

Abstract
Idiopathic erythrocytosis (IE) is a rare condition in which there is an increase in red cell mass and hematocrit. As it is typically driven by elevated or inappropriately normal erythropoietin (Epo) levels, it has the potential to reveal the identities of proteins involved in the oxygen sensing pathway that regulates the transcription factor, Hypoxia Inducible Factor (HIF), and hence Epo production in humans. One example of this is provided by Chuvash polycythemia, a form of erythrocytosis due to a mutation in the von Hippel Lindau tumor suppressor protein (VHL), a component of an E3 ubiquitin ligase complex that targets hydroxylated HIF for degradation. A recent report of familial erythrocytosis now implicates a different protein, Prolyl Hydroxylase Domain protein 2 (PHD2), which is an enzyme that hydroxylates HIF.
Keywords: HIF, Hypoxia Inducible Factor, PHD2, EGLN1, HPH2, prolyl hydroxylation, idiopathic erythrocytosis, erythropoietin
Idiopathic Erythrocytosis
Under normal conditions, red cell production is controlled by an exquisitely sensitive physiological mechanism, leading to the daily replacement of 1% of the total red cells in the body. Red cell production is driven by the glycoprotein hormone erythropoietin, which is synthesized by the kidney in response to hypoxia. An absolute erythrocytosis occurs by definition when the red cell mass is greater than 125% of that predicted, and this is associated with a raised hematocrit 1. Such an erythrocytosis can be associated with a range of serum Epo levels and can be broadly divided into three groups based upon clinical features. The myeloproliferative group consists of individuals with an absolute erythrocytosis who have the primary bone marrow clonal disorder polycythemia vera (PV), which is characterized by an expansion of the erythroid, myeloid and megakaryocytic cell lineages.
The individuals who do not fulfil the criteria for PV, and hence are excluded from the first group, fall generally into two categories: those with low Epo levels who may be suspected to have a defect in the Epo signaling pathway, and those with inappropriately normal or raised Epo who may have a defect in oxygen sensing. Although the former is typified by mutations in the EpoR leading to constitutive Epo-independent signaling 2, 3, this is a rare event and in many such individuals the molecular defect remains unknown. In the latter group, numerous cases with elevated Epo have a secondary cause such as a high affinity hemoglobin, an Epo-producing tumour, or lung disease due to smoking. Within this category, there remains a cohort of individuals in whom no secondary cause has been identified. This heterogeneous sub-group, which includes familial and sporadic cases, is referred to as IE.
Erythropoietin Gene Regulation and Oxygen Sensing
Over the past ten years or so, substantial progress has been made in understanding the molecular mechanism by which Epo gene transcription is regulated. The essential features of this pathway, which is a classic negative feedback loop, are as follows. Decreased oxygen delivery to the kidney due to any of a number of causes, such as anemia, results in the activation in specialized renal cortical cells of the transcription factor HIF. Hypoxia Inducible Factor is a heterodimeric complex composed of an α-subunit, either HIF-1α or HIF-2α, and a β-subunit, which is the Aryl Hydrocarbon Nuclear Translocator 4. Protein concentrations of the α-subunit, in contrast to those of the β-subunit, are extremely sensitive to oxygen tension and provide the central means by which the HIF complex is regulated by hypoxia.
More specifically, under normoxic conditions, the α-subunit of HIF is hydroxylated at two prolyl residues by a family of three PHDs (also known as HIF Prolyl Hydroxylases and EGLNs) 5-7. In the case of HIF-1α, this occurs at Pro-402 and Pro-564 8-11. This site-specific hydroxylation allows recognition by the von Hippel Lindau tumor suppressor protein, a component of an E3 ubiquitin ligase complex that then targets HIF-1α for constitutive degradation by the ubiquitin-proteasome pathway 12. Under hypoxic conditions, the prolyl hydroxylation is inhibited, thereby allowing escape of the α subunit from degradation and its stabilization. In a parallel pathway, Factor Inhibiting HIF (FIH), which is an asparaginyl hydroxylase, constitutively hydroxylates the α subunit of HIF in its transactivation domain (Asn-803 in the case of HIF-1α), and this modification blocks the interaction between HIF and the transcriptional coactivator CBP/p300 13-16. Under hypoxic conditions, this modification is inhibited, thereby promoting the interaction between HIF and CBP/p300.
The PHDs and FIH utilize molecular oxygen in the hydroxylation reaction, providing an appealing and simple mechanism by which changes in oxygen tension can be transduced to changes in protein modification and hence, protein level. However, it should also be noted that recent results also implicate reactive oxygen species in the pathway by which hypoxia activates HIF 17-19, and the relative contributions of intrinsic oxygen utilization versus reactive oxygen species remains an area of active investigation. The stabilized HIF complex then activates an enhancer in the 3’ end of the Epo gene, leading to increased Epo transcription and hence, Epo production 20-22.
While the Epo pathway is restricted to specialized renal cortical cells and also to hepatocytes, it serves as a paradigm for a universal oxygen sensing pathway, since the central components of the pathway, which include the PHDs, FIH, the HIFs, and VHL are preserved in an extremely wide range of tissues and cell types, in which they serve to regulate a broader hypoxic transcriptional response that activates glycolysis, glucose uptake, angiogenesis, and iron moblization. For more detailed information on this pathway, the reader is referred to several excellent recent reviews 12, 23-25. As should be apparent, while IE with inappropriately normal or high Epo levels is a rare disease, it has the potential to reveal insight into the mechanism by which humans regulate Epo, and more generally, respond to changes in oxygen tension.
Validation of this has been provided by a form of erythrocytosis, autosomal recessive Chuvash polycythemia, that is endemic in the Chuvash Republic of the former Soviet Union and is associated with normal to elevated Epo levels 26. Linkage studies eliminated EpoR as a candidate gene. Instead, it was discovered to be associated with a region on chromosome 3 27. Screening candidate genes in this region indicated the presence of a mutation (R200W) in the VHL gene, and functional studies revealed that it renders VHL partially defective in the ubiquitination of HIF 28. Importantly, this mutation, which results in early mortality from cerebrovascular and thrombotic events 29, does not lead to the clinical phenotype of VHL syndrome, which is characterized by renal cell carcinoma, pheochromocytoma, and CNS hemangioblastoma.
Registry for Idiopathic Erythrocytosis
Over the last ten years, British and Irish IE patients have been referred to us for further investigation allowing the development of a registry of clinical data with matching stored DNA samples. The inclusion and exclusion criteria for the registry are based on the published guidelines 1, 30, but basically patients are included if they do not fulfil the criteria for PV and do not have an identified secondary cause. Only patients with a raised red cell mass greater than 125% of that predicted and/or a raised hematocrit (>0.52 in males and >0.48 in females), no splenomegaly and no identified cause of erythrocytosis are included on the registry.
To date more than 120 individuals have been recruited to the IE registry and around 15% have a family history of a first degree relative with erythrocytosis. On entry to the registry, individuals are routinely screened for defects in the genes encoding for the cytoplasmic region of the Epo receptor (EpoR)—which includes the negative regulatory domains, VHL, and most recently, the catalytic domain of PHD2 (for reasons detailed below). In addition recently, those individuals with low Epo levels are screened for the JAK2 V617F mutation. Since this mutation is associated with acquired clonal myeloproliferative disorders, it is then possible to exclude individuals from the registry who have the clonal disorder of PV 31. Such a clone was present in 1.6% of IE individuals tested.
Chuvash Polycythemia and the P582S polymorphism in HIF-1α
Screening individuals from the IE registry has detected in several families originating from the Indian sub-continent the same VHL R200W mutation as originally described in Chuvash polycythemia 32, 33; to date, 10 such families have been identified. Simultaneously, investigation of the VHL gene in non-Chuvash childhood erythrocytosis also revealed this same VHL defect 34. Thus, the VHL R200W mutation was found to exhibit a wide geographical distribution and is not solely confined to Chuvashia, which suggests it is of greater significance than initially thought. Haplotype analysis of Chuvash, Asian and Caucasian individuals reported one founder mutation, which was an ancient event 35. Subsequently, a second occurrence for the VHL R200W mutation that arose independently of the Chuvash mutation was identified in a child of Turkish origin 36.
Further studies have revealed other different VHL mutations are associated with erythrocytosis, which can be present in either the homozygous or compound heterozygous state 36-40. More recently, a separate cluster of Chuvash erythrocytosis patients has been identified in the island of Ischia, Italy with a haplotype that matched the Chuvash cluster 41. Intriguingly, there are a growing number of erythrocytosis individuals who are heterozygous for the VHL R200W mutation but also express the wild type allele 36, 41, 42, or mutations at otherVHL sites 34, 36, 38, 39. This is contrary to the recessive mode of inheritance established by most cases with VHL-associated erythrocytosis, thus raising the possibility of an undefined second molecular defect.
VHL mutations appear to be the most identified cause of erythrocytosis, but there remains a significant cohort of patients which are negative for VHL mutations. They could be hypothesized to possess defects in the oxygen sensing pathways since their Epo levels were either inappropriately normal or elevated. Towards this end, we investigated exon 12 of the HIF-1α, which encodes for the primary site of prolyl hydroxylation, Pro-564. This study identified a heterozygous base change of C to T at nucleotide 1744, resulting in the replacement of proline at codon 582 with serine. Although this proline is conserved within mammalian species, it is not in present in the protein from Xenopus laevis, and screening a group of control samples indicated that it is a common polymorphism 43. Further studies revealed that there was no statistically significant association between the HIF-1α P582S variant and erythrocytosis, and functional studies failed to detect any discernable effect on hydroxylation at Pro-564. Therefore, this polymorphism appears unlikely to be the cause of IE in this patient population.
Occurrence of the P582S polymorphism was not found to be associated with renal cell carcinoma 44. However, the P582S polymorphism has been found to be associated with other disease phenotypes, including decreased coronary artery collateralization in ischemic heart disease patients, lower exercise-induced oxygen consumption in patients age 60 and older, prostate carcinoma, head and neck cancer, and type 2 diabetes 45-50.
A Mutation in PHD2
We recently described a family with erythrocytosis with autosomal dominant transmission of a mutation in the coding sequence of PHD2 that is predicted to change Pro-317 to Arg 51. In this family, the mutation was present in a heterozygous state in the father, daughter, and son, all of whom displayed erythrocytosis. The mother, who is hematologically normal, did not display this mutation. Mutations were not seen in the coding sequences for either PHD1, PHD3 , VHL, or EpoR, or in exon 12 of HIF-1α.
The PHD2 mutation is predicted to alter an evolutionarily conserved residue in the active site of this enzyme, and indeed, mutant PHD2 showed markedly deficient binding to HIF-1α and HIF-2α peptides, diminished prolyl hydroxylase activity, and reduced ability to inhibit hypoxia- or HIF-overexpression induced activation of a Hypoxia Response Element (HRE) reporter gene. In transient overexpression assays, the mutant PHD2 was unable to reverse wild type PHD2 inhibition of an HRE reporter or activate it by itself, making a dominant negative effect unlikely, and thereby making near haploinsufficiency the most probable cause of the erythrocytosis. This would imply that PHD1 and PHD3, the sequences of which were normal, are not sufficient to compensate for this near-haploinsufficiency. At the same time, it remains formally possible that this mutation might lead to a gain of function—e.g. acquired activity towards a novel substrate, that might also contribute to the phenotype.
Parallels between PHD2 and VHL
The recent findings place PHD2 and VHL in the pathway regulating Epo production in humans (Figure 1), and raise interesting parallels between VHL and PHD2. Mutations in VHL can yield two distinct syndromes—cancer predisposition and erythrocytosis. The former is inherited in an autosomal dominant manner, and is due to a germline mutation in one VHL allele, most commonly leading to markedly impaired binding of either HIF or elongin C 12. In conformation with Knudson's two hit hypothesis, a second somatic mutation affects the second VHL allele, impairing or abolishing its function, and then predisposes to cancer. VHL-associated erythrocytosis was originally described in the context of Chuvash polycythemia, which in contrast to classic VHL syndrome is inherited in an autosomal recessive manner, and it is due to a mutation, R200W, that resides in region of VHL distinct from its HIF or Elongin C-binding sites and results in only a partial loss of activity 28. Additional VHL mutations leading to erythrocytosis have subsequently been described, and most cluster in the same region in the three dimensional structure of VHL as the Chuvash mutation 40. One might therefore hypothesize the following: first, that the phenotypes are due to loss of function, and second that the degree of loss of function correlates with phenotype—partial loss of function, at least in those cells producing Epo, leading to erythrocytosis; complete loss of function in a cell-type dependent manner predisposing to cancer.
Figure 1.
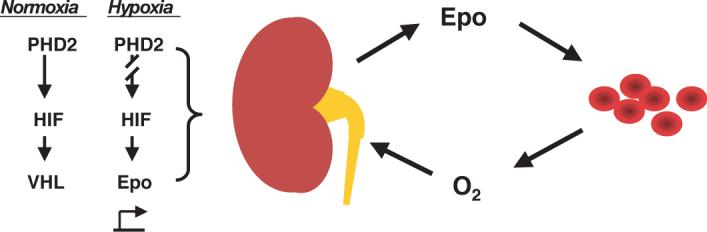
Control of Epo production by the PHD2:HIF:VHL pathway. Under normoxic conditions, the α subunit of HIF is constitutively hydroxylated by PHD2 and hence degraded in a VHL-dependent manner. Under hypoxic conditions, PHD2-induced hydroxylation is inhibited, thereby allowing HIF to bind to the enhancer of the Epo gene. Epo production by the kidney then stimulates red blood cell production, leading to increased oxygen delivery to the kidney and providing a negative feedback loop that then downregulates Epo production.
A mutation in PHD2 leading to near haploinsufficiency has now been demonstrated to yield erythrocytosis, raising the possibility that PHD2 may follow the same paradigm as VHL, namely that partial loss of function leads to erythrocytosis, and complete loss of function predisposing to cancer—i.e. PHD2 may in certain cell contexts behave as a tumor suppressor protein. Indeed, recent reports have identified sporadic PHD2 mutations and loss of heterozygosity in uterine cancer 52, and it will be of interest to see whether and to what extent this applies to other tumors. At the same time, it should be recognized that pathways dependent on PHD2 but not VHL, and conversely ones dependent on VHL but not PHD2 may exist, implying that deficiencies in PHD2 and VHL would not necessarily phenocopy one another.
In addition, it is clear that there is considerably more complexity to this model. First, and as already mentioned, there are patients heterozygous for the Chuvash mutation who nonetheless have erythrocytosis, suggesting that there may be a defect in the control of the wild type allele or yet some other undefined defect 36, 38, 41, 42. Second, the hypomorphic phenotype of Chuvash polycythemia yields erythrocytosis while haploinsufficiency of VHL only rarely yields erythrocytosis; in the latter case, this is typically due to Epo production by the VHL-associated tumor itself 53. In this regard, it is conceivable that the mutations have effects distinct from enzymatic activity. In the case of VHL, for example, effects on extracellular matrix have been documented 54, 55, and numerous VHL-associated proteins with activities outside of the HIF pathway identified 56. Third, and similarly, near-haploinsufficiency of PHD2 in the family recently reported yields erythrocytosis while haploinsufficiency of VHL only rarely yields erythrocytosis. One potential explanation here is differences in sensitivity to dose reductions. It suggests that PHD2 levels in the Epo-producing cells of the kidney are not saturating with respect to HIF hydroxylation, perhaps less saturating than VHL levels are with respect to capture of hydroxylated HIF. Finally, although loss of function remains the most appealing explanation for Chuvash polycythemia and the PHD2 P317R mutation, it is conceivable that gain of function effects may also be present.
Future Directions
Recent studies have provided compelling evidence that study of erythrocytosis can yield rich insight into the oxygen sensing pathway leading to Epo regulation in humans. The identification of VHL and, now, PHD2 as mutational gene targets strongly implicates the gene products as central to the control of Epo. It will certainly be of interest to determine whether other erythrocytosis-associated PHD2 mutations exist, and also whether HIF mutations can be identified in this condition, as would be suggested by the current understanding of this pathway. More broadly, it should be noted that the majority of patients with erythrocytosis still have no identifiable molecular defect, indicating that that the continuing study of these individuals holds the promise of yielding additional insights into this fundamental homeostatic pathway in humans.
Acknowledgements
Work in the authors’ laboratories was supported by the Northern Ireland Leukaemia Research Fund, and National Institutes of Health grant R01 CA090261 (FSL). We thank Professor Terence Lappin for a critical reading of the manuscript.
Abbreviations
- Epo
erythropoietin
- FIH
Factor Inhibiting HIF
- HIF
Hypoxia Inducible Factor
- HRE
Hypoxia Response Element
- IE
Idiopathic Erythrocytosis
- PHD
Prolyl Hydroxylase Domain protein
- PV
Polycythemia vera
- VHL
von Hippel Lindau tumor suppressor protein
References
- 1.McMullin MF, Bareford D, Campbell P, Green AR, Harrison C, Hunt B, Oscier D, Polkey MI, Reilly JT, Rosenthal E, Ryan K, Pearson TC, Wilkins B. Guidelines for the diagnosis, investigation and management of polycythaemia/erythrocytosis. Br J Haematol. 2005;130:174–95. doi: 10.1111/j.1365-2141.2005.05535.x. [DOI] [PubMed] [Google Scholar]
- 2.de la Chapelle A, Traskelin AL, Juvonen E. Truncated erythropoietin receptor causes dominantly inherited benign human erythrocytosis. Proc Natl Acad Sci U S A. 1993;90:4495–9. doi: 10.1073/pnas.90.10.4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McMullin MF, Percy MJ. Erythropoietin receptor and hematological disease. Am J Hematol. 1999;60:55–60. doi: 10.1002/(sici)1096-8652(199901)60:1<55::aid-ajh9>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 4.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-pas heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92:5510–4. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O'Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ. C. Elegans egl-9 and mammalian homologs define a family of dioxygenases that regulate hif by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 6.Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify hif. Science. 2001;294:1337–40. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 7.Ivan M, Haberberger T, Gervasi DC, Michelson KS, Gunzler V, Kondo K, Yang H, Sorokina I, Conaway RC, Conaway JW, Kaelin WG., Jr Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor. Proc Natl Acad Sci U S A. 2002;99:13459–64. doi: 10.1073/pnas.192342099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr Hifα targeted for vhl-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science. 2001;292:464–68. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 9.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim Av A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Targeting of hif-α to the von hippel-lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–72. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 10.Masson N, Willam C, Maxwell PH, Pugh CW, Ratcliffe PJ. Independent function of two destruction domains in hypoxia-inducible factor-α chains activated by prolyl hydroxylation. Embo J. 2001;20:5197–206. doi: 10.1093/emboj/20.18.5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu F, White SB, Zhao Q, Lee FS. Hif-1α binding to vhl is regulated by stimulus-sensitive proline hydroxylation. Proc Natl Acad Sci U S A. 2001;98:9630–5. doi: 10.1073/pnas.181341498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaelin WG., Jr The von hippel-lindau protein, hif hydroxylation, and oxygen sensing. Biochem Biophys Res Commun. 2005;338:627–38. doi: 10.1016/j.bbrc.2005.08.165. [DOI] [PubMed] [Google Scholar]
- 13.Mahon PC, Hirota K, Semenza GL. Fih-1: A novel protein that interacts with hif-1α and vhl to mediate repression of hif-1 transcriptional activity. Genes Dev. 2001;15:2675–86. doi: 10.1101/gad.924501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the hif transactivation domain a hypoxic switch. Science. 2002;295:858–61. doi: 10.1126/science.1068592. [DOI] [PubMed] [Google Scholar]
- 15.Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. Fih-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16:1466–71. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hewitson KS, McNeill LA, Riordan MV, Tian YM, Bullock AN, Welford RW, Elkins JM, Oldham NJ, Bhattacharya S, Gleadle JM, Ratcliffe PJ, Pugh CW, Schofield CJ. Hypoxia-inducible factor (hif) asparagine hydroxylase is identical to factor inhibiting hif (fih) and is related to the cupin structural family. J Biol Chem. 2002;277:26351–5. doi: 10.1074/jbc.C200273200. [DOI] [PubMed] [Google Scholar]
- 17.Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U, Schumacker PT. Mitochondrial complex iii is required for hypoxia-induced ros production and cellular oxygen sensing. Cell Metab. 2005;1:401–8. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 18.Mansfield KD, Guzy RD, Pan Y, Young RM, Cash TP, Schumacker PT, Simon MC. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic hif-α activation. Cell Metab. 2005;1:393–9. doi: 10.1016/j.cmet.2005.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brunelle JK, Bell EL, Quesada NM, Vercauteren K, Tiranti V, Zeviani M, Scarpulla RC, Chandel NS. Oxygen sensing requires mitochondrial ros but not oxidative phosphorylation. Cell Metab. 2005;1:409–14. doi: 10.1016/j.cmet.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 20.Semenza GL, Nejfelt MK, Chi SM, Antonarakis SE. Hypoxia-inducible nuclear factors bind to an enhancer element located 3' to the human erythropoietin gene. Proc Natl Acad Sci U S A. 1991;88:5680–4. doi: 10.1073/pnas.88.13.5680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beck I, Ramirez S, Weinmann R, Caro J. Enhancer element at the 3'-flanking region controls transcriptional response to hypoxia in the human erythropoietin gene. J Biol Chem. 1991;266:15563–6. [PubMed] [Google Scholar]
- 22.Pugh CW, Tan CC, Jones RW, Ratcliffe PJ. Functional analysis of an oxygen-regulated transcriptional enhancer lying 3' to the mouse erythropoietin gene. Proc Natl Acad Sci U S A. 1991;88:10553–7. doi: 10.1073/pnas.88.23.10553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schofield CJ, Ratcliffe PJ. Signalling hypoxia by hif hydroxylases. Biochem Biophys Res Commun. 2005;338:617–26. doi: 10.1016/j.bbrc.2005.08.111. [DOI] [PubMed] [Google Scholar]
- 24.Dann CE, 3rd, Bruick RK. Dioxygenases as O(2)-dependent regulators of the hypoxic response pathway. Biochem Biophys Res Commun. 2005;338:639–47. doi: 10.1016/j.bbrc.2005.08.140. [DOI] [PubMed] [Google Scholar]
- 25.Hirota K, Semenza GL. Regulation of hypoxia-inducible factor 1 by prolyl and asparaginyl hydroxylases. Biochem Biophys Res Commun. 2005;338:610–6. doi: 10.1016/j.bbrc.2005.08.193. [DOI] [PubMed] [Google Scholar]
- 26.Sergeyeva A, Gordeuk VR, Tokarev YN, Sokol L, Prchal JF, Prchal JT. Congenital polycythemia in Chuvashia. Blood. 1997;89:2148–54. [PubMed] [Google Scholar]
- 27.Ang SO, Chen H, Gordeuk VR, Sergueeva AI, Polyakova LA, Miasnikova GY, Kralovics R, Stockton DW, Prchal JT. Endemic polycythemia in Russia: Mutation in the VHL gene. Blood Cells Mol Dis. 2002;28:57–62. doi: 10.1006/bcmd.2002.0488. [DOI] [PubMed] [Google Scholar]
- 28.Ang SO, Chen H, Hirota K, Gordeuk VR, Jelinek J, Guan Y, Liu E, Sergueeva AI, Miasnikova GY, Mole D, Maxwell PH, Stockton DW, Semenza GL, Prchal JT. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat Genet. 2002;32:614–21. doi: 10.1038/ng1019. [DOI] [PubMed] [Google Scholar]
- 29.Gordeuk VR, Sergueeva AI, Miasnikova GY, Okhotin D, Voloshin Y, Choyke PL, Butman JA, Jedlickova K, Prchal JT, Polyakova LA. Congenital disorder of oxygen sensing: Association of the homozygous Chuvash polycythemia VHL mutation with thrombosis and vascular abnormalities but not tumors. Blood. 2004;103:3924–32. doi: 10.1182/blood-2003-07-2535. [DOI] [PubMed] [Google Scholar]
- 30.Pearson TC, Messinezy M. The diagnostic criteria of polycythaemia rubra vera. Leuk Lymphoma. 1996;22(Suppl 1):87–93. [PubMed] [Google Scholar]
- 31.Percy MJ, Jones FGC, Green AR, Reilly JT, McMullin MF. The incidence of the JAK2V617F mutation in patients with idiopathic erythrocytosis. Haematologica. 2006;2006 In press. [PubMed] [Google Scholar]
- 32.Percy MJ, McMullin MF, Jowitt SN, Potter M, Treacy M, Watson WH, Lappin TR. Chuvash-type congenital polycythemia in 4 families of Asian and western European ancestry. Blood. 2003;102:1097–9. doi: 10.1182/blood-2002-10-3246. [DOI] [PubMed] [Google Scholar]
- 33.Percy MJ, Beard ME, Carter C, Thein SL. Erythrocytosis and the Chuvash von hippel-lindau mutation. Br J Haematol. 2003;123:371–2. doi: 10.1046/j.1365-2141.2003.04631.x. [DOI] [PubMed] [Google Scholar]
- 34.Pastore YD, Jelinek J, Ang S, Guan Y, Liu E, Jedlickova K, Krishnamurti L, Prchal JT. Mutations in the VHL gene in sporadic apparently congenital polycythemia. Blood. 2003;101:1591–5. doi: 10.1182/blood-2002-06-1843. [DOI] [PubMed] [Google Scholar]
- 35.Liu E, Percy MJ, Amos CI, Guan Y, Shete S, Stockton DW, McMullin MF, Polyakova LA, Ang SO, Pastore YD, Jedlickova K, Lappin TR, Gordeuk V, Prchal JT. The worldwide distribution of the VHL 598C>T mutation indicates a single founding event. Blood. 2004;103:1937–40. doi: 10.1182/blood-2003-07-2550. [DOI] [PubMed] [Google Scholar]
- 36.Cario H, Schwarz K, Jorch N, Kyank U, Petrides PE, Schneider DT, Uhle R, Debatin KM, Kohne E. Mutations in the von hippel-lindau (VHL) tumor suppressor gene and VHL-haplotype analysis in patients with presumable congenital erythrocytosis. Haematologica. 2005;90:19–24. [PubMed] [Google Scholar]
- 37.Gordeuk VR, Stockton DW, Prchal JT. Congenital polycythemias/erythrocytoses. Haematologica. 2005;90:109–16. [PubMed] [Google Scholar]
- 38.Randi ML, Murgia A, Putti MC, Martella M, Casarin A, Opocher G, Fabris F. Low frequency of VHL gene mutations in young individuals with polycythemia and high serum erythropoietin. Haematologica. 2005;90:689–91. [PubMed] [Google Scholar]
- 39.Bento MC, Chang KT, Guan Y, Liu E, Caldas G, Gatti RA, Prchal JT. Congenital polycythemia with homozygous and heterozygous mutations of von hippel-lindau gene: Five new caucasian patients. Haematologica. 2005;90:128–9. [PubMed] [Google Scholar]
- 40.Pastore Y, Jedlickova K, Guan Y, Liu E, Fahner J, Hasle H, Prchal JF, Prchal JT. Mutations of von hippel-lindau tumor-suppressor gene and congenital polycythemia. Am J Hum Genet. 2003;73:412–9. doi: 10.1086/377108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perrotta S, Nobili B, Ferraro M, Migliaccio C, Borriello A, Cucciolla V, Martinelli V, Rossi F, Punzo F, Cirillo P, Parisi G, Zappia V, Rotoli B, Ragione FD. Von hippel-lindau-dependent polycythemia is endemic on the island of Ischia: Identification of a novel cluster. Blood. 2006;107:514–9. doi: 10.1182/blood-2005-06-2422. [DOI] [PubMed] [Google Scholar]
- 42.Percy MJ, Jones FGC, Lappin TRJ, McMullin MF. Mutations in the VHL gene are the major identified cause of inherited erythrocytosis. Blood. 2005;106:169a. [Google Scholar]
- 43.Percy MJ, Mooney SM, McMullin MF, Flores A, Lappin TR, Lee FS. A common polymorphism in the oxygen-dependent degradation (odd) domain of hypoxia inducible factor-1alpha (hif-1α) does not impair pro-564 hydroxylation. Mol Cancer. 2003;2:31. doi: 10.1186/1476-4598-2-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clifford SC, Astuti D, Hooper L, Maxwell PH, Ratcliffe PJ, Maher ER. The pvhl-associated scf ubiquitin ligase complex: Molecular genetic analysis of elongin b and c, rbx1 and hif-1α in renal cell carcinoma. Oncogene. 2001;20:5067–74. doi: 10.1038/sj.onc.1204602. [DOI] [PubMed] [Google Scholar]
- 45.Fu XS, Choi E, Bubley GJ, Balk SP. Identification of hypoxia-inducible factor-1alpha (hif-1α) polymorphism as a mutation in prostate cancer that prevents normoxia-induced degradation. Prostate. 2005;63:215–21. doi: 10.1002/pros.20190. [DOI] [PubMed] [Google Scholar]
- 46.Prior SJ, Hagberg JM, Phares DA, Brown MD, Fairfull L, Ferrell RE, Roth SM. Sequence variation in hypoxia-inducible factor 1alpha (hif1α): Association with maximal oxygen consumption. Physiol Genomics. 2003;15:20–6. doi: 10.1152/physiolgenomics.00061.2003. [DOI] [PubMed] [Google Scholar]
- 47.Tanimoto K, Yoshiga K, Eguchi H, Kaneyasu M, Ukon K, Kumazaki T, Oue N, Yasui W, Imai K, Nakachi K, Poellinger L, Nishiyama M. Hypoxia-inducible factor-1α polymorphisms associated with enhanced transactivation capacity, implying clinical significance. Carcinogenesis. 2003;24:1779–83. doi: 10.1093/carcin/bgg132. [DOI] [PubMed] [Google Scholar]
- 48.Yamada N, Horikawa Y, Oda N, Iizuka K, Shihara N, Kishi S, Takeda J. Genetic variation in the hypoxia-inducible factor-1α gene is associated with type 2 diabetes in japanese. J Clin Endocrinol Metab. 2005;90:5841–7. doi: 10.1210/jc.2005-0991. [DOI] [PubMed] [Google Scholar]
- 49.Resar JR, Roguin A, Voner J, Nasir K, Hennebry TA, Miller JM, Ingersoll R, Kasch LM, Semenza GL. Hypoxia-inducible factor 1α polymorphism and coronary collaterals in patients with ischemic heart disease. Chest. 2005;128:787–91. doi: 10.1378/chest.128.2.787. [DOI] [PubMed] [Google Scholar]
- 50.Anastasiadis AG, Ghafar MA, Salomon L, Vacherot F, Benedit P, Chen MW, Shabsigh A, Burchardt M, Chopin DK, Shabsigh R, Buttyan R. Human hormone-refractory prostate cancers can harbor mutations in the O(2)-dependent degradation domain of hypoxia inducible factor-1α (hif-1α). J Cancer Res Clin Oncol. 2002;128:358–62. doi: 10.1007/s00432-002-0346-1. [DOI] [PubMed] [Google Scholar]
- 51.Percy MJ, Zhao Q, Flores A, Harrison C, Lappin TR, Maxwell PH, McMullin MF, Lee FS. A family with erythrocytosis establishes a role for prolyl hydroxylase domain protein 2 in oxygen homeostasis. Proc Natl Acad Sci U S A. 2006;103:654–9. doi: 10.1073/pnas.0508423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kato H, Inoue T, Asanoma K, Nishimura C, Matsuda T, Wake N. Induction of human endometrial cancer cell senescence through modulation of hif-1α activity by egln1. Int J Cancer. 2006;118:1144–53. doi: 10.1002/ijc.21488. [DOI] [PubMed] [Google Scholar]
- 53.Wiesener MS, Seyfarth M, Warnecke C, Jurgensen JS, Rosenberger C, Morgan NV, Maher ER, Frei U, Eckardt KU. Paraneoplastic erythrocytosis associated with an inactivating point mutation of the von hippel-lindau gene in a renal cell carcinoma. Blood. 2002;99:3562–5. doi: 10.1182/blood.v99.10.3562. [DOI] [PubMed] [Google Scholar]
- 54.Clifford SC, Cockman ME, Smallwood AC, Mole DR, Woodward ER, Maxwell PH, Ratcliffe PJ, Maher ER. Contrasting effects on hif-1α regulation by disease-causing pvhl mutations correlate with patterns of tumourigenesis in von hippel-lindau disease. Hum Mol Genet. 2001;10:1029–38. doi: 10.1093/hmg/10.10.1029. [DOI] [PubMed] [Google Scholar]
- 55.Hoffman MA, Ohh M, Yang H, Klco JM, Ivan M, Kaelin WG., Jr Von hippel-lindau protein mutants linked to type 2c vhl disease preserve the ability to downregulate hif. Hum Mol Genet. 2001;10:1019–27. doi: 10.1093/hmg/10.10.1019. [DOI] [PubMed] [Google Scholar]
- 56.Kaelin WG., Jr The von hippel-lindau gene, kidney cancer, and oxygen sensing. J Am Soc Nephrol. 2003;14:2703–11. doi: 10.1097/01.asn.0000092803.69761.41. [DOI] [PubMed] [Google Scholar]