.1073/pnas.93.14.6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bourbon NA, Yun J, Kester M. J. Biol. Chem. 2000;275:35617–35623. doi: 10.1074/jbc.M007346200. [DOI] [PubMed] [Google Scholar]
- 53.Dobrowsky RT, Hannun YA. J. Biol. Chem. 1992;267:5048–5051. [PubMed] [Google Scholar]
- 54.Obeid LM, Linardic CM, Karolak LA, Hannun YA. Science. 1993;259:1769–1771. doi: 10.1126/science.8456305. [DOI] [PubMed] [Google Scholar]
- 55.Kitatani K, Akiba S, Hayama M, Sato T. Arch. Biochem. Biophys. 2001;395:208–214. doi: 10.1006/abbi.2001.2573. [DOI] [PubMed] [Google Scholar]
- 56.Sultan I, Senkal CE, Ponnusamy S, Bielawski J, Szulc Z, Bielawska A, Hannun YA, Ogretmen B. Biochem. J. 2006;393:513–521. doi: 10.1042/BJ20051083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang E, Norred WP, Bacon CW, Riley RT, Merrill AH., Jr. J. Biol. Chem. 1991;266:14486–14490. [PubMed] [Google Scholar]
- 58.Xu R, Jin J, Hu W, Sun W, Bielawski J, Szulc Z, Taha T, Obeid LM, Mao C. FASEB J. 2006;20:1813–1825. doi: 10.1096/fj.05-5689com. [DOI] [PubMed] [Google Scholar]
- 59.Delgado A, Casas J, Llebaria A, Abad JL, Fabrias G. Chem. Med. Chem. 2007;2:580–606. doi: 10.1002/cmdc.200600195. [DOI] [PubMed] [Google Scholar]
- 60.Miyake Y, Kozutsumi Y, Nakamura S, Fujita T, Kawasaki T. Biochem. Biophys. Res. Commun. 1995;211:396–403. doi: 10.1006/bbrc.1995.1827. [DOI] [PubMed] [Google Scholar]
- 61.Ohta H, Yatomi Y, Sweeney EA, Hakomori S, Igarashi Y. FEBS Lett. 1994;355:267–270. doi: 10.1016/0014-5793(94)01218-0. [DOI] [PubMed] [Google Scholar]
- 62.Suzuki E, Handa K, Toledo MS, Hakomori S. Proc. Natl. Acad. Sci. U. S. A. 2004;101:14788–14793. doi: 10.1073/pnas.0406536101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nikolova-Karakashian M, Morgan ET, Alexander C, Liotta DC, Merrill AH., Jr. J. Biol. Chem. 1997;272:18718–18724. doi: 10.1074/jbc.272.30.18718. [DOI] [PubMed] [Google Scholar]
- 64.Meacci E, Vasta V, Farnararo M, Bruni P. Biochem. Biophys. Res. Commun. 1996;221:1–7. doi: 10.1006/bbrc.1996.0534. [DOI] [PubMed] [Google Scholar]
- 65.Cuvillier O, Edsall L, Spiegel S. J. Biol. Chem. 2000;275:15691–15700. doi: 10.1074/jbc.M000280200. [DOI] [PubMed] [Google Scholar]
- 66.Bass BL. Cell. 2000;101:235–238. doi: 10.1016/s0092-8674(02)71133-1. [DOI] [PubMed] [Google Scholar]
- 67.Swanton C, Marani M, Pardo O, Warne PH, Kelly G, Sahai E, Elustondo F, Chang J, Temple J, Ahmed AA, Brenton JD, Downward J, Nicke B. Cancer Cell. 2007;11:498–512. doi: 10.1016/j.ccr.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 68.Tani M, Igarashi Y, Ito M. J. Biol. Chem. 2005;280:36592–36600. doi: 10.1074/jbc.M506827200. [DOI] [PubMed] [Google Scholar]
- 69.Kraveka JM, Li L, Szulc ZM, Bielawski J, Ogretmen B, Hannun YA, Obeid LM, Bielawska A. J. Biol. Chem. 2007;282:16718–16728. doi: 10.1074/jbc.M700647200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pettus BJ, Kitatani K, Chalfant CE, Taha TA, Kawamori T, Bielawski J, Obeid LM, Hannun YA. Mol. Pharmacol. 2005;68:330–335. doi: 10.1124/mol.104.008722. [DOI] [PubMed] [Google Scholar]
- 71.Bionda C, Portoukalian J, Schmitt D, Rodriguez-Lafrasse C, Ardail D. Biochem. J. 2004;382:527–533. doi: 10.1042/BJ20031819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zeidan YH, Hannun YA. J. Biol. Chem. 2007;282:11549–11561. doi: 10.1074/jbc.M609424200. [DOI] [PubMed] [Google Scholar]
- 73.Finkel T, Holbrook NJ. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 74.Kamata H, Hirata H. Cell. Signal. 1999;11:1–14. doi: 10.1016/s0898-6568(98)00037-0. [DOI] [PubMed] [Google Scholar]
- 75.Dbaibo GS, El-Assaad W, Krikorian A, Liu B, Diab K, Idriss NZ, El-Sabban M, Driscoll TA, Perry DK, Hannun YA. FEBS Lett. 2001;503:7–12. doi: 10.1016/s0014-5793(01)02625-4. [DOI] [PubMed] [Google Scholar]
- 76.Heller RA, Kronke M. J. Cell Biol. 1994;126:5–9. doi: 10.1083/jcb.126.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xia P, Wang L, Gamble JR, Vadas MA. J. Biol. Chem. 1999;274:34499–34505. doi: 10.1074/jbc.274.48.34499. [DOI] [PubMed] [Google Scholar]
- 78.Zhang Y, Yao B, Delikat S, Bayoumy S, Lin XH, Basu S, McGinley M, Chan-Hui PY, Lichenstein H, Kolesnick R. Cell. 1997;89:63–72. doi: 10.1016/s0092-8674(00)80183-x. [DOI] [PubMed] [Google Scholar]
- 79.Heinrich M, Wickel M, Schneider-Brachert W, Sandberg C, Gahr J, Schwandner R, Weber T, Saftig P, Peters C, Brunner J, Kronke M, Schutze S. EMBO J. 1999;18:5252–5263. doi: 10.1093/emboj/18.19.5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chalfant CE, Kishikawa K, Mumby MC, Kamibayashi C, Bielawska A, Hannun YA. J. Biol. Chem. 1999;274:20313–20317. doi: 10.1074/jbc.274.29.20313. [DOI] [PubMed] [Google Scholar]
- 81.Galadari S, Kishikawa K, Kamibayashi C, Mumby MC, Hannun YA. Biochemistry. 1998;37:11232–11238. doi: 10.1021/bi980911+. [DOI] [PubMed] [Google Scholar]
- 82.Dobrowsky RT, Kamibayashi C, Mumby MC, Hannun YA. J. Biol. Chem. 1993;268:15523–15530. [PubMed] [Google Scholar]
- 83.Ruvolo PP. Pharmacol. Res. 2003;47:383–392. doi: 10.1016/s1043-6618(03)00050-1. [DOI] [PubMed] [Google Scholar]
- 84.Kitatani K, Idkowiak-Baldys J, Hannun YA. J. Biol. Chem. 2007;282:20647–20656. doi: 10.1074/jbc.M609162200. [DOI] [PubMed] [Google Scholar]
- 85.Becker KP, Hannun YA. J. Biol. Chem. 2003;278:52747–52754. doi: 10.1074/jbc.M305228200. [DOI] [PubMed] [Google Scholar]
- 86.Idkowiak-Baldys J, Becker KP, Kitatani K, Hannun YA. J. Biol. Chem. 2006;281:22321–22331. doi: 10.1074/jbc.M512540200. [DOI] [PubMed] [Google Scholar]
- 87.Ogretmen B, Schady D, Usta J, Wood R, Kraveka JM, Luberto C, Birbes H, Hannun YA, Obeid LM. J. Biol. Chem. 2001;276:24901–24910. doi: 10.1074/jbc.M100314200. [DOI] [PubMed] [Google Scholar]
- 88.Ogretmen B, Kraveka JM, Schady D, Usta J, Hannun YA, Obeid LM. J. Biol. Chem. 2001;276:32506–32514. doi: 10.1074/jbc.M101350200. [DOI] [PubMed] [Google Scholar]
- 89.Dai Q, Liu J, Chen J, Durrant D, McIntyre TM, Lee RM. Oncogene. 2004;23:3650–3658. doi: 10.1038/sj.onc.1207430. [DOI] [PubMed] [Google Scholar]
- 90.Brady RO. Annu. Rev. Biochem. 1978;47:687–713. doi: 10.1146/annurev.bi.47.070178.003351. [DOI] [PubMed] [Google Scholar]
- 91.Futerman AH, van Meer G. Nat. Rev. Mol. Cell Biol. 2004;5:554–565. doi: 10.1038/nrm1423. [DOI] [PubMed] [Google Scholar]
- 92.Kacher Y, Futerman AH. FEBS Lett. 2006;580:5510–5517. doi: 10.1016/j.febslet.2006.08.041. [DOI] [PubMed] [Google Scholar]
- 93.Kolter T, Sandhoff K. Biochim. Biophys. Acta. 2006;1758:2057–2079. doi: 10.1016/j.bbamem.2006.05.027. [DOI] [PubMed] [Google Scholar]
- 94.Park JH, Schuchman EH. Biochim. Biophys. Acta. 2006;1758:2133–2138. doi: 10.1016/j.bbamem.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 95.Eliyahu E, Park JH, Shtraizent N, He X, Schuchman EH. FASEB J. 2007;21:1403–1409. doi: 10.1096/fj.06-7016com. [DOI] [PubMed] [Google Scholar]
- 96.Xu YH, Quinn B, Witte D, Grabowski GA. Am. J. Pathol. 2003;163:2093–2101. doi: 10.1016/s0002-9440(10)63566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dinur T, Osiecki KM, Legler G, Gatt S, Desnick RJ, Grabowski GA. Proc. Natl. Acad. Sci. U. S. A. 1986;83:1660–1664. doi: 10.1073/pnas.83.6.1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tybulewicz VL, Tremblay ML, LaMarca ME, Willemsen R, Stubblefield BK, Winfield S, Zablocka B, Sidransky E, Martin BM, Huang SP. Nature. 1992;357:407–410. doi: 10.1038/357407a0. [DOI] [PubMed] [Google Scholar]
- 99.Horinouchi K, Erlich S, Perl DP, Ferlinz K, Bisgaier CL, Sandhoff K, Desnick RJ, Stewart CL, Schuchman EH. Nat. Genet. 1995;10:288–293. doi: 10.1038/ng0795-288. [DOI] [PubMed] [Google Scholar]
- 100.Marchesini N, Luberto C, Hannun YA. J. Biol. Chem. 2003;278:13775–13783. doi: 10.1074/jbc.M212262200. [DOI] [PubMed] [Google Scholar]