

Abstract
Some trypsin-like proteases are endowed with Na+-dependent allosteric enhancement of catalytic activity, but this important mechanism has been difficult to engineer in other members of the family. Replacement of nineteen amino acids in Streptomyces griseus trypsin targeting the active site and the Na+ binding site were found necessary to generate efficient Na+ activation. Remarkably, this property was linked to the acquisition of a new substrate selectivity profile similar to that of factor Xa, a Na+-activated protease involved in blood coagulation. The X-ray crystal structure of the mutant trypsin solved to 1.05 Å resolution defines the engineered Na+ site and active site loops in unprecedented detail. The results demonstrate that trypsin can be engineered into an efficient allosteric protease and that Na+ activation is interwoven with substrate selectivity in the trypsin scaffold.
Keywords: Trypsin, allostery, monovalent cation activation, crystal structure, sodium
Introduction
Serine proteases of the S1 family are responsible for critical physiological functions such as digestion, salt homeostasis, fibrinolysis, immunity, fertilization, development, and blood coagulation 1; 2. Of these, several members of the complement system and blood coagulation cascade belong to the large family of monovalent cation activated enzymes 3; 4 and their catalytic activity is enhanced by Na+ binding in an allosteric manner 5. A remarkable dichotomy in the distribution of residue 225 (chymotrypsinogen numbering) acts as a predictor of allosteric behavior 5; 6; 7. Serine proteases of the S1 family carry either Pro or Tyr at this position with very few outliers 5; 6. Dichotomous codon choice at residue 225 is noteworthy because the amino acid codons of Pro and Tyr do not interconvert by a single nucleotide substitution.
Composition of residue 225 impacts the architecture of the primary specificity pocket of trypsin-like proteases. Pro is found in nearly all digestive and degradative proteases, but Tyr is found in proteases of the complement system and blood coagulation 5; 6. Presence of Pro-225 restricts the carbonyl O atom of residue 224, one Na+ ligand 8; 9, to point toward the carboxylate of Asp-189. The ensuing H-bond stabilizes the side chain of Asp-189 to define the bottom of a cavity connecting the primary specificity pocket to the active site 10. In contrast, Na+-activated proteases carry a Tyr (or Phe) residue at position 225 relieving the constraint on the carbonyl O atom of residue 224 and enabling direct coordination of Na+ 5; 9; 10. The H-bond between that carbonyl O atom and the carboxylate of Asp-189 in Pro-225 bearing proteases is replaced by an intervening water molecule connecting the bound Na+ to Asp-189. Stabilization of Asp-189 becomes a function of the ligation state of the Na+ site and generates a powerful mechanism controlling substrate binding 9; 11; 12.
Mutagenesis of Y225P thrombin, factor Xa, factor VIIa and activated protein C abrogates Na+ binding 5; 13; 14; 15, but the reverse substitution P225Y in proteases devoid of Na+ binding is not sufficient to enable Na+ activation 16; 17. Engineering allostery 18; 19 and documenting its structural signatures 20; 21; 22 are currently topics of substantial general interest. Because the majority of serine proteases are devoid of Na+-dependent allosteric regulation 5; 6, studies aimed at introducing this property in the trypsin scaffold are both timely and important.
Results & Discussion
Preliminary attempts to engineer Na+-dependent allostery into the trypsin scaffold illustrate the complexity of this task 17. Streptomyces griseus trypsin (SGT) was recently converted into a Na+-activated protease by swapping the 170-, 186- and 220- loops with those of clotting factor Xa (FXa) 17. Unfortunately, the resulting mutant SGT (FX170) was a poorly active protease (activity <1% compared to wild-type) and structural insight could not be garnered on the spatial architecture of the engineered loops. Inclusion of five amino acid substitutions to introduce the entire 99-loop of FXa into FX170 produces a construct, FX99, dramatically more active than FX170 carrying a total of nineteen replacements and the entire structural determinants for Na+ binding and recognition of the S1–S3 positions of substrate found in FXa. Selective activation of FX99 by Na+ (Figure 1A) brings the kcat/Km for hydrolysis of H-d-Arg-Gly-Arg-p-nitroanilide within 6% of that of wild-type SGT with a significant (>50-fold) improvement relative to the Na+-activated mutant of SGT, FX170 17. Affinity of FX99 for Na+ (KA=6.1±0.1 M−1) is almost 2-fold higher than that of FX170 17 and comparable to that of FXa 23. Activity toward chromogenic substrates carrying substitutions at the P1, P2 and P3 positions 24 is significantly higher than that of FX170 and is at least as high as that of FXa (Figure 1B). The gain in activity comes with a small but noteworthy gain in selectivity. FXa differs from SGT in the ability to discriminate residues at the P1–P3 positions of substrate. Both proteases prefer H-d-Arg vs. H-d-Phe at the P3 position and Arg vs. Lys at the P1 position, though FXa shows >10-fold better discrimination. Furthermore, the Pro vs. Gly preference at the P2 position in SGT is reversed in FXa (Figure 2). Comparison of FX170 with FX99 reveals a resemblance of the former construct with SGT and of the latter construct with FXa (Figure 2). FX99 has successfully captured Na+ activation, high activity and substrate selectivity of FXa in the SGT scaffold.
Figure 1.

(A) Monovalent cation specificity profile of FX99 reveals the specific Na+ effect on the hydrolysis of the chromogenic substrate RGR. Values of s=kcat/Km in the presence of 200 mM salt as indicated are expressed relative to the value measured in the presence of 200 mM choline (Ch) chloride. (B) Na+ activation of FX99 (■) improves the value of s=kcat/Km for the hydrolysis of RGR. Even in the absence of Na+, FX99 is more active than the Na+-activated mutant of SGT (○), FX170, reported previously 17. Continuous lines were drawn using the linkage equation 9; 11, where s0 and s1 are the values of s at [Na+]=0 and [Na+]=∞, and KA is the apparent Na+ affinity, with best-fit parameter values: (■) KA=6.1±0.1 M−1, s0=0.19±0.01 µM−1s−1 and s1=3.0±0.1 µM−1s−1; (○) KA=3.6±0.3 M−1, s0=0.0036±0.0002 µM−1s−1 and s1=0.073±0.003 µM−1s−1. Experimental conditions are: 25 mM Tris, 10 mM CaCl2, 0.1 % PEG 8000, pH 8.0 at 25 ° C.
Figure 2.
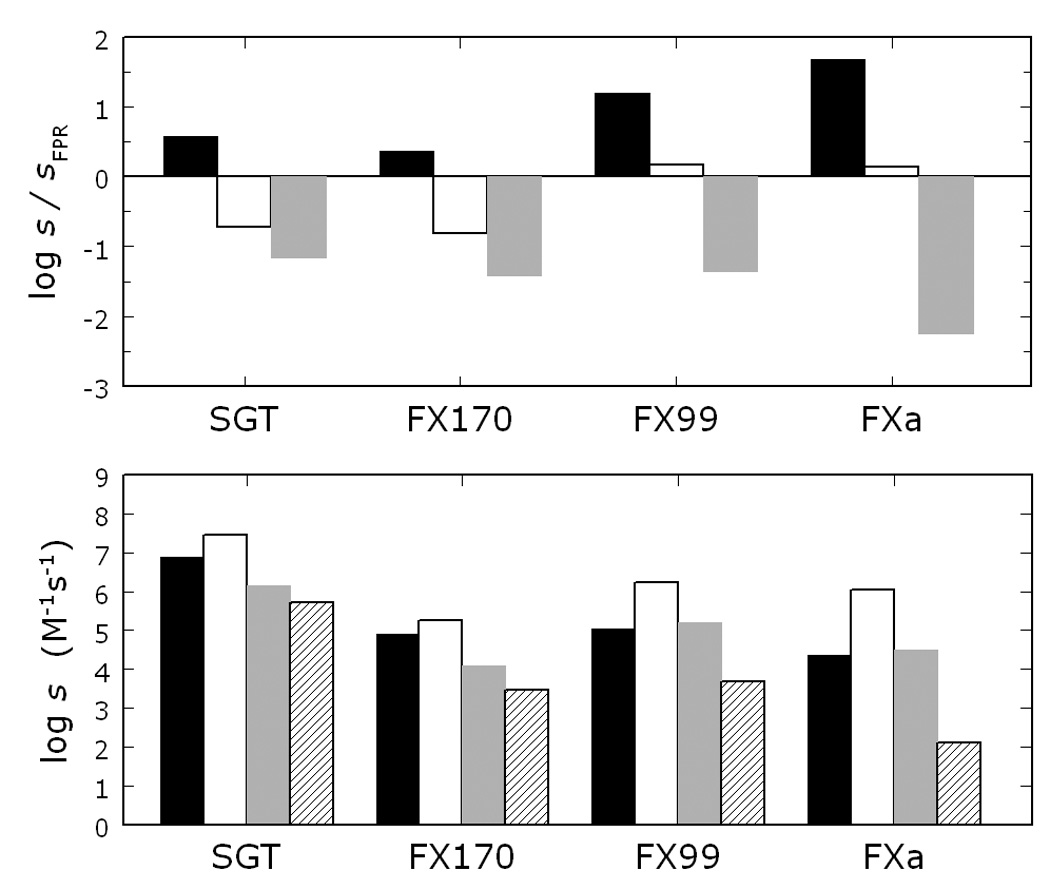
Substrate selectivity profile of SGT and its mutants FX170 and FX99, compared to that of FXa. The bottom panel shows the values of s=kcat/Km for the hydrolysis of FPR (black bar), RPR (white bar), FGR (gray bar) and FPK (hatched bar) under experimental conditions of 25 mM Tris, 200 mM NaCl, 10 mM CaCl2, 0.1 % PEG 8000, pH 8.0 at 25 °C. The top panel illustrates the relative specificity of RPR, FGR and FPK compared to the reference substrate FPR, and directly shows the H-d-Phe vs. H-d-Arg preference at P3 (black bar), the Pro vs. Gly preference at P2 (white bar) and the Arg vs. Lys preference at P1 (gray bar). Note how Na+ activation in FX99 results in a construct as active as FXa, and significantly more active than FX170 (bottom panel). Furthermore, FX99 features a substrate selectivity profile almost identical to that of FXa, as opposed to FX170 that retains the selectivity of wild-type SGT (top panel).
Spatial arrangement of the substituted loops is clearly illustrated by the X-ray crystal structure of FX99 solved at 1.05 Å resolution in the presence of Na+ and the active site inhibitor benzamidine. Although crystallization conditions and cell dimensions are similar to those reported for wild-type and mutants of SGT 17; 25; 26, the structure of FX99 bears two molecules in the asymmetric unit with a space group of P2121 21 that are nearly identical (rmsd 0.221 Å). As expected from its kinetic properties, FX99 structurally resembles FXa in substituted regions (Figure 3A). A notable exception is the conformation of the engineered 99-loop, where relative positions of Glu-97 and Tyr-99 are flipped compared to FXa (Figure 3B). As a result, hydrophobicity of the quaternary amine site in FXa 27 is reduced and may explain why FX99 lacks inhibition by choline or tetramethylammonium salts like FXa (data not shown). However, this structural alteration does not impact the Gly vs. Pro preference at the P2 position of substrate, which is the same for both FX99 and FXa (Figure 2). Architecture of the active site and primary specificity pocket reveals the basis of the high catalytic activity of FX99. Catalytic triad, oxyanion hole, substrate recognition strand involving residues 214–216, and Asp-189 at the base of the S1 pocket are structurally identical to wild-type SGT and FXa.
Figure 3.
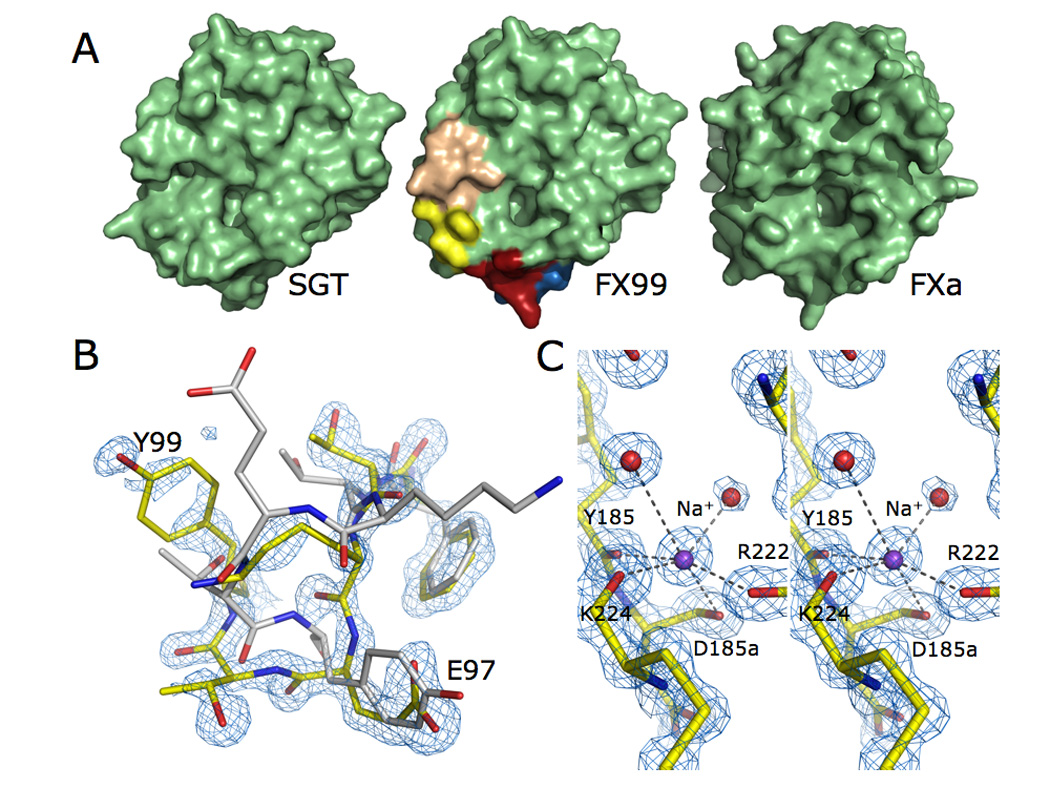
(A) Surface representation the FX99 mutant. Mutagenesis of the 99- (wheat), 170- (yellow), 186- (blue), and 220- (red) loops remodels the active site to more closely resemble that of FXa (shown on the right) than the wild-type scaffold (on left). (B) The 99-loop in the FX99 mutant of SGT (shown in yellow) is flipped relative to FXa (shown in white, PDB ID 2BOK). Weaker electron density for this region suggests flexibility of the loop. However, the polypeptide backbone and majority of side chains can be traced in the electron density (shown is the 2F0-Fc map contoured at 2.0 σ). (C) Excellent quality diffraction data (see also Table 1) resolves the bound Na+ in FX99 and reveals an identical geometry to that observed in FXa. Na+ coordination is mediated by four carbonyl O atoms from Tyr-185, Asp-185a, Arg-222, and Lys-224 and two buried water molecules (present at full occupancy with B-factors of 16.54 Å2 and 30.56 Å2 in molecule A, 19.31 Å2 and 30.56 Å2 in molecule B). The electron density 2F0-Fc map is contoured at 1.75 σ. Na+ ions are present at full occupancy with temperature factors of 17.54 Å2 and 20.47 Å2 in molecules A and B, respectively.
Successful engineering of an allosteric Na+ site is directly demonstrated by the bound cation, which is engaged in octahedral coordination by four carbonyl O atoms from the polypeptide backbone donated by Tyr-185, Asp-185a, Arg-222, and Lys-224, and two buried water molecules (Figure 3C). The coordination shell is identical to that of FXa 28 and confirms the differing contribution of protein backbone to Na+ binding between FXa and thrombin 9; 28;29. Geometry of the Na+ site of FX99 is notable in view of its atomic resolution, which is unmatched in the current structural database of monovalent cation binding sites in proteins 3. The six Na+-O distances in the octahedral coordination shell are close to the ideal value of 2.46 Å 4; 30; 31, which translates into an expected valence of 1.00 at the Na+ peak 31; 32; 33. Values of the valence at the assigned Na+ peaks in the two molecules of the asymmetric unit are 1.01 and 1.02. The carbonyl O atom of Lys-224 is correctly oriented to bind Na+, as expected for proteases carrying Tyr-225 5; 10. The critical change in the coordination shell is brought about by replacement of the entire environment around the Na+ site provided by the 186- and 220- loops. Incidentally, these are the same loops altered in the conversion of the primary selectivity of trypsin to chymotrypsin 34; 35; 36. Orientation of the carbonyl O atom of residue 224 is a critical determinant of Na+ binding in trypsin-like proteases 5. Additional changes outside the coordination shell enable Na+-dependent activation in trypsin. Rotation of the carbonyl O atom of residue 224 induced by Tyr-225 has long-range impact on catalytic activity of the enzyme through a network of buried water molecules connecting the primary specificity site to the 170-loop and the 214–216 β-strand responsible for substrate recognition.
High resolution structures of FXa 28 and FX99 present an identical constellation of buried water molecules, which is brokered by replacement of critical loops in SGT with those of FXa (Figure 4). The network of water molecules overlays the position of Tyr-172 in SGT and other digestive trypsins and illustrates the importance of the side-chain of this residue towards specificity and activity 34; 35; 36. Previous work highlighted the flexibility of the 170-loop when similar active site mutations were introduced into bovine or rat trypsin 37; 38; 39; 40; 41. In particular, the crystal structure of one active site mutant in the presence of benzamidine presented the engineered Phe-174 side-chain buried in the position of the water network. In turn, the intermediate helix at the base of the 170-loop adopts an unnatural conformation and the S4 pocket is not suitable for substrate discrimination 20. The structure of FX99 similarly contains benzamidine, yet the 170-loop and intermediate helix is correctly oriented as observed in FXa. Na+-dependent allostery therefore imparts an organizing influence on the extended substrate binding site, including the intermediate helix, through water mediated interactions. Both structural and kinetic analyses demonstrate that replacement of active site and Na+ site introduce Na+-binding, allosteric activation, high activity and substrate selectivity of FXa in SGT.
Figure 4.
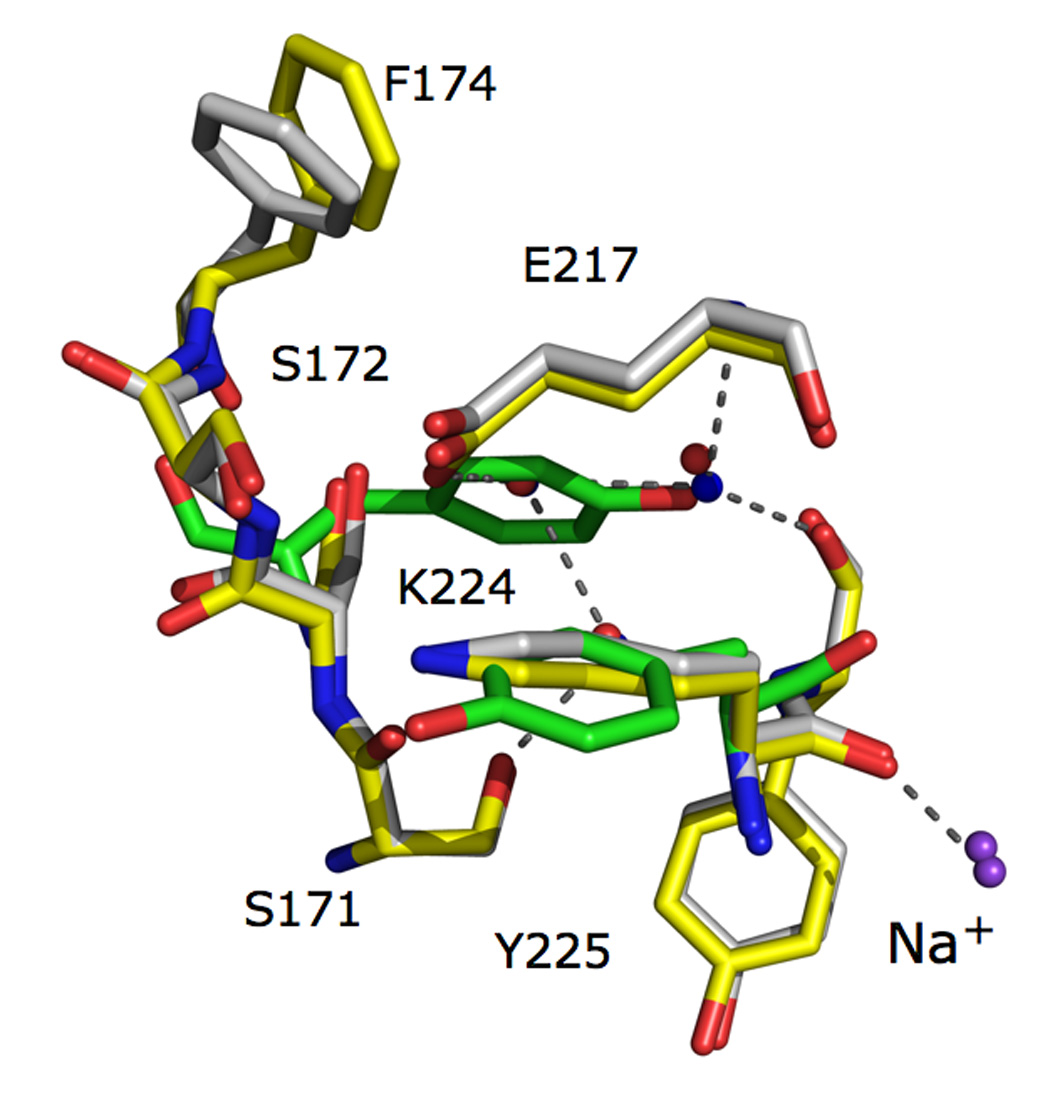
The 170-loop defines the extended substrate binding site in trypsin-like proteases and is identical in the FX99 trypsin mutant (shown in yellow) to that in FXa (shown in white, PDB ID 2BOK). Three buried water molecules (red spheres = FXa, blue spheres = FX99) underneath the substrate recognition ®-strand link the Na+-site to the active site via Glu-217 and the 170-loop through Ser-171 (Na+ shown in purple spheres). Such an H-bonding pattern is not satisfied if the carbonyl O atom of Lys-224 is not displaced downward as in Tyr-224 of wild-type SGT (shown in green). In wild-type SGT and many other digestive trypsins, the side chain of Tyr-172 occupies the identical position to the buried water network to place the OH atom in the exact position of one of the three water molecules.
Previous engineering studies on trypsin-like proteases have shown that active site mutations fail to alter substrate selectivity 37; 38; 39; 40; 41. Results from the FX99 mutant of SGT demonstrate that, when mutations in the 99-loop are combined with those necessary to generate a Na+-activated protease, the resulting construct acquires both Na+ activation and new substrate specificity profile. Linkage therefore exists among different protein domains participating in substrate recognition, allosteric regulation and catalytic activity, and explains the difficulties encountered when engineering new properties in the trypsin scaffold utilizing only “local” substitutions. Recent spectroscopic studies of thrombin demonstrate that Na+ binding influences the structure of the enzyme globally 42. Reciprocity of linkage demands many functions of the protease to be influenced by Na+ binding 43. Not surprisingly, then, engineered Na+ activation in the trypsin scaffold is interwoven with high catalytic activity and also substrate selectivity. A new paradigm is therefore defined whereby monovalent cation dependent activation and allostery are critical ingredients in the general strategy to engineer catalytic properties in enzymes.
Materials & Methods
Recombinant Protein Production, Purification and Characterization
The FX99 mutant was constructed using nineteen replacements in wild-type SGT with residues present in FXa, i.e., N95T/G96K/T97E/+T98/+Y99 in the 99-loop (+ = insertion), A171S/Y172S/G173S/N174F/E175I/E180M in the 170-loop, ΔP185/G186K/G187Q/V188E (Δ = deletion) in the 186-loop and Y217E/P222K/Y224K/P225Y in the 220-loop. This construct corresponds to the Na+-activated mutant of SGT reported previously 17, FX170, with five additional substitutions in the 99-loop. The FX99 construct was expressed as soluble, active form in B. subtilis and secreted directly into the extracellular environment by recombinant bacteria. Yields were 20-fold lower than typically observed with wild-type SGT 25, yet sufficient for purification to homogeneity followed by kinetic and structural analyses. Protein purity was >98% by active site titration using PPACK. Oligopeptide chromogenic peptides used for kinetic analysis were synthesized by Midwest Biotech.
Crystallization
Hanging-drop crystallization was used to prepare diffraction quality crystals of the FX99 mutant of SGT. Protein (1.5 µL of 8 mg/mL in 10 mM calcium acetate, 50 mM NaCl, 10 mM benzamidine, pH 6.0) was mixed with an equal volume of reservoir buffer containing 1.6 M ammonium sulphate. Diffraction quality crystals appeared within three days at 25 °C. Crystals were soaked briefly in artificial mother liquor containing 20% glycerol and flash frozen in liquid nitrogen prior to data collection. X-ray diffraction data were recorded on an ADSC Quantum 315 CCD at Beamline 14-BM-C at BIOCARS (Argonne, IL). Two passes were made at the same wavelength; the first pass was 200° with steps of 1° processed to 1.6 Å resolution, and the second pass was 200° with steps of 0.5° processed to 1.05 Å resolution. Integration and scaling of diffraction data were carried out with HKL-2000 44. The structure of FX99 was solved by molecular replacement using the CCP4 suite 45. Initial positional and isotropic temperature factor refinement was done in REFMAC 46. Alternating cycles of anisotropic temperature factor refinement with SHELXL 47 and model-building with COOT 48 were performed until the final model was produced. Statistics of the final model are summarized in Table 1. Valence of the two Na+ ions was calculated from the distances of nearest bonding partners as described previously 30. The six interaction distances yield valence values of 1.01 and 1.02 for the two coordinated Na+ ions near the expected value of 1.00.
Table 1.
Crystallographic data for FX99 (PDB ID 3BEU). Values for the highest resolution shell are noted in parentheses.
| Data collection: | |
| Wavelength (Å) | 0.9 |
| Space Group | P212121 |
| Unit cell dimension (Å) | a=41.79, b=72.43, c=122.49 |
| α=90.0°, β=90.0°, γ=90.0° | |
| Molecules/asymmetric unit | 2 |
| Resolution range (Å) | 50.0–1.05 |
| Observations | 1509645 |
| Unique observations | 173357 |
| Completeness (%) | 99.6 (94.5) |
| Rsym (%) | 6.6 (38.4) |
| I/σ(I) | 26.8 (3.6) |
| Refinement: | |
| Resolution (Å) | 10.0–1.05 |
| |F|/σ(|F|) | >0 |
| Rcryst, Rfree | 0.119, 0.146 |
| Reflections (working/test) | 164162/8643 |
| Protein atoms | 3345 |
| Solvent molecules | 605 |
| Rmsd bond lengthsa (Å) | 0.016 |
| Rmsd anglesa (°) | 2.4 |
| Rmsd ΔB (Å2) (mm/ms/ss)b | 1.7 / 2.6 / 5.2 |
| <B> protein (Å2) | 14.04 |
| <B> solvent (Å2) | 28.62 |
| Ramachandran plot: | |
| Most favored (%) | 90.6 |
| Generously allowed (%) | 9.4 |
| Disallowed (%) | 0.0 |
Root-mean-squared deviation (Rmsd) from ideal bond lengths and angles and Rmsd in B-factors of bonded atoms.
mm, main chain-main chain; ms, main chain-side chain; ss, side chain-side chain.
Accession Numbers
Atomic coordinates and structure factors have been deposited in the Protein Data Bank (PDB ID 3BEU).
Acknowledgments
This work was supported by a Postdoctoral Fellowship from the American Heart Association (to M.J.P.) and by NIH research grants HL49413, HL58141 and HL73813 (to E.D.C.).
Abbreviations used
- RGR
H-d-Arg-Gly-Arg-p-nitroanilide
- FPR
H-d-Phe-Pro-Arg-p-nitroanilide
- RPR
H-d-Arg-Pro-Arg-p-nitroanilide
- FGR
H-d-Phe-Gly-Arg-p-nitroanilide
- FPK
H-d-Phe-Pro-Lys-p-nitroanilide
- PPACK
H-d-Phe-Pro-Arg-CH2Cl
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hedstrom L. Serine protease mechanism and specificity. Chem Rev. 2002;102:4501–4524. doi: 10.1021/cr000033x. [DOI] [PubMed] [Google Scholar]
- 2.Page MJ, Di Cera E. Serine peptidases: classification, structure and function. Cell Mol Life Sci. 2008 doi: 10.1007/s00018-008-7565-9. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Di Cera E. A structural perspective on enzymes activated by monovalent cations. J Biol Chem. 2006;281:1305–1308. doi: 10.1074/jbc.R500023200. [DOI] [PubMed] [Google Scholar]
- 4.Page MJ, Di Cera E. Role of Na+ and K+ in enzyme function. Physiol Rev. 2006;86:1049–1092. doi: 10.1152/physrev.00008.2006. [DOI] [PubMed] [Google Scholar]
- 5.Dang QD, Di Cera E. Residue 225 determines the Na(+)-induced allosteric regulation of catalytic activity in serine proteases. Proc Natl Acad Sci U S A. 1996;93:10653–10656. doi: 10.1073/pnas.93.20.10653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krem MM, Di Cera E. Molecular markers of serine protease evolution. Embo J. 2001;20:3036–3045. doi: 10.1093/emboj/20.12.3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krem MM, Rose T, Di Cera E. The C-terminal sequence encodes function in serine proteases. J Biol Chem. 1999;274:28063–28066. doi: 10.1074/jbc.274.40.28063. [DOI] [PubMed] [Google Scholar]
- 8.Di Cera E, Guinto ER, Vindigni A, Dang QD, Ayala YM, Wuyi M, Tulinsky A. The Na+ binding site of thrombin. J Biol Chem. 1995;270:22089–22092. doi: 10.1074/jbc.270.38.22089. [DOI] [PubMed] [Google Scholar]
- 9.Pineda AO, Carrell CJ, Bush LA, Prasad S, Caccia S, Chen W, Mathews FS, Di Cera E. Molecular dissection of Na+ binding to thrombin. J Biol Chem. 2004;279:31842–31853. doi: 10.1074/jbc.M401756200. [DOI] [PubMed] [Google Scholar]
- 10.Guinto ER, Caccia S, Rose T, Futterer K, Waksman G, Di Cera E. Unexpected crucial role of residue 225 in serine proteases. Proc Natl Acad Sci U S A. 1999;96:1852–1857. doi: 10.1073/pnas.96.5.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Cera E, Page MJ, Bah A, Bush-Pelc LA, Garvey LC. Thrombin allostery. Phys Chem Chem Phys. 2007;9:1292–1306. doi: 10.1039/b616819a. [DOI] [PubMed] [Google Scholar]
- 12.Prasad S, Cantwell AM, Bush LA, Shih P, Xu H, Di Cera E. Residue Asp-189 controls both substrate binding and the monovalent cation specificity of thrombin. J Biol Chem. 2004;279:10103–10108. doi: 10.1074/jbc.M312614200. [DOI] [PubMed] [Google Scholar]
- 13.He X, Rezaie AR. Identification and characterization of the sodium-binding site of activated protein C. J Biol Chem. 1999;274:4970–4976. doi: 10.1074/jbc.274.8.4970. [DOI] [PubMed] [Google Scholar]
- 14.Petrovan RJ, Ruf W. Role of residue Phe225 in the cofactor-mediated, allosteric regulation of the serine protease coagulation factor VIIa. Biochemistry. 2000;39:14457–14463. doi: 10.1021/bi0009486. [DOI] [PubMed] [Google Scholar]
- 15.Rezaie AR, He X. Sodium binding site of factor Xa: role of sodium in the prothrombinase complex. Biochemistry. 2000;39:1817–1825. doi: 10.1021/bi992006a. [DOI] [PubMed] [Google Scholar]
- 16.Vindigni A, Di Cera E. Role of P225 and the C136–C201 disulfide bond in tissue plasminogen activator. Protein Sci. 1998;7:1728–1737. doi: 10.1002/pro.5560070807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Page MJ, Bleackley MR, Wong S, MacGillivray RTA, Di Cera E. Conversion of trypsin into a Na+ activated enzyme. Biochemistry. 2006;45:2987–2993. doi: 10.1021/bi052481a. [DOI] [PubMed] [Google Scholar]
- 18.Hardy JA, Wells JA. Searching for new allosteric sites in enzymes. Curr Opin Struct Biol. 2004;14:706–715. doi: 10.1016/j.sbi.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 19.Swain JF, Gierasch LM. The changing landscape of protein allostery. Curr Opin Struct Biol. 2006;16:102–108. doi: 10.1016/j.sbi.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 20.Gunasekaran K, Ma B, Nussinov R. Is allostery an intrinsic property of all dynamic proteins? Proteins: Structure, Function and Genetics. 2004;57:433–443. doi: 10.1002/prot.20232. [DOI] [PubMed] [Google Scholar]
- 21.Pellicena P, Kuriyan J. Protein-protein interactions in the allosteric regulation of protein kinases. Curr Opin Struct Biol. 2006;16:702–709. doi: 10.1016/j.sbi.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 22.Gandhi PS, Chen Z, Mathews FS, Di Cera E. Structural identification of the pathway of long-range communication in an allosteric enzyme. Proc Natl Acad Sci USA. 2008;105:1832–1837. doi: 10.1073/pnas.0710894105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Underwood MC, Zhong D, Mathur A, Heyduk T, Bajaj SP. Thermodynamic linkage between the S1 site, the Na+ site, and the Ca2+ site in the protease domain of human coagulation factor xa. Studies on catalytic efficiency and inhibitor binding. J Biol Chem. 2000;275:36876–36884. doi: 10.1074/jbc.M001386200. [DOI] [PubMed] [Google Scholar]
- 24.Vindigni A, Dang QD, Di Cera E. Site-specific dissection of substrate recognition by thrombin. Nat Biotechnol. 1997;15:891–895. doi: 10.1038/nbt0997-891. [DOI] [PubMed] [Google Scholar]
- 25.Page MJ, Wong SL, Hewitt J, Strynadka NC, MacGillivray RT. Engineering the primary substrate specificity of Streptomyces griseus trypsin. Biochemistry. 2003;42:9060–9066. doi: 10.1021/bi0344230. [DOI] [PubMed] [Google Scholar]
- 26.Read RJ, James MN. Refined crystal structure of Streptomyces griseus trypsin at 1.7 A resolution. J Mol Biol. 1988;200:523–551. doi: 10.1016/0022-2836(88)90541-4. [DOI] [PubMed] [Google Scholar]
- 27.Monnaie D, Arosio D, Griffon N, Rose T, Rezaie AR, Di Cera E. Identification of a binding site for quaternary amines in factor Xa. Biochemistry. 2000;39:5349–5354. doi: 10.1021/bi9926781. [DOI] [PubMed] [Google Scholar]
- 28.Scharer K, Morgenthaler M, Paulini R, Obst-Sander U, Banner DW, Schlatter D, Benz J, Stihle M, Diederich F. Quantification of cation-pi interactions in protein-ligand complexes: crystal-structure analysis of Factor Xa bound to a quaternary ammonium ion ligand. Angew Chem Int Ed Engl. 2005;44:4400–4404. doi: 10.1002/anie.200500883. [DOI] [PubMed] [Google Scholar]
- 29.Zhang E, Tulinsky A. The molecular environment of the Na+ binding site of thrombin. Biophys Chem. 1997;63:185–200. doi: 10.1016/s0301-4622(96)02227-2. [DOI] [PubMed] [Google Scholar]
- 30.Harding MM. Metal-ligand geometry relevant to proteins and in proteins: sodium and potassium. Acta Crystallogr D Biol Crystallogr. 2002;58:872–874. doi: 10.1107/s0907444902003712. [DOI] [PubMed] [Google Scholar]
- 31.Brown ID, Wu KK. Empirical parameters for calculating cation-oxygen bond valence. Acta Crystallogr B. 1976;32:1957–1959. [Google Scholar]
- 32.Nayal M, Di Cera E. Predicting Ca(2+)-binding sites in proteins. Proc Natl Acad Sci U S A. 1994;91:817–821. doi: 10.1073/pnas.91.2.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nayal M, Di Cera E. Valence screening of water in protein crystals reveals potential Na+ binding sites. J Mol Biol. 1996;256:228–234. doi: 10.1006/jmbi.1996.0081. [DOI] [PubMed] [Google Scholar]
- 34.Hedstrom L, Perona JJ, Rutter WJ. Converting trypsin to chymotrypsin: residue 172 is a substrate specificity determinant. Biochemistry. 1994;33:8757–8763. doi: 10.1021/bi00195a017. [DOI] [PubMed] [Google Scholar]
- 35.Hedstrom L, Farr-Jones S, Kettner CA, Rutter WJ. Converting trypsin to chymotrypsin: ground-state binding does not determine substrate specificity. Biochemistry. 1994;33:8764–8769. doi: 10.1021/bi00195a018. [DOI] [PubMed] [Google Scholar]
- 36.Hedstrom L, Szilagyi L, Rutter WJ. Converting trypsin to chymotrypsin: the role of surface loops. Science. 1992;255:1249–1253. doi: 10.1126/science.1546324. [DOI] [PubMed] [Google Scholar]
- 37.Rauh D, Klebe G, Stubbs MT. Understanding protein-ligand interactions: the price of protein flexibility. J Mol Biol. 2004;335:1325–1341. doi: 10.1016/j.jmb.2003.11.041. [DOI] [PubMed] [Google Scholar]
- 38.Rauh D, Klebe G, Sturzebecher J, Stubbs MT. ZZ made EZ: influence of inhibitor configuration on enzyme selectivity. J Mol Biol. 2003;330:761–770. doi: 10.1016/s0022-2836(03)00617-x. [DOI] [PubMed] [Google Scholar]
- 39.Rauh D, Reyda S, Klebe G, Stubbs MT. Trypsin mutants for structure-based drug design: expression, refolding and crystallisation. Biol Chem. 2002;383:1309–1314. doi: 10.1515/BC.2002.148. [DOI] [PubMed] [Google Scholar]
- 40.Di Fenza A, Heine A, Koert U, Klebe G. Understanding Binding Selectivity toward Trypsin and Factor Xa: the Role of Aromatic Interactions. ChemMedChem. 2007;2:297–308. doi: 10.1002/cmdc.200600185. [DOI] [PubMed] [Google Scholar]
- 41.Reyda S, Sohn C, Klebe G, Rall K, Ullmann D, Jakubke HD, Stubbs MT. Reconstructing the binding site of factor Xa in trypsin reveals ligand-induced structural plasticity. J Mol Biol. 2003;325:963–977. doi: 10.1016/s0022-2836(02)01337-2. [DOI] [PubMed] [Google Scholar]
- 42.Bah A, Garvey LC, Ge J, Di Cera E. Rapid kinetics of Na+ binding to thrombin. J Biol Chem. 2006;281:40049–40056. doi: 10.1074/jbc.M608600200. [DOI] [PubMed] [Google Scholar]
- 43.Di Cera E. Site-specific analysis of mutational effects in proteins. Adv Protein Chem. 1998;51:59–119. doi: 10.1016/s0065-3233(08)60651-8. [DOI] [PubMed] [Google Scholar]
- 44.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected by oscillation methods. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 45.Bailey S. The CCP4 suite. Programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 46.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 47.Sheldrick G, Schneider T. SHELXL: High-resolution refinement. Methods Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- 48.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]