

Abstract
The use of live cell microscopy has made a number of contributions to the study of apoptosis. Many of the tools and techniques are available that allow us to image the key events that occur during cell death including mitochondrial outer membrane permeabilization, mitochondrial transmembrane potential changes, translocation of Bcl-2 family members, caspase activation, phosphatidylserine flip and plasma membrane rupture. We discuss these techniques here and highlight the advantages and drawbacks of using such approaches to study apoptosis.
Keywords: Apoptosis, mitochondria, time-lapse confocal microscopy, cytochrome c, caspase
1. Introduction
During apoptosis, many pro-death stimuli converge on the mitochondrial pathway. This leads to permeabilization of the outer mitochondrial membrane and the release of proteins that reside in the mitochondrial inter membrane space including cytochrome c, Omi and Smac. Diffusion of cytochrome c to the cytosol results in the activation of caspases, the subsequent cleavage of specific cellular substrates and death of the cell (1). The use of live cell microscopy has led to many advances in the field of apoptosis especially with respect to studying the mitochondrial pathway.
The first descriptions of apoptosis described the morphological changes that consistently occur during this form of cell death – namely cell shrinkage, nuclear condensation, blebbing and the formation of apoptotic bodies as the cell’s membrane collapses (2). More recently, we and others have used live cell confocal and wide-field fluorescence microscopy to assess the dynamic nature of these events (3–5). Through the use of fluorescently labeled proteins and organelle specific dyes we can visualize, in real time, the events that occur during mitochondrial apoptosis including mitochondrial outer membrane permeabilization (MOMP), loss of mitochondrial transmembrane potential, caspase activation, plasma membrane reorganization and deterioration.
2. Single Cell Analysis of the Events that Occur During Apoptosis
2.1. Visualizing cytochrome c release
When stably expressed in cells, cytochrome c fused to green fluorescent protein (cyt c-GFP) has been shown to behave similarly to endogenous cytochrome c (4, 6). It localizes predominantly to the mitochondria and is released when the cells are treated with a pro-apoptotic stimulus to assume a diffuse cytosolic pattern. When these cells were observed using time-lapse confocal microscopy the exact duration of cyt c-GFP release in single cells could be measured and was found to take approximately 5 min irrespective of stimulus and the release was complete and kinetically invariant (3, 4).
Cyt c-GFP can be introduced into a variety of cell lines such as Hela by retroviral transduction. This allows multiple copies of cyt c-GFP to be expressed in each cell, which boosts the signal of GFP facilitating the detection of the protein by wide-field fluorescence or confocal microscopy. Often the cells must be sorted on the GFP intensity to obtain a more homogeneous population or to select clones with the correct mitochondrial localization. The extent of cyt c-GFP release after treatment with a proapoptotic stimulus that engages the mitochondrial pathway, such as actinomycin D or UV, can be measured in these cells by counting the number of cells with a diffuse GFP signal, indicating release, versus those with a punctate or mitochondrial distribution of cytochrome c-GFP. These cells can also be used to monitor the release of cytochrome c during apoptosis in real time using time-lapse microscopy (see Box 1) and the kinetics of the release can be expressed graphically (Figure 2, Box 3).
Box 1: General Protocol for Imaging Cells in Real Time
1. Plate the cells in glass-bottom dishes or multi-well plates (Mattek Corp). Dishes can be pre-coated with collagen, fibronectin or poly-L-lysine to improve the attachment of the cells.
2. Supplement media with 20mM Hepes to buffer the pH or cells can be kept in CO2 for the duration of the experiment (this can be achieved if the microscope has an attached incubator). Use of phenol-red free media at this stage can be included to reduce artifacts due to autofluorescence although it is not essential. Addition of 0.5 mM dithiothreitol (DTT) or 55 µM 2-mercaptoethanol is necessary to prevent unwanted production of ROS that can be toxic to the cells.
3. Overlay the media with mineral oil to prevent evaporation of the media from the dish. This step can be omitted if the microscope has an incubator with a humidified CO2 workhead.
4. Cells should be maintained at constant temperature (usually 37°C) for the duration of the time-lapse experiment by placing them in a temperature controller on the microscope stage or preferably on a microscope with an incubator enclosure. The cells should be allowed to equilibrate to the desired temperature before focusing on the cells. Even small changes in temperature can lead to significant focal drift.
4. Control experiments should be carried out either simultaneously or using the same conditions on untreated cells to ensure that the imaging conditions are not inducing phototoxicity and mitosis can ensue.
Figure 2.
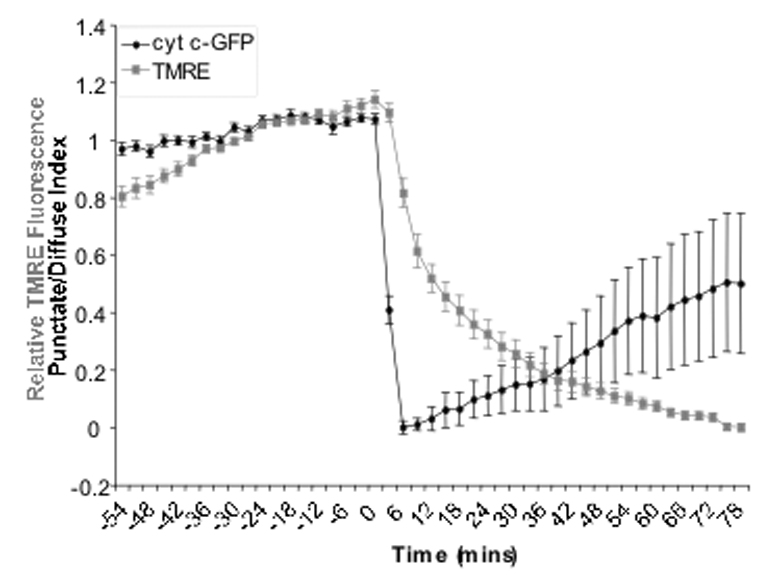
Time-lapse analysis of the relative brightness of TMRE compared to the punctate diffuse index of cytochrome c-GFP. Hela cyt c-GFP cells were exposed to Act D (0.5µM) and the caspase inhibitor qVD-OPH (20µM) for 13 hours. Each point on the graph represents the average punctate/diffuse index or average relative TMRE fluorescence of 32 cells at 3 min intervals. Error bars represent the SEM.
Box 3: Analysis of Confocal Time-Lapse Data
The duration or extent of release of cytochrome c and other intermembrane space proteins from the mitochondria can be expressed graphically by the punctate/diffuse index. The punctate /diffuse index is the standard deviation of the average brightness of all the pixels in an individual cell and can be measured using Metamorph or a similar software program. A high standard deviation value represents a cell with a punctate distribution of GFP, because there are many bright green pixels adjacent to black (non-fluorescent) pixels. Conversely a low standard deviation value represents a cell with diffuse or released cyt c-GFP since the brightness of the pixels in the cell is evenly distributed. The punctate/diffuse index of a cell is measured in each frame of the movie and the release of cyt c-GFP is seen as a sudden drop in the punctate/diffuse index. The duration of release is the time it takes between the maximum point in the graph and the lowest point when release is complete.
The release of cytochrome c from a number of cells can be averaged by correcting the punctate/diffuse index for the maximum and minimum values such that the maximum for each cell is one and the minimum is zero (Figure 2).
Loss of ΔΨm can be similarly measured and displayed graphically (Figure 2). However unlike cytochrome c-GFP, which changes distribution after MOMP, the signal due to TMRE is simply lost. Therefore the cells are measured for changes in the average intensity of the cell, loss of which is representative of the loss of ΔΨm.
Such analyses can give very accurate representations of the changes that occur at a single cell level during apoptosis. However, any non-specific microscopic aberrations such as focal drift or photobleaching can lead to spurious results when calculating the statistics. Such problems must be taken into account and controlled for in each separate experiment.
The size of the GFP moiety may alter the localization or function of a protein, such as cytochrome c, leading to an incorrect interpretation of the behavior of the endogenous protein. In some cases, therefore, it may be desirable to use an alternative method to study, for instance, the possible size restraints of the MOMP-responsible pore. Tsien and colleagues have developed a method to visualize proteins in real time through the addition of a small tag that binds cell-permeable fluorophores (7, 8). This tag of 12 amino acids comprises a core tetracysteine motif that can bind biarsenical dyes, and it is thus named “tetracysteine” or TC tag. Two of these dyes, FlAsH and ReAsH (fluorescein/resorufin arsenical helix binder), are commercially available (Invitrogen) and are green and red emitting, respectively. Cells can stably or transiently express the tagged protein of interest, and then are stained with the dye (Box 2). The main problem of this technique is a high level of background due to the non-specific staining of different areas around the cell. Regardless, this technique has been successfully used to monitor the release of cytochrome c during apoptosis in NCI-H1299 and Hela cells, and Smac, Omi, Adenylate kinase 2 and AIF in Hela cells (3, 9).
Box 2: FlASH/ReASH Staining Protocol
1. Prepare a solution of 10mM 1,2-ethanedithiol (EDT) by dilution in 1ml DMSO. (Note: EDT is toxic so prepare every solution containing EDT in the fume hood. Leave the gloves used for opening the stock in the hood for several minutes before throwing them away). Vortex to dissolve.
2. Add 1ul of this solution to a tube containing 50ul of HBSS.
3. Add 1ul of ReASH or FlASH (2.5mM) to this solution. Mix and incubate for 5–10 min at room temperature
4. Dilute the 50ul in 1ml of culture medium
5. Incubate the cells for 1–2 hrs in this medium
6. Rinse the cells twice with HBSS to eliminate the dye.
7. Prepare a 100 µM solution of BAL (2,3-dimercapto-1-propanol) in HBSS.
8. Add this solution to the cells. BAL is the de-staining reagent needed in order to eliminate background staining. The concentration and the incubation time of this reagent can be varied in order to improve the signal/noise ratio.
9. Incubate the cells at room temperature for 10–15 min
10. Wash the cells carefully with culture medium twice and incubate in regular medium before changing to the appropriate medium for imaging. Do not add 2-mercaptoethanol or DTT to the imaging medium as they can reduce the dye-tetracysteine complexes.
2.2. Loss of Mitochondrial Transmembrane Potential
Apart from its role in apoptosis, cytochrome c also plays an integral role in the electron transport chain that produces ATP and provides energy to the cell. The electron transport chain is comprised of a number of protein complexes (I–V) that are localized to the inner mitochondrial membrane. As electrons are transported from one complex to the next protons are produced and pumped from the mitochondrial matrix to the intermitochondrial space. This creates an electrochemical potential across the membrane known as the mitochondrial transmembrane potential (ΔΨm). Upon MOMP and cytochrome c release the ΔΨm is lost (10).
This loss of ΔΨm can by measured in single cells by measuring the loss of TMRE or TMRM (Tetramethyl rhodamine ethyl-or methyl-ester) over time. These fluorescent dyes bind to mitochondria with high ΔΨm and the dye is released from the mitochondria when ΔΨm dissipates.
Loss of TMRE has been observed to occur at the same time as the release of cytochrome c-GFP and is often used as a good approximation of the occurrence of MOMP (Figure 1). However, careful observation of TMRE loss versus cytochrome c release in cells undergoing apoptosis has demonstrated that the two events are not simultaneous (11). Loss of ΔΨm actually occurs slightly later than release of cytochrome c. Furthermore it is possible to observe loss of ΔΨm without cytochrome c release. The electron transport chain uncouplers FCCP or CCP immediately induce loss of TMRE when added to cells but do not cause cytochrome c release. Also when caspases are inhibited ΔΨm is decreased but is restored over time, while cytochrome c release is normal (11).
Figure 1.
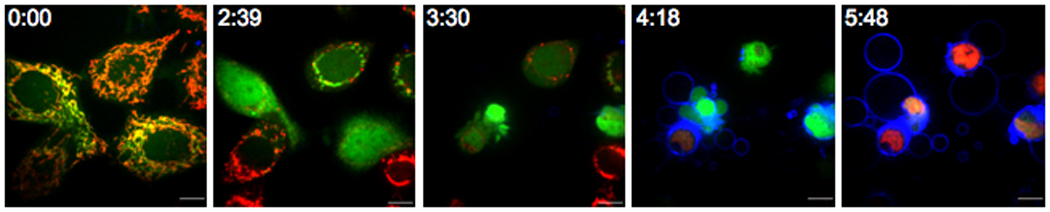
Time-lapse analysis of the morphological changes that occur during apoptosis. Hela cyt c-GFP cells were stained with TMRE (50nM, red), Annexin V-APC (1% (v/v), blue) and PI (0.4µg/ml, pink) and treated with actinomycin D (1µM). The time after exposure to actinomycin D is at the top left of each panel. Scale bars (10µm) are at the bottom right of each panel.
There are other dyes available that also measure ΔΨm each with their own advantages and drawbacks (12). Rhodamine 123 is similar to TMRE but quenches at high concentrations and is also quite toxic to most cells. CMXROS has the advantage of being fixable. JC1 is a dual color dye that is green when it is cytosolic but shifts to red fluorescence when it binds to the mitochondria. These dyes do not rapidly efflux, however, when ΔΨm dissipates and therefore are less useful for single cell imaging.
2.3. Translocation of Bcl-2 family proteins
MOMP is primarily regulated by the Bcl-2 family of proteins. Some of these proteins are constitutively located on mitochondria or other intracellular membranes. Others, however, translocate to mitochondria during apoptosis. A GFP-Bax fusion protein has been used to study Bax translocation in real time, and these studies show that Bax markedly changes its intracellular localization at a time coincident with mitochondrial outer membrane permeabilization (13, 14).
In many cell types, caspase-8 cleaves Bid, and this is a requisite for mitochondrial permeabilization in response to death ligands. Translocation of the cleaved fragment to the mitochondria has been followed by expression of a GFP-fusion of full length Bid (15). Bid cleavage can also be detected in real time. Onuki et al. generated a YFP-Bid-CFP molecule that can be used for fluorescence resonance energy transfer (FRET, see below) analysis as well as for translocation studies (16). Using this approach, Ward et al. found that, Bid cleavage, Bid translocation and mitochondrial depolarization occur co-ordinately during death receptor–induced apoptosis (17).
2.4. Caspase Activation
The key event during apoptosis that is common to all pathways is the activation of caspases. Unfortunately this event has proven to be the most difficult to visualize in single cells. The use of FRET probes for caspase activation show some promise for this type of analysis. These probes are composed of a pair of GFP variants (the most popular being the CFP/YFP pair) linked by a caspase cleavage site (usually DEVD). During FRET the donor molecule (CFP) can directly transfer its excited state energy to the acceptor molecule (YFP). In the absence of caspase-mediated cleavage, the linked donor/acceptor pair remains in close proximity and they undergo FRET. When these two proteins are dissociated due to caspase-mediated cleavage, they are no longer able to undergo FRET. Such a probe has been used to determine the kinetics of caspase activation at the single cell level as a rapid process reaching completion in a matter of minutes (18). It has also been demonstrated that, through the use of two different FRET pairs, it is possible to measure simultaneously caspase-mediated cleavage of two different substrates (19, 20). However due to a considerable overlap in the substrate specificities of each caspase it is difficult to interpret from such experiments which specific caspases have been activated.
Another drawback of these FRET probes for caspase activation is the poor sensitivity of this technique mainly due to the small dynamic range of the donor-acceptor pair. An optimized CFP-YFP pair called CyPet-YPet has been developed that shows greatly enhanced sensitivity for the detection of caspase activation by flow cytometry and may similarly yield superior results in confocal microscopy (21).
These sensitized-emission FRET analyses are also limited because spectrally distinct fluorescent proteins must be used as the acceptor-donor pair, which often impedes the analysis of multiple events simultaneously. Crosstalk due to direct acceptor excitation as well as filter bleed-through requires the capture of numerous reference images. The detectable decrease in donor or increase in acceptor signal is usually not significant therefore ratiometric measurements are required. Fluorescence lifetime imaging microscopy (FLIM) is a technique that, although requiring additional instrumentation, enables FRET efficiency determination solely by measuring the change in lifetime of the donor molecule, irrespective of concentration and signal strength, thereby limiting these confounding variables in FRET analysis.
2.5. Phosphatidylserine Flip
Some of the consequences of caspase activation during apoptosis are the changes that occur in the plasma membrane – namely the redistribution of the lipid phosphatidylserine (PS) from the inner leaflet of the membrane to the outer, which serves as a trigger for recognition of apoptotic cells by phagocytes. The protein Annexin V has a high affinity for phosphatidylserine in the presence of Ca2+ ions and thus serves as a reliable probe for this caspase-dependent event (22). Annexin V is commercially available conjugated to a variety of fluorophores of different wavelengths. To visualize Annexin V binding to cells undergoing apoptosis by microscopy it is simply added to the media of the cells in the presence of 2.5mM CaCl2 and imaged using the appropriate wavelength (Figure 1).
A disadvantage of using available fluorescently conjugated versions of Annexin V is that many of them photobleach upon laser or mercury lamp excitation. This means that in time-lapse experiments a limited number of images can be taken. Since the process of apoptosis in a population occurs over an extended time period the images must be spaced out by approximately 5 mins. However the individual events that occur once a cell has committed to apoptosis, including PS externalization, occur quite rapidly and it would be more informative to be able to reduce the delay between images. A recent paper attempted to overcome this problem by conjugating Annexin V to quantum dots (Qdots). Qdots are fluorescent nanoparticles that are very bright and quite resistant to photobleaching (23). Qdot-AnnexinV was vastly superior to organic dyes with respect to photobleaching issues, thus enabling more frequent and higher resolution images of the cells to be taken during live cell imaging.
2.6. Loss of Plasma Membrane Integrity
Following PS flip, apoptotic cells in culture lose their plasma membrane integrity. This can be measured by the addition of cell impermeable nuclear dyes to the cell culture. The most common of these is propidium iodide (PI). Upon permeabilization of the plasma membrane, the cell becomes permeable to PI resulting in a nuclear localized fluorescent stain. Due to the nuclear condensation that occurs during this stage of apoptosis the apoptotic morphology of the nucleus will be evident upon staining (4). PI is most informatively used alongside Annexin V because PI also stains cells that have undergone necrosis so use of PI alone during live cell microscopy will not distinguish between the two types of cell death. During apoptosis Annexin V binding always precedes PI uptake but during necrosis the plasma membrane ruptures and PI is taken up in the absence of, or prior to Annexin V binding in real time (Figure 1).
The relatively broad excitation and emission spectrum of PI means that it is often detected in multiple channels of a confocal or wide-field fluorescence microscope. For this reason, it is not always the most suitable dye to use when simultaneously analyzing a number of fluorophores. An alternative to PI is TO-PRO-3, which emits in the far-red, allowing the rest of the spectrum to be utilized for other probes. However, TO-PRO-3 is very susceptible to photobleaching. YO-PRO-1 is a cell-impermeant dye that emits in the green spectra. When used at low concentrations, cells become slightly permeable to the dye after mitochondrial permeabilization and PS exposure. A few minutes or hours later, there is a sudden and massive incorporation of YO-PRO-1 in the DNA probably corresponding to loss of plasma membrane integrity, which happens several minutes or even hours after the initial incorporation of the probe (24). Thus the use of YO-PRO-1 is a useful method of distinguishing between apoptotic and necrotic cells.
2.7 Other applications
There are other events such as mitochondrial fission and fusion, mitochondrial permeability transition (mPT), reactive oxygen species generation and calcium flux that while not always necessary for apoptosis to occur are often associated with apoptotic pathways. Similar to the hallmark apoptotic events described above, these associated events can also be monitored by time-lapse confocal microscopy.
Mitochondrial fission occurs during apoptosis around the time of cytochrome c release. One straight-forward method to observe fusion and fission is the transfection of cells with matrix-targeted fluorescent proteins such as mito-YFP or mito-DsRed (Clontech, (25)). A more quantifiable method is the use of a matrix-targeted photoactivable GFP (PAGFP) (14). After excitation of a region of interest with 488-nm light, the green signal (originally present in the excited mitochondria) will slowly distribute over the mitochondrial network if mitochondria are undergoing fusion.
mPT involves the opening of a large pore in the mitochondria leading to mitochondrial swelling and rupture of the outer mitochondrial membrane. Although it can be associated with apoptosis, mPT is considered to be a later event that is more likely to induce necrosis. mPT can be measured using calcein-acetoxmethyl ester (calcein-AM) that is taken up into all cellular compartments upon incubation. Subsequent incubation with CoCl2 quenches the extramitochondrial calcein. Upon induction of mPT calcein leaks out of the mitochondria and can be measured over time as a loss of fluorescence (26).
There are a number of dyes and proteins available to measure calcium flux in the cell and also the generation of reactive oxygen species (ROS), two events that are associated with apoptosis. Ratiometric dyes, such as fura 2, and various protein-based calcium indicators can be used to measure intracellular calcium (27), while hydroethidine has been successfully used to measure ROS generation by confocal microscopy (28).
3. General Considerations for Time-Lapse Experimental Design
When preparing any of the aforementioned techniques for time-lapse microscopy, it is critical to optimize the image acquisition parameters to gain the highest quality data without damaging the cells. Unfortunately, time-lapse fluorescence microscopy has strong potential to perturb biological systems through the introduction of phototoxicity. Some critical elements in the optical system to consider include: bright and photostable fluorophores, cells grown on No. 1.5 coverslips, appropriate filter sets that match the fluorophores, high numerical aperture (NA) objective lenses with the lowest magnification needed, excitation light that can be adequately attenuated when imaging and turned off when not, and highly efficient detectors of fluorescence emission light (for a good review see (29)).
Careful consideration of each one of these elements and of the efficient movement of photons through the imaging system will pay dividends in cell health and data quality. However, one must bear in mind that, even when the imaging parameters are optimized for minimal phototoxic effects, the collected data will generally be of much lower signal-to-noise than data collected from fixed samples or at fixed time-points. It is further recommended to run control experiments where the cells are only imaged at the start and end of the experiment to fully control for phototoxic effects.
The addition of a precision motorized XY stage with one’s chosen imaging system enables the repeated study several fields of view over a given time-course. This allows untreated controls to be imaged alongside treated samples in a multi-well chamber slide. Thus each sample type is imaged not only at high optical resolution but also with a large sample size for a given experiment, yielding high-powered statistical analysis.
4. Advantages of live cell microscopy over conventional methods of studying apoptosis
Real-time single-cell analysis is the best method to study kinetic events. Other methods can lead to erroneous conclusions due to two different causes: the asynchronicity of events occurring in individual cells, and the possibility of occurrence of different forms of death in the same population. Indeed, many inducers of apoptosis can trigger both necrosis and apoptosis depending on the dose. It is also possible that an inducer can engage different pathways of apoptosis in individual cells: for example, UV or nucleotide analogs may induce death receptor-dependent or –independent apoptosis. Single cell analysis is an invaluable tool to avoid making incorrect interpretations of events that occur in mixed populations.
An obvious advantage of using microscopy instead of flow cytometric analysis is the possibility of discriminating between whole-cell and organelle-specific properties, such as pH or transmembrane potential. Many conclusions about the causes and consequences of ΔΨm loss have been drawn from studies where this potential was measured by flow cytometry with dyes such as rhodamine 123, that have inconsistent specificity, or with dyes such as DiOC6(3) that are also sensitive to plasma-membrane changes, or can stain other organelles such as Golgi and endoplasmic reticulum (30). This may explain some conflicting results regarding the caspase-dependency of m loss during apoptosis. Performing single-cell analysis, Dussmann et al. determined that loss of ΔΨm after treatment with staurosporine was caspase-independent (measured by TMRE), while caspases were responsible for plasma-membrane depolarization (measured by Dibac4(3) (31)
5. Disadvantages of live cell microscopy
One handicap of the real-time, microscope-based techniques is that, in principle, we can only follow cells that grow attached to the substrate (i.e. the glass coverslip) and do not move out of the focal plane or the field. In some cases, non-adherent cells can attach to dishes covered with poly-L-lysine, collagen or fibronectin. However, many non-adherent cell lines cannot attach even to coated dishes. Goldstein et al studied cytochrome c release in Jurkat and FL5.12 lines by ‘trapping’ the cells in an agar matrix (3). This matrix is made by mixing the cells with warm low melting point agar and 2x culture medium. Cells become immobilized in a field of view but keep rotating freely. Another tool that has been used to study apoptosis, in this case in U937 cells, is the trapping of cells in a microfluidic device. A chip made of polydimethyl-siloxane (PDMS) is attached to a standard glass-bottom culture dish, and cells are injected into the channels of this chip. The shallow channels (25 µm) restrict the height of the area of interest for the microscope, thereby assuring a continuous focusing of the cells, while still allowing some freedom of movement for suspension cells (24).
Most cells detach in late stages of apoptosis and will thus leave the focal plane during time-lapse microscopy. The use of caspase inhibitors in the experiment usually prevents detachment, allowing the study of events, such as MOMP that precede caspase activation. However these compounds, obviously, preclude the study of downstream caspase-dependent features of apoptosis. For this reason, many researchers have assessed post-mitochondrial events in response to staurosporine, which does not trigger detachment of the cells. Better methods are needed to study the execution phase of apoptosis in real time.
It is also possible to apply a Z-stack to follow the upward movement of cells as they detach. However this adds the disadvantage of having to take more images per time point and exposing the cells to increased phototoxicity. One way of overcoming this is by increasing the interval between frames thus taking less images and decreasing the cumulative exposure of the cells but again this decreases the amount of information that can be obtained from each experiment.
6. Perspectives
Confocal and wide-field fluorescence time-lapse microscopy methods, such as those described here, have led to great advances in the understanding of the events that occur during apoptosis. The ability to study single cells in real time offers numerous advantages over many of the conventional techniques that we use to study apoptosis however we need to continue improving the tools we have (such as those listed here) while developing superior methods to investigate the kinetics of the key events that occur. One of the major hindrances of these techniques it that it is often difficult to study multiple events simultaneously due to the overlapping spectra of fluorescent proteins or fluorescent dyes and the low brightness of some. The recent development of superior mutants of GFP such as the CFP derivative Cerulean or the red fluorescent mCherry and tdTomato may help to overcome many of these problems (32, 33). The use of brighter, less toxic and more photostable probes will allow us to image cells during apoptosis more often and with less delay between images allowing us to interpret apoptotic events in an increasingly accurate fashion.
Table 1.
Concentrations of Standard Reagents
| Reagent | Concentration | Incubation Time at 37°C | Excitation/Emmision Max. |
|---|---|---|---|
| Mitochondrial membrane potential | |||
| TMRE | 50–100nM | 20 min | 550/573 |
| CMXROS | 20–500nM | 20 min | 579/599 |
| JC1 | 5–10ug/ml | 20 min | Green 510/527 Red aggregate 485–585/590 |
| DiOC6(3) | 40nM | 20 min | 484/500 |
| Rhodamine 123 | 2–5uM | 20 min | 507/529 |
| Dibac4(3) | 1µM | 1h | 493/516 |
| Plasma membrane changes | |||
| Annexin V | 0.5–1%(v/v) + 2.5mM CaCl2 | 10 min | As per conjugated fluor |
| PI | 0.4ug/ml | 5 min | 535/617 |
| YO-PRO-1 | 500nM | 10 min | 491/509 |
| TO-PRO-3 | 250nM | 10 min | 642/661 |
| mPT | |||
| Calcein-AM | 1uM | 10min | 488/520 |
| CoCl2 | 1nM | 1h | Quenches Calcein-AM |
Appendices
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bouchier-Hayes L, Lartigue L, Newmeyer DD. J Clin Invest. 2005;115:2640–2647. doi: 10.1172/JCI26274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kerr JF, Wyllie AH, Currie AR. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldstein JC, Munoz-Pinedo C, Ricci JE, Adams SR, Kelekar A, Schuler M, Tsien RY, Green DR. Cell Death Differ. 2005;12:453–462. doi: 10.1038/sj.cdd.4401596. [DOI] [PubMed] [Google Scholar]
- 4.Goldstein JC, Waterhouse NJ, Juin P, Evan GI, Green DR. Nat Cell Biol. 2000;2:156–162. doi: 10.1038/35004029. [DOI] [PubMed] [Google Scholar]
- 5.Rehm M, Dussmann H, Prehn JH. J Cell Biol. 2003;162:1031–1043. doi: 10.1083/jcb.200303123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heiskanen KM, Bhat MB, Wang HW, Ma J, Nieminen AL. J Biol Chem. 1999;274:5654–5658. doi: 10.1074/jbc.274.9.5654. [DOI] [PubMed] [Google Scholar]
- 7.Griffin BA, Adams SR, Tsien RY. Science. 1998;281:269–272. doi: 10.1126/science.281.5374.269. [DOI] [PubMed] [Google Scholar]
- 8.Martin BR, Giepmans BNG, Adams SR, Tsien RY. Nat Biotech. 2005;23:1308. doi: 10.1038/nbt1136. [DOI] [PubMed] [Google Scholar]
- 9.Munoz-Pinedo C, Guio-Carrion A, Goldstein JC, Fitzgerald P, Newmeyer DD, Green DR. Proc Natl Acad Sci U S A. 2006;103:11573–11578. doi: 10.1073/pnas.0603007103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ricci JE, Waterhouse N, Green DR. Cell Death Differ. 2003;10:488–492. doi: 10.1038/sj.cdd.4401225. [DOI] [PubMed] [Google Scholar]
- 11.Waterhouse NJ, Goldstein JC, von Ahsen O, Schuler M, Newmeyer DD, Green DR. J Cell Biol. 2001;153:319–328. doi: 10.1083/jcb.153.2.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galluzzi L, Zamzami N, de La Motte Rouge T, Lemaire C, Brenner C, Kroemer G. Apoptosis. 2007;12:803–813. doi: 10.1007/s10495-007-0720-1. [DOI] [PubMed] [Google Scholar]
- 13.Wolter KG, Hsu Y-T, Smith CL, Nechushtan A, Xi X-G, Youle RJ. J. Cell Biol. 1997;139:1281–1292. doi: 10.1083/jcb.139.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karbowski M, Arnoult D, Chen H, Chan DC, Smith CL, Youle RJ. J Cell Biol. 2004;164:493–499. doi: 10.1083/jcb.200309082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zha J, Weiler S, Oh KJ, Wei MC, Korsmeyer SJ. Science. 2000;290:1761–1765. doi: 10.1126/science.290.5497.1761. [DOI] [PubMed] [Google Scholar]
- 16.Onuki R, Nagasaki A, Kawasaki H, Baba T, Uyeda TQP, Taira K. PNAS. 2002;99:14716–14721. doi: 10.1073/pnas.232177599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ward MW, Rehm M, Duessmann H, Kacmar S, Concannon CG, Prehn JH. J Biol Chem. 2006;281:5837–5844. doi: 10.1074/jbc.M511562200. [DOI] [PubMed] [Google Scholar]
- 18.Rehm M, Dussmann H, Janicke RU, Tavare JM, Kogel D, Prehn JH. J Biol Chem. 2002;277:24506–24514. doi: 10.1074/jbc.M110789200. [DOI] [PubMed] [Google Scholar]
- 19.Kawai H, Suzuki T, Kobayashi T, Mizuguchi H, Hayakawa T, Kawanishi T. Biochim Biophys Acta. 2004;1693:101–110. doi: 10.1016/j.bbamcr.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 20.Kawai H, Suzuki T, Kobayashi T, Sakurai H, Ohata H, Honda K, Momose K, Namekata I, Tanaka H, Shigenobu K, Nakamura R, Hayakawa T, Kawanishi T. J Pharmacol Sci. 2005;97:361–368. doi: 10.1254/jphs.fp0040592. [DOI] [PubMed] [Google Scholar]
- 21.Nguyen AW, Daugherty PS. Nat Biotechnol. 2005;23:355–360. doi: 10.1038/nbt1066. [DOI] [PubMed] [Google Scholar]
- 22.Martin SJ, Reutelingsperger CP, McGahon AJ, Rader JA, van Schie RC, LaFace DM, Green DR. J Exp Med. 1995;182:1545–1556. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Le Gac S, Vermes I, van den Berg A. Nano Lett. 2006;6:1863–1869. doi: 10.1021/nl060694v. [DOI] [PubMed] [Google Scholar]
- 24.Munoz-Pinedo C, Green DR, van den Berg A. Lab Chip. 2005;5:628–633. doi: 10.1039/b503770k. [DOI] [PubMed] [Google Scholar]
- 25.Karbowski M, Lee YJ, Gaume B, Jeong SY, Frank S, Nechushtan A, Santel A, Fuller M, Smith CL, Youle RJ. J Cell Biol. 2002;159:931–938. doi: 10.1083/jcb.200209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petronilli V, Miotto G, Canton M, Brini M, Colonna R, Bernardi P, Di Lisa F. Biophys J. 1999;76:725–734. doi: 10.1016/S0006-3495(99)77239-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rudolf R, Mongillo M, Rizzuto R, Pozzan T. Nat Rev Mol Cell Biol. 2003;4:579–586. doi: 10.1038/nrm1153. [DOI] [PubMed] [Google Scholar]
- 28.Dussmann H, Kogel D, Rehm M, Prehn JH. J Biol Chem. 2003;278:12645–12649. doi: 10.1074/jbc.M210826200. [DOI] [PubMed] [Google Scholar]
- 29.Waters JC. Methods Cell Biol. 2007;81:115–140. doi: 10.1016/S0091-679X(06)81007-1. [DOI] [PubMed] [Google Scholar]
- 30.Salvioli S, Ardizzoni A, Franceschi C, Cossarizza A. FEBS Letters. 1997;411:77. doi: 10.1016/s0014-5793(97)00669-8. [DOI] [PubMed] [Google Scholar]
- 31.Dussmann H, Rehm M, Kogel D, Prehn JH. J Cell Sci. 2003;116:525–536. doi: 10.1242/jcs.00236. [DOI] [PubMed] [Google Scholar]
- 32.Rizzo MA, Springer GH, Granada B, Piston DW. Nat Biotechnol. 2004;22:445–449. doi: 10.1038/nbt945. [DOI] [PubMed] [Google Scholar]
- 33.Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Nat Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]