

Abstract
Previous studies have shown that VEGF expression in forebrain increases after experimental manipulations that increase neuronal activity [6,12]. One question is whether this also occurs in motor neurons. If so, it could be potentially advantageous from a therapeutic perspective, because VEGF prevents motor neuron degeneration [2,10,24]. Therefore, we asked whether endogenous VEGF expression in motor neurons could be modulated. We also asked what VEGF exposure would do to motor neurons using electrophysiology.
Immunocytochemistry showed that motor neuron VEGF expression increased after a stimulus that increases neuronal and motor activity, i.e., convulsions. The increase in VEGF immunoreactivity occured in all motor neuron populations that were examined 24 hrs later. This effect was unlikely to be due to seizure-induced toxicity, because silver degeneration stain did not show the typical appearance of a dying or dead neuron.
To address the effects of VEGF on motor neuron function, VEGF was applied directly to motor neurons while recording intracellularly, using a brainstem slice preparation. Exposure to exogenous VEGF (200 ng/ml) in normal conditions depressed stimulus-evoked depolarization of hypoglossal motor neurons. There was no detectable effect of VEGF on membrane properties or firing behavior. We suggest that the VEGF is upregulated in neurons when they are activated strongly, and VEGF depresses neuronal excitation as a compensatory mechanism. Failure of this mechanism may contribute to diseases that involve a dysregulation of VEGF, excessive excitation of motor neurons, and lead to motor neuron loss, such as amyotrophic lateral sclerosis (ALS).
Keywords: Amyotrophic Lateral Sclerosis (ALS), facial nucleus, hypoglossal nucleus, seizures, spinal cord, status
Introduction
The ability to produce endogenous vascular endothelial growth factor (VEGF) may be important for maintaining the health of motor neurons in the brainstem and spinal cord. A decrease in endogenous production of VEGF has been linked to motor neuron degeneration in motor system diseases such as amyotrophic lateral sclerosis (ALS) and Kennedy disease [2,7,10,14,23,24,31]. Also, impairing VEGF production through genetic manipulation results in degeneration of lower motor neurons in adulthood [14,23]. VEGF has also been shown to be neuroprotective in animal models of ischemia or other types of insult or injury [6,25].
To optimize a potential therapeutic approach, it would be advantageous to clarify the ways motor neuron VEGF can be manipulated, and the effects of altered expression on motor neuron function. To address the first issue, we asked whether a robust stimulus that increases neuronal activity and motor behavior, seizures, would increase VEGF in motor neurons. To test this hypothesis, seizures were induced by the muscarinic agonist pilocarpine, a convulsant that is commonly used to induce severe motor seizures that are continuous and prolonged (status epilepticus or “status”) [27,28]. During pilocarpine-induced status, there are regular, involuntary motor contractions of all limbs (myoclonic jerks), as well as involuntary facial and oral movements (mastication) [27,28]. Therefore, descending and peripheral inputs would be likely to activate motor neurons more than would occur under normal conditions. Animals were evaluated for changes in VEGF expression 24 hrs after the onset of status, because this time appeared to be associated with increased VEGF expression in forebrain [6]. It also appears to be the time when activity-dependent changes in other growth factors are maximal [20]
To predict any clinical potential for VEGF therapy, it would be useful to clarify its effects on motor neuron function. Therefore, a second aim of this study was to apply exogenous VEGF to motor neurons and evaluate any physiological effects on membrane properties or synaptic transmission. Physiological effects might be expected given the evidence for direct effects of VEGF on motor neuron survival in recent studies [2,14]. In addition, there could be indirect effects mediated by the influence of VEGF on the vasculature or signal transduction pathways [13]. However, no studies of motor neuron physiology after exposure to VEGF have been reported to date.
Materials and Methods
I. General methods
Experiments were conducted according to the guidelines set by the New York State Department of Health and the National Institutes of Health, and were approved by the Institutional Animal Care and Use Committee of Helen Hayes Hospital.
Male Sprague-Dawley rats (Taconic Farms) were provided food and water ad libitum and housed using a 12 hr light/dark cycle. All chemicals were purchased from Sigma-Aldrich unless otherwise noted.
II. Seizure induction
Animals were housed in clear cages to optimize observation of seizures. Each animal was injected with atropine methylbromide (1 mg/kg, s.c.) followed by pilocarpine hydrochloride (380 mg/kg, s.c.) 30 min later. The dose was chosen to maximize the chance of inducing status epilepticus while minimizing mortality. After pilocarpine injection, animals were then observed continuously for evidence of behavioral seizures using the Racine scale [16]. Most animals developed mild (stages 1–3) and then severe (stages 4–5) limbic seizures, and eventually a stage 4 or 5 seizure occurred which does not stop (status epilepticus or “status”). The stage 4 or 5 seizure that was signaled the onset of status was followed immediately by loss of postural tone, and animals subsequently displayed rhythmic muscular contractions, typically while lying on their side. The onset of that stage 4 or 5 seizure was defined as the onset of status, and typically occurred within 60 min of pilocarpine administration.
One hour after the onset of status, and animals were injected with diazepam (5 mg/kg i.p.). After this time, animals continued to have subtle motor contractions for several hours. In addition to the motor contractions of the limbs, animals exhibited movements of the facial area, as indicated by movements of the whiskers, the mouth and tongue. This is noted because it could be relevant to the increase in VEGF expression in facial and hypoglossal motor neurons described in the Results. After approximately 5–6 hrs, animals received 5% dextrose-lactate Ringer’s solution (2.5 ml, s.c.).
Control animals were treated identically, i.e., atropine, diazepam, dextrose-lactate Ringer’s solution, but were administered 3 ml/kg phosphate-buffered saline (i.p.) instead of pilocarpine. They were administered diazepam approximately 2 hr after saline injection, similar to the timing of diazepam injection in rats that had status.
III. Anatomical methods
Animals were anesthetized with an overdose of urethane (2.5mg/ kg, i.p.), and then transcardially-perfused with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). Brains and spinal cords were left in situ and refrigerated overnight. On the following day, brains and spinal cords were removed and postfixed in 4 % paraformaldhyde in 0.1 M phosphate buffer for 1 week, and 50 µm-thick coronal sections were made with a Vibratome (Ted Pella Inc.). After processing, sections were viewed with an Olympus BX-51 microscope and digital photography (Camera: Optronics; Frame grabber: Foresight Imaging; Software: Microbrightfield Inc.)
A. VEGF immunocytochemistry
Sections from control and treated animals were processed concurrently, using free-floating sections. Sections were placed in one compartment of a 8-compartment plexiglas tray that was rinsed with 0.5% bovine albumin. Each compartment was filled with approximately 6 ml of reagents. In each compartment, 5 sections were selected from a given animal, so that areas that included motor neuron nuclei were well-represented. In addition, there were at least 12 sections from cervical spinal cord. Similar sections, but from other animals, were placed in separate compartments. Sections were processed in the same order (compartment 1 followed by compartment 2, etc.) so that duration of exposure to reagents was similar for all sections. Sections were first immersed in 0.1 M TRIS buffer (pH 7.4) and placed on a rotator for 5 min. This was repeated twice (i.e., total, 3 × 5 min). Sections were then transferred to 1% H2O2 in 0.1 M TRIS buffer for 30 min, followed by 0.1 M TRIS buffer (3 × 5 min), and then transferred to 0.1% Triton X-100 in 0.1 M TRIS buffer (TRIS A) for 10 min, followed by Triton X-100 and 0.005% bovine serum albumin in 0.1 M TRIS buffer (TRIS B; 10 min). Sections were then incubated in 10 % normal goat serum in TRIS B for 1 hr, followed by incubation in anti-mouse VEGF antibody, made in goat (1:500; R & D Systems). Incubation was conducted as follows: the compartmentalized tray was placed on a rotator at room temperature overnight. In some cases, incubation was conducted for 2 days in the cold (4°C). The two procedures led to results that were indistinguishable so they are presented together in the Results. Sections were then washed in TRIS A for 10 min, TRIS B for 10 min, and incubated in in biotinylated horse anti-goat IgG diluted in TRIS B (1:400, Vector Laboratories) for 45 min. Sections were then washed in TRIS A for 10 min followed by 0.1 %Triton X-100 and 0.005 % bovine serum albumin in 0.5 M TRIS buffer (TRIS D; 10 min). This was followed by incubation in avidin-biotin horseradish peroxidase complex (ABC), made in TRIS D, for 2 hrs (ABC Standard kit; 1:1000; Vector Laboratories). Sections were then washed in 0.1 M TRIS buffer (3 × 5 min), and developed in 0.022% 3,3’-diaminobezidine (DAB), and 1 mM NiCl2 in TRIS buffer. The development was stopped by consecutive TRIS washes (3 × 5 min). Sections were mounted on subbed slides, dehydrated in a graded series of alcohols, cleared in xylene and coverslipped in Permount (Fisher).
B. Silver stain
Silver stain was used instead of fluorojade B, because fluorojade B is ineffective in staining degenerating motor neurons [1]. The silver stain procedure was modified from one previously reported [22]. Sections were mounted on subbed slides, washed in distilled H2O (3 × 5 min), and then pretreated with a solution composed of the following, in equal volumes: 9% aqueous sodium hydroxide and 1.2% aqueous ammonium nitrate. Sections were then exposed to 60 ml of 9% NaOH, 40 ml of 16% ammonium nitrate, and 0.6 ml of 50% silver nitrate (all solutions were diluted in distilled water). Sections were then washed for 10 minutes 1 ml of 1.2% ammonium nitrate, 300 ml 95% ethyl alcohol, and 0.05% sodium carbonate. Finally, sections were developed in a 1:1 aqueous solution with the following constituents: 1 ml 1.2% ammonium nitrate, 100 ml 95% ethyl alcohol, 15 ml 37% formalin, and 0.005% citric acid monohydrate (pH of 5.8). Sections were mounted in 0.1M TRIS buffer and fingernail polish was used to cement coverslips in place. Slides were stored in the dark.
C. Electrophysiology
1) Slice Preparation
Untreated male Sprague Dawley rats (19–28 days old, 50–75 g) were anesthetized by inhalation of CO2 and decapitated after loss of consciousness. The brain was rapidly removed, and was immersed in ice-cold sucrose-based artificial cerebrospinal fluid (“sucrose-ACSF,” in mM: 126 sucrose, 5.0 KCl, 2.0 CaCl2, 2.0 MgSO4, 1.25 NaH2PO4, 26 NaHCO3, 10 d-glucose; pH = 7.4). The brainstem severed from the forebrain by a coronal cut at the anterior tip of the cerebellum, and the anterior surface glued to a Teflon-coated tray. The tray was immersed in ice-cold sucrose ACSF and 400 µm-thick slices were cut using a Vibroslice (World Precision Instruments). Slices containing the hypoglossal nucleus were transferred immediately to an interface recording chamber, where they were maintained on a nylon net at 30–32°C, oxygenated (5% CO2, 95% O2), and immersed in sucrose-ACSF except for the upper surface. After 30 min, sucrose-ACSF was replaced by ACSF containing NaCl substituted equimolar for sucrose (“saline-ACSF”), and perfusion with saline-ACSF continued for the remainder of the recordings (flow rate: approximately 1 ml/min, controlled by a peristaltic pump). Recordings started approximately 30 min after the onset of saline-ACSF perfusion.
2). Recording and stimulation
Intracellular recording electrodes were made of borosilicate glass containing a capillary fiber in the lumen (0.75 mm inner diameter, 1.0 mm outer diameter; World Precision Instruments), were pulled horizontally (Flaming-Brown Model P97, Sutter Instruments), filled with 1M potassium acetate, and resistances were 70–110 MΩ. Data were recorded using an intracellular amplifier with a bridge circuit (Axoclamp 2B, Molecular Devices), and the bridge was balanced whenever current was passed. Recordings were monitored on a digital oscilloscope (Pro10, Nicolet Instruments), and DAT Recorder (4 channel, Microdata Instruments), and saved on floppy disk for subsequent offline analysis using OriginLab 7.0 software (OriginLabs). In some experiments, 4% Neurobiotin in 1 M potassium acetate was used to backfill electrodes; the shaft was filled with 1 M potassium acetate alone. Methods to inject Neurobiotin, and process the slice after Neurobiotin injection, are described elsewhere[18].
A bipolar electrode was used for electrical stimulation. It was made from Teflon-coated stainless steel wire (75 µm diameter, including Teflon, A & M Systems). The tips of the poles rested on the slice surface. One of the poles was placed dorsal to the location of the solitary nucleus, and the other pole was placed ventral to the nucleus. A rectangular current pulse (100 µA, 10–200 µsec) was used as an electrical stimulus, and was triggered digitally (Pulsemaster, World Precision Instruments) using a stimulus isolator (AMPI). Stimulus frequency was 0.05 Hz. Stimulation and recording electrodes were not moved throughout the baseline period and exposure to test reagents.
D. Drug Application
The 165 amino acid isoform of recombinant human VEGF protein (22 kDa) was supplied by Regeneron Pharmacueticals. It was derived from transfected Chinese hamster ovary cells and supplied in 20 µl aliquots of 2.61 mg/ml solution in 15 mM citrate containing 0.02% CHAPS (pH 5.5), which were maintained at −20°C until use. Aliquots were diluted with 0.05% bovine serum albumin (BSA) and added to the saline-ACSF perfusate at a final concentration of 200 ng/ml VEGF and <0.001% BSA. For control experiments, BSA was used at concentrations up to 0.0015%, or heat-inactivated VEGF was used. For heat-inactivation, VEGF placed in a container that was immersed in boiling water for 30 minutes.
IV. Data analysis
A. Electrophysiology
1) Intrinsic properties
Terminology and measurements of intrinsic properties have been described elsewhere [18]. In brief, resting membrane potential (RMP) was defined as the difference between the intracellular potential and the potential recorded after the electrode was withdrawn from the cell. Input resistance was determined as the slope of the steepest part of the steady state I–V curve, based on steady-state responses to ± 0.1 − 1.0 nA, 200 msec duration rectangular current pulses, injected intracellularly at RMP. Time constant (τ) was defined as the latency from the onset of the response to a −0.1 nA, 200 msec rectangular current step to the point that reached 0.63% of the peak voltage deflection. A −0.1 nA step was chosen because the voltage response was similar across cells and was too weak to activate rectifying currents. Action potentials (APs) parameters were measured from an AP evoked at threshold, using a 200 msec current pulse that generated a single AP in response to approximately 50% of current pulses. AP amplitude was measured from RMP to peak. Dv/dt was defined as the maximum slope of the rising phase of the AP divided by the maximum slope of the falling phase; maximal slope was determined from the derivative of the AP in Origin 7.0 (OriginLabs). Afterhyperpolarization (AHP) was defined as the negative deflection following an AP evoked at threshold by current injection, and measured from the membrane potential preceding the fast rise of the AP to the maximal hyperpolarization following the decay of the AP.
2) Synaptic responses
Measurements of synaptic potentials were made from an average of three or more evoked responses, using a low stimulus frequency to avoid frequency-dependent effects (0.01–0.02 Hz). For each time the response was evaluated (every 5 min before VEGF and every 10 min after VEGF was added to the perfusate), at least 3 responses to a stimulus were averaged. The stimulus required to elicit a maximal subthreshold depolarization during the baseline (pre-VEGF) period was used for the entire experiment. Holding potentials for all cells were similar (approximately −60 mV).
Measurements of peak amplitude and maximal rising slope was measured from the averaged trace described above. Peak amplitude was defined as the voltage difference between the pre-stimulus potential and the peak of the synaptic potential. Maximum slope was defined as the maximum rate of change during the rising phase of the synaptic potential, determined by a derivative function of the potential waveform, computed by Origin 7.0 (OriginLabs). EPSP half-duration was determined as the time from the onset of the EPSP to the time during the decay phase when the amplitude had declined to 50% of its peak amplitude.
B. Statistics
Comparisons of intrinsic properties before VEGF exposure relative to the time after VEGF exposure (pre vs. post VEGF) used a paired Student’s t-test. Statistics used to evaluate the time course of the change during the entire period of VEGF exposure used a mixed-design repeated measures ANOVA. Significance was defined by p < 0.05.
RESULTS
I. Prolonged motor seizures increase motor neuron VEGF expression
Pilocarpine-treated rats that had status (n=18 total) were compared to saline-treated controls (n=9 total). 24 hrs after status, VEGF expression was evident in motor neurons in all areas examined (hypoglossal, facial, spinal cord; Figure 1; n=12). In saline controls, VEGF expression was relatively weak (Figure 1; n=9), but localized to the same areas, the cytoplasm and proximal dendrites (Figure 1).
Figure 1. VEGF expression is increased in motor neurons after status.

A1. A coronal section through the brainstem showing the hypoglossal nucleus (HYPO). This section was stained with an antibody to VEGF and shows weak VEGF expression.
2. A coronal section through the same rostro-caudal level of the brainstem as A, but the section was from a rat that had 1 hr of status the previous day. Neurons in the hypoglossal nucleus show increased VEGF expression (arrows). The sections in A and B were processed concurrently.
B1. Motor neurons in the facial nucleus demonstrated weak VEGF immunoreactivity in control rats.
2. Arrows point to facial motor neurons with strong strong VEGF immunoreactivity in a section from a rat that had 1 hr of status the previous day.
C1. A section from the spinal cord of a rat that was a saline control.
2. VEGF immunoreactivity was present in many cells of the spinal cord 1 day after status (arrows). A large arrowhead points to a large cluster of VEGF-immunoreactive cells. Arrows point to large neurons, presumably motor neurons.
D1. VEGF expression in ventral horn from a saline-control rat.
2. VEGF expression in large neurons, presumably motor neurons, of the ventral horn (arrows) in a rat that was perfused 1 day after status. Calibration: A, 250 µm; B, 70 µm; C, 150 µm; D, 35 µm.
II. VEGF upregulation: a response to seizure-induced injury to motor neurons?
One potential reason why motor neurons increased expression of VEGF after seizures was that they were dying, because status leads to cell death in several neuronal populations in forebrain [27,28]. Thus, using the methods in the current study, previous studies have shown that status, even when truncated by anticonvulsants, damages neurons in forebrain [15,21]. An alternate explanation is that the motor neurons expressed VEGF as a response to insult or injury. This explanation is suggested by the studies of traumatic injury and ischemia, which induce growth factor expression in many types of neurons (for review, see [19]. In addition, hypoxia-inducible factor 1α (HIF1α) is known to induce VEGF expression [5]. To address this issue, silver degeneration stain was used to evaluate whether status led to degeneration of motor neurons.
As shown in Figure 2, control animals (n=3) and animals that had convulsions (n=6) did not demonstrate degeneration of motor neurons. Thus, no neurons had silver uptake in their cytoplasm. However, upon closer examination, there was a difference between motor neurons in control rats and motor neurons in rats that had status. In control rats, silver stain was present in the nucleolus of motor neurons (Figure 2). In animals that experienced status and were examined 24 hrs later (n=6), silver stain was present not only in the nucleolus, but also the nucleus itself (Figure 2). Results were similar regardless of the motor neuron populations that were examined (facial motor neurons, hypoglossal motor neurons, and spinal cord motor neurons; Figure 2). Silver uptake was similar to controls (nucleolus only) when animals that had status were examined three days later (n=2) or one week later (n=3; Figure 2).
Figure 2. Silver uptake in motor neurons after status.

A–B. Silver stained sections from a saline control (A) and animal that was perfused 1 day after status (B) show increased staining in the hypoglossal nucleus of the animal that had status (arrows in B).
C–D. At higher power, the silver staining in the saline control in A appears to be confined to the nucleolus (arrow in C) or is not evident, but it is in the nucleus after status (arrows in D)
E–F. High magnification and increased contrast is used to illustrate the difference in silver uptake shown in C–D more clearly. In F, an exceptional neuron that does not show silver uptake is marked.
G–H. Silver uptake in spinal cord motor neurons was similar qualitatively to the pattern of staining in hypoglossal motor neurons: the nucleolus was stained in motor neurons from saline-treated controls (arrows in G), and the entire nucleus was stained in motor neurons from animals that had status (arrow in H).
I. A neuron in the facial nucleus with silver stain throughout the nucleolus shows the same distribution of silver as other motor neurons from animals that had status the previous day.
J. Silver uptake in spinal motor neurons 1 week after status epilepticus shows a pattern of silver uptake similar to control rats. Calibration: A–B, 320 µm; C–D, 80 µm; E–J, 40 µm.
III. Physiological effects of VEGF
A. Identification of hypoglossal motoneurons
Recordings were made from hypoglossal motor neurons for a number of reasons. First, they are a defined population of motor neurons, which are relevant to our understanding of VEGF [9,26,30]. In addition, VEGF was upregulated in hypoglossal motor neurons after status. They are also practical to study, because the hypoglossal nucleus can be reliably located with a dissecting microscope, based on landmarks such as the central canal. Another reason to record from hypoglossal motoneurons is the ability to elicit robust evoked responses in slices, providing an opportunity to evaluate afferent input. Finally, hypoglossal motoneurons are the primary cell type in hypoglossal nucleus, so that virtually all neurons sampled are likely to be motor neurons.
To confirm that hypoglossal motor neurons were sampled in our recordings within the hypoglossal nucleus, recorded cells were filled with the marker Neurobiotin so they could be evaluated morphologically. As shown in Figure 3, the morphology of recorded cells suggested that they were motor neurons. In addition, the intrinsic properties and firing behavior was also consistent with motor neurons (see below). Although we can not exclude that other cell types were sampled, the data suggest it is unlikely.
Figure 3. An intracellularly-labeled hypoglossal motorneuron.

A. A hypoglossal neuron that was filled intracellularly with Neurobiotin in shown. The slice was 400 µm thick, and then resectioned, so many branches of the dendrites are cut. Arrows point to the ventral border of the hypoglossal nucleus. An asterisk marks the central canal.
B. A montage of three sections of the cell shown in A illustrates the location of many of the dendrites. Arrows point to the axon, which descended ventrally and exited the nucleus. Calibration (in B): 70 µm (A), 35 µm (B).
B. Intrinsic properties
As shown in Table 1, there were no significant differences in resting membrane potential, input resistance, time constant (τ), AP parameters (amplitude, slope, or duration), or the AHP in hypoglossal neurons that were exposed to 200 ng/ml VEGF (n=6). Comparisons were made between the mean value for the period prior to VEGF exposure and the mean value after 40 min of exposure to VEGF (Table 1; paired Student’s t-tests; p> 0.05). Prior to VEGF exposure, slices were examined every 5 minutes for 15 minutes. After VEGF exposure, slices were examined every 10 minutes, and 40 minutes was chosen for statistical comparisons because the effects of VEGF on evoked responses appeared to be maximal at that time.
Table 1.
Intrinsic properties of hypoglossal motor neurons before and after exposure to VEGF.
| RMP | Rin | τ | -AP amplitude- | ---- AP slope------ | --AP duration-- | AHP | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| total amp. | from thresh. | max. rise | max. decay | dv/dt ratio | total dur. | 1/2 width | |||||
| (mV) | (MΩ) | (msec) | (mV) | (mV) | (V/S) | (V/S) | (msec) | (msec) | (mV) | ||
| Control | |||||||||||
| Mean | −60.1 | 15.6 | 2.2 | 59.1 | 57.3 | 267.1 | 161.9 | 1.7 | 1.7 | 1.3 | 8.98 |
| SEM | 1.2 | 1.9 | 0.4 | 2.6 | 2.5 | 35.7 | 24.7 | 0.1 | 0.3 | 0.1 | 1.1 |
| VEGF | |||||||||||
| Mean | −60.8 | 16.9 | 2.1 | 58.4 | 57.1 | 250.2 | 142.8 | 1.8 | 1.7 | 1.3 | 9.08 |
| SEM | 1.7 | 1.4 | 0.3 | 3.5 | 4.0 | 44.0 | 24.0 | 0.1 | 0.3 | 0.3 | 2.3 |
| p | 0.8 | 0.3 | 0.8 | 0.8 | 0.9 | 0.2 | 0.3 | 0.5 | 0.9 | 0.9 | 0.9 |
Intrinsic properties of hypoglossal neurons are shown. Measurements were made during a 15 min period before VEGF and averaged (Control) or at a time after VEGF exposure (40–60 min after VEGF was added to the perfusate) when synaptic potentials were depressed. There were no significant differences (paired Student’s t-tests). Abbreviations: amp. = amplitude; dur. = duration; max. = maximum; RMP = resting membrane potential; Rin = input resistance; τ = time constant; dv/dt ratio = ratio of the maximal slope of the rising phase/ maximal slope of the decay phase of an AP at threshold; AHP = afterhyperpolarization.
Figure 4 illustrates the lack of effect of VEGF on the response to hyperpolarizing current commands. VEGF did not influence rectification elicited by hyperpolarizing current commands (“sag;” Figure 4). Neither the initial or steady-state response to hyperpolarizing current changed upon exposure to VEGF for 60 min (Figure 4).
Figure 4. VEGF does not alter motor neuron intrinsic properties.
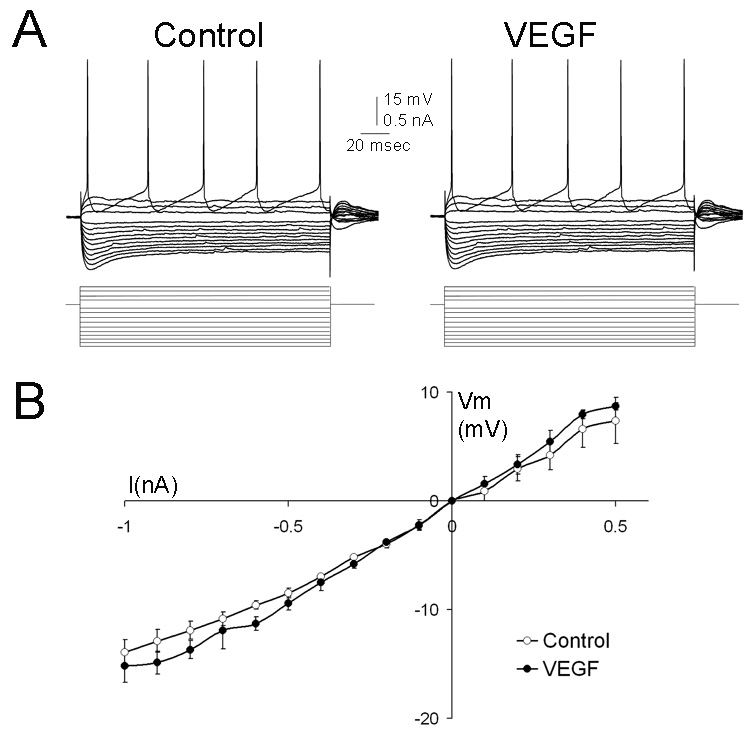
A. Representative traces from a hypoglossal motoneuron show no detectable change in input resistance, time constant, or firing behavior before (control) or 60 min after VEGF administration (VEGF).
B. Composite I–V curve based on responses from 6 cells illustrates the mean steady-state voltage response to a family of current steps, evaluated before and 40–60 min after addition of VEGF to the perfusate. Comparison of maximal slopes of these curves showed no significant effect of VEGF (paired Student’s t-test, p>0.05). Abbreviations: current: I, membrane potential: Vm.
C. Firing behavior
Figure 4A illustrates the firing pattern elicited by suprathreshold depolarizing current. The same current pulse was tested throughout exposure to VEGF (200 ng/ml, 60 min) and the firing pattern was similar: cells continued to fire in trains. In addition, f-I curves demonstrated no changes during VEGF exposure in the frequency of the first two APs during a train as a function of injected current (Figure 4C). The maximal slope of the f-I curve was not statistically different before and after VEGF treatment (Student’s t-test; p>0.05). Spike frequency adaptation also appeared to be unaffected by VEGF, as shown in Figure 4D. To compare adaptation, a current pulse that evoked 5 APs for a 200 msec pulse was compared (n=4 cells). The mean interspike intervals during the train of 5 APs for these 4 cells were not influenced by VEGF (Figure 4D).
D. Evoked responses
As illustrated in Figure 5, depolarizations that were elicited by stimulation of the slice were decreased after VEGF was added to the perfusate. Using a stimulus that elicited a maximal, subthreshold depolarization, the amplitude, slope and duration were measured during a 10–15 min period before exposure to VEGF, to establish stability of the response. It was then monitored every 10 min after 200 ng/ml VEGF was added to the saline-ACSF. After VEGF was added, there was a gradual decrease in the slope of the evoked depolarization, and amplitude, but not the half-duration (n=6; Figure 5). In two of these experiments, heat-inactivated VEGF was tested prior to VEGF (Figure 5), and had no detectable effect. However, the same cells exhibited a robust response when VEGF was subsequently added to the perfusate (Figure 5). Comparison of the average response that was evoked during the baseline period (pre-VEGF) to the average response after exposure to VEGF for 60 min (post-VEGF) demonstrated that the amplitude and slope declined (Student’s t-test, baseline vs. VEGF+ 60 min, p < 0.05), but not the half-duration (Student’s t-test, baseline vs. VEGF + 60 min, p > 0.05). To evaluate the experiments in their entirety, a mixed design ANOVA was conducted for each measure with perfusate (VEGF vs. heat-inactivated VEGF) as the between-subjects factor, and time after treatment (10 – 60 minutes after addition to the bath) as the within-subjects factor. This analysis showed that VEGF significantly reduced the slope (F(1, 30) = 15.72, p = 0.0074), and amplitude (F(1, 30) = 6.19, p = 0.047) of the synaptic response, but not the duration (F(1, 30) = 0.40, p = 0.548).
Figure 5. VEGF reduces depolarizations in hypoglossal motor neurons evoked by electrical stimulation of the slice.

A. Responses of a hypoglossal neuron to an electrical stimulus. Electrodes were placed in the solitary nucleus. Responses are shown before VEGF was added (Control), after 30 min perfusion with heat-inactivated VEGF (Denatured VEGF) and following exposure to VEGF for 30 min (VEGF). Each trace is the response to a fixed stimulus at the same holding potential (−70 mV; a dot marks the stimulus artifact, which is truncated). The effect of heat- inactivated VEGF was not detectable, but subsequent exposure to VEGF decreased the evoked response.
B. Responses from A are superimposed to illustrate the stability of the response to stimulation during the Control period (Control) and after exposure to heat-inactivated VEGF (D VEGF), and the reduction in the response after VEGF was added to the perfusate.
C. Data from all experiments are normalized and pooled to show that VEGF significantly reduced slope and amplitude of evoked depolarizations, but not duration. The mean response during the baseline period was defined as 100%. Effects were induced by 200 ng/ml VEGF (black circles) but not heat-inactivated VEGF (white boxes).
DISCUSSION
Summary
The main findings of the present study are as follows: 1) VEGF is increased in many populations of motor neurons after severe seizures, and 2) VEGF reduces depolarizing input to hypoglossal motor neurons in vitro without an apparent influence on intrinsic properties, or the intrinsic pattern of action potential discharge.
VEGF upregulation after seizures: a response to insult or injury, excess afferent input, or other factors?
VEGF was increased in diverse types of motor neurons after seizures. A likely explanation is the increase in afferent input to motor neurons. This would be consistent with the ability of many growth factors to be increased by neuronal activity or seizures [4,17,19]. Increased activation of motor neurons could have been due to descending cortical input, which would be expected to be greater than normal during generalized seizures; in addition, increased afferent input may have developed because of muscular contractions. Pilocarpine itself is unlikely to have been a stimulus, given the fact that animals administered pilocarpine that did not have status failed to demonstrate changes relative to saline-treated controls (data not shown).
Increased expression of VEGF could also be a response to injury. The fact that silver uptake was confined to the nucleus, and did not invade the cytoplasm, suggests that motor neurons were not degenerating. It is unlikely that these neurons would have died if longer periods of survival had been used, because at 3 or 7 days after status, silver uptake was similar to controls.
Silver uptake into different subcellular compartments has not been reported to our knowledge, so it is hard to interpret the functional relevance of silver uptake into the nucleus vs. nucleolus. It may be that the uptake into the nucleus reflects a reaction to intense neuronal activation. VEGF upregulation could be analogous to heat shock protein expression, which occurs in neurons that are exposed to insults but do not die. Given that VEGF protects neurons from insults such as experimental ischemia or seizure-induced damage [6,11,29], and motorneurons [10,25,31], it is possible that the upregulation of VEGF is an endogenous protective mechanism.
Our data are consistent with the demonstration that motor neurons increase VEGF expression after hypoxia or spinal cord injury [3,14]. It is important to consider that hypoxia, spinal cord injury, and seizures may share a common mechanism, one that is well-established to regulate VEGF: the intermediary hypoxia inducible factor-1 (HIF-1α). It may be that activity generated during seizures led to an increase in oxygen demand, which activated HIF-1α, especially in motor neurons.
VEGF suppression of evoked responses
The data suggest that VEGF has an effect on motor neurons that is independent of intrinsic properties or the vasculature: VEGF suppresses depolarizations evoked by afferent input, presumably excitatory afferent input. The ability of VEGF to suppress depolarizing inputs to brainstem motor neurons is consistent with the ability of VEGF to suppress excitatory input to pyramidal and granule neurons in the hippocampus [11].
In hippocampus, a glial mechanism was suggested to explain the effects of VEGF on synaptic transmission, because similar changes were observed on glutamatergic and GABAergic transmission, without changes in intrinsic properties [11]. In addition, the primary pattern of expression of VEGF in hippocampus appears to be punctate immunoreactivity outlining glia [6]. Glia are known to express VEGF receptors, and glia are known to modulate neurotransmission [11,29]. Therefore, a mechanism that is consistent with these data would be an action of VEGF at glial VEGF receptors which would decrease synaptic transmission. However, future experiments will be required to prove this hypothesis.
Significance
The data presented here suggest that VEGF expression is plastic in motor neurons throughout the brainstem and spinal cord, and it is upregulated after insults. In addition, VEGF exerts effects on synaptic transmission. The results provide new insight into the ways that VEGF may be important in motor neuron survival and function. The ability of VEGF to increase its synthesis after insults, and depress excitation of motorneurons, could be relevant to ALS, where excitotoxicity and deficits in VEGF are suggested to be contributing factors [8,30].
Acknowledgements
This work was supported by National Institutes of Health Grant NS 35762 (H.E.S.), and the Helen Hayes Hospital Foundation. We thank Regeneron Pharmaceuticals for the generous gift of VEGF. We thank Dr. Susan Croll for discussion, and are grateful to Annmarie Curcio and Catherine Snodgrass for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Anderson K, Fugaccia I, Scheff S. Fluoro-jade B stains quiescent and reactive astrocytes in the rodent spinal cord. J Neurotrauma. 2003;20:1223–1231. doi: 10.1089/089771503770802899. [DOI] [PubMed] [Google Scholar]
- 2.Azzouz M, Ralph GS, Storkebaum E, Walmsley LE, Mitrophanous KA, Kingsman SM, Carmeliet P, Mazarakis ND. VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature. 2004;429:413–417. doi: 10.1038/nature02544. [DOI] [PubMed] [Google Scholar]
- 3.Bartholdi D, Rubin B, Schwab M. VEGF mRNA induction correlates with changes in the vascular architecture upon spinal cord damage in the rat. Eur J Neurosci. 1997;9:2549–2560. doi: 10.1111/j.1460-9568.1997.tb01684.x. [DOI] [PubMed] [Google Scholar]
- 4.Binder DK, Scharfman HE. Growth Factors and Epilepsy. New York: Nova Sciences; 2005. [Google Scholar]
- 5.Choi KS, Bae MK, Jeong JW, Moon HE, Kim KW. Hypoxia-induced angiogenesis during carcinogenesis. J Biochem Mol Biol. 2003;36:120–127. doi: 10.5483/bmbrep.2003.36.1.120. [DOI] [PubMed] [Google Scholar]
- 6.Nicoletti J, Sachin S, McCloskey DP, Goodman JH, Scharfman HE, Croll SD. Vascular endothelial growth factor (VEGF) is upregulated after status epilepticus and protects against seizure-induced neuronal loss in hippocampus. Neuroscience. doi: 10.1061/j.neuroscience.2007.09.083. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Devos D, Moreau C, Lassalle P, Perez T, De Seze J, Brunaud-Danel V, Destee A, Tonnel AB, Just N. Low levels of the vascular endothelial growth factor in CSF from early ALS patients. Neurology. 2004;62:2127–2129. doi: 10.1212/01.wnl.0000129913.44351.a3. [DOI] [PubMed] [Google Scholar]
- 8.Heath PR, Shaw PJ. Update on the glutamatergic neurotransmitter system and the role of excitotoxicity in amyotrophic lateral sclerosis. Muscle Nerve. 2002;26:438–458. doi: 10.1002/mus.10186. [DOI] [PubMed] [Google Scholar]
- 9.Lambrechts D, Storkebaum E, Carmeliet P. VEGF: necessary to prevent motoneuron degeneration, sufficient to treat ALS? Trends Mol. Med. 2004;10:275–282. doi: 10.1016/j.molmed.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 10.Lambrechts D, Storkebaum E, Morimoto M, Del-Favero J, Desmet F, Marklund SL, Wyns S, Thijs V, Andersson J, Van Marion I, Al-Chalabi A, Bornes S, Musson R, Hansen V, Beckman L, Adolfsson R, Pall HS, Prats H, Vermeire S, Rutgeerts P, Katayama S, Awata T, Leigh N, Lang-Lazdunski L, Dewerchin M, Shaw C, Moons L, Vlietinck R, Morrison KE, Robberecht W, Van Broeckhoven C, Collen D, Andersen PM, Carmeliet P. VEGF is a modifier of amyotrophic lateral sclerosis in mice and humans and protects motoneurons against ischemic death. Nature Gen. 2003;34:383–394. doi: 10.1038/ng1211. [DOI] [PubMed] [Google Scholar]
- 11.McCloskey DP, Croll SD, Scharfman HE. Depression of synaptic transmission by vascular endothelial growth factor in adult rat hippocampus and evidence for increased efficacy after chronic seizures. J Neurosci. 2005;25:8889–8897. doi: 10.1523/JNEUROSCI.2577-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Newton SS, Collier EF, Hunsberger J, Adams D, Terwilliger R, Selvanayagam E, Duman RS. Gene profile of electroconvulsive seizures: induction of neurotrophic and angiogenic factors. J Neurosci. 2003;23:10841–10851. doi: 10.1523/JNEUROSCI.23-34-10841.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling - in control of vascular function. Nat Rev Mol Cell Biol. 2006;7:359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 14.Oosthuyse B, Moons L, Storkebaum E, Beck H, Nuyens D, Brusselmans K, Van Dorpe J, Hellings P, Gorselink M, Heymans S, Theilmeier G, Dewerchin M, Laudenbach V, Vermylen P, Raat H, Acker T, Vleminckx V, Van Den Bosch L, Cashman N, Fujisawa H, Drost M, Sciot R, Bruyninckx F, Hicklin D, Ince C, Gressens P, Lupu F, Plate K, Robberecht W, Herbert J, Collen D, Carmeliet P. Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nature Gen. 2001;28:107–108. doi: 10.1038/88842. [DOI] [PubMed] [Google Scholar]
- 15.Pitkanen A, Schwartzkroin PA, Moshe SL. Models of Seizures and Epilepsy. New York: Elsevier; 2005. [Google Scholar]
- 16.Racine RJ. Modification of seizure activity by electrical stimulation: II. Motor seizure. Electroencephal Clin Neurophys. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- 17.Scarisbrick IA, Isackson PJ, Windebank AJ. Differential expression of brain-derived neurotrophic factor, neurotrophin-3, and neurotrophin-4/5 in the adult rat spinal cord: regulation by the glutamate receptor agonist kainic acid. J Neurosci. 1999;19:7757–7769. doi: 10.1523/JNEUROSCI.19-18-07757.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scharfman HE. Electrophysiological diversity of pyramidal shaped neurons at the granule cell layer hilus border of the rat dentate gyrus recorded in vitro. Hippocampus. 1995;5:287–305. doi: 10.1002/hipo.450050403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scharfman HE. Epilepsy as an example of neural plasticity. The Neuroscientist. 2002;8:154–173. doi: 10.1177/107385840200800211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scharfman HE. Seizure-induced neurogenesis in the dentate gyrus and its dependence on growth factors and cytokines. In: Binder DK, Scharfman HE, editors. Growth Factors and Epilepsy. New York: Nova Science Publishers; 2006. pp. 1–40. [Google Scholar]
- 21.Scharfman HE, Sollas AL, Smith KL, Jackson MB, Goodman JH. Structural and functional asymmetry in the normal and epileptic rat dentate gyrus. J Comp Neurol. 2002;454:424–439. doi: 10.1002/cne.10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sloviter RS. A simplified Timm stain procedure compatible with formaldehyde fixation and routine paraffin embedding of rat brain. Brain Res. Bull. 1982;8:771–774. doi: 10.1016/0361-9230(82)90104-6. [DOI] [PubMed] [Google Scholar]
- 23.Sopher BL, Thomas PS, LaFevre-Bernt MA, Holm IE, Wilke SA, Ware CB, Jin LW, Libby RT, Ellerby LM, La Spada AR. Androgen receptor YAC transgenic mice recapitulate SBMA motor neuronopathy and implicate VEGF164 in the motor neuron degeneration. Neuron. 2004;41:687–699. doi: 10.1016/s0896-6273(04)00082-0. [DOI] [PubMed] [Google Scholar]
- 24.Storkebaum E, Lambrechts D, Dewerchin M, Moreno-Murciano M-P, Appelmans S, Oh H, Van Damme P, Rutten B, Man WY, De Mol M, Wyns S, Manka D, Vermeulen K, Van Den Bosch L, Mertens N, Schmitz C, Robberecht W, Conway EM, Collen D, Moons L, Carmeliet P. Treatment of motoneuron degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS. Nat Neurosci. 2005;8:85–92. doi: 10.1038/nn1360. [DOI] [PubMed] [Google Scholar]
- 25.Sun Y, JIn K, Xie L, Childs J, Mao XO, Logvinova A, Greenberg DA. VEGF-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. J Clin Invest. 2003;111:1843–1851. doi: 10.1172/JCI17977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takahashi H, Shibuya M. The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clin Sci (Lond) 2005;109:227–241. doi: 10.1042/CS20040370. [DOI] [PubMed] [Google Scholar]
- 27.Turski L, Ikonomidou C, Turski WA, Bortolotto ZA, Cavalheiro EA. Review: cholinergic mechanisms and epileptogenesis. The seizures induced by pilocarpine: a novel experimental model of intractable epilepsy. Synapse. 1989;3:154–171. doi: 10.1002/syn.890030207. [DOI] [PubMed] [Google Scholar]
- 28.Turski WA, Cavalheiro EA, Schwarz M, Czuczsar SJ, Kleinrok Z, Turski L. Limbic seizures produced by pilocarpine in rats: behavioural, electroencephalographic and neuropathological study. Behav Brain Res. 1983;9:315–335. doi: 10.1016/0166-4328(83)90136-5. [DOI] [PubMed] [Google Scholar]
- 29.Croll S, Goodman J, Scharfman H. Vascular endothelial growth factor (VEGF) in seizures: a double-edged sword. Adv Exp Medi Biol. 2004;548:57–68. doi: 10.1007/978-1-4757-6376-8_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Damme P, Dewil M, Robberecht W, Van Den Bosch L. Excitotoxicity and amyotrophic lateral sclerosis. Neurodegen Dis. 2005;2:147–159. doi: 10.1159/000089620. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y, Jin K, Mao XO, Xie L, Banwait S, Marti HH, Greenberg DA. Vascular endothelial growth factor overexpression delays neurodegeneration and prolongs survival in amyotrophic lateral sclerosis mice. J Neurosci. 2007;27:304–307. doi: 10.1523/JNEUROSCI.4433-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]