

Abstract
Streptococcus pyogenes is one of the most frequent human pathogens. Recent studies have identified dendritic cells (DCs) as important contributors to host defense against S. pyogenes. The objective of this study was to identify the receptors involved in immune recognition of S. pyogenes by DCs. To determine whether Toll-like receptors (TLRs) were involved in DC sensing of S. pyogenes, we evaluated the response of bone marrow-derived DCs obtained from mice deficient in MyD88, an adapter molecule used by almost all TLRs, following S. pyogenes stimulation. Despite the fact that MyD88−/− DCs did not differ from wild-type DCs in the ability to internalize and kill S. pyogenes, the up-regulation of maturation markers, such as CD40, CD80, and CD86, and the production of inflammatory cytokines, such as interleukin-12 (IL-12), IL-6, and tumor necrosis factor alpha, were dramatically impaired in S. pyogenes-stimulated MyD88−/− DCs. These results suggest that signaling through TLRs is the principal pathway by which DCs sense S. pyogenes and become activated. Surprisingly, DCs deficient in signaling through each of the TLRs reported as potential receptors for gram-positive cell components, such as TLR1, TLR2, TLR4, TLR9, and TLR2/6, were not impaired in the secretion of proinflammatory cytokines and the up-regulation of costimulatory molecules after S. pyogenes stimulation. In conclusion, our results exclude a major involvement of a single TLR or the heterodimer TLR2/6 in S. pyogenes sensing by DCs and argue for a multimodal recognition in which a combination of several different TLR-mediated signals is essential for a rapid and effective response to the pathogen.
Streptococcus pyogenes (group A streptococcus) is an important human pathogen that causes a wide spectrum of diseases, ranging from mild infections, such as pharyngitis and superficial skin infections, to severe invasive diseases, such as necrotizing fasciitis and sepsis (23). Invasive streptococcal infection is a significant cause of morbidity and mortality (27). Protection from primary streptococcal infection results from innate rather than adaptive immune responses (11, 24), and we have recently shown that dendritic cells (DCs) play an important role in host defense against S. pyogenes (22).
DCs are important components of the innate immune system that sense microbial products and initiate immune responses against pathogens (33). Upon recognition of microbes, DCs undergo a maturation process characterized by the production of inflammatory cytokines and chemokines as well as by the up-regulation of costimulatory molecules (3). In vitro studies examining the interaction of S. pyogenes with DCs have clearly demonstrated the rapid induction of maturation and activation of human (41, 42) as well as murine (22) DCs after exposure to this pathogen. DCs are the main in vivo source of interleukin-12 (IL-12) during infection, and this response is critical for the development of protective immune responses against infection with S. pyogenes (22). Despite the important contribution of DCs to the initiation of innate immune responses against S. pyogenes, nothing is known about the mechanism of immune recognition of S. pyogenes by these important antigen-presenting cells.
Recognition of microbes by DCs is mediated by pattern recognition receptors, including the Toll-like receptor (TLR) family (19, 20, 34). TLRs recognize the presence of microbial pathogens via the detection of conserved microbial structures called pathogen-associated molecular patterns (1, 2, 5, 36, 37). For example, TLR4 has been reported to function as a receptor for lipopolysaccharide (LPS), an integral component of the outer membranes of gram-negative bacteria (31, 38). TLR2 is reported to be specialized to recognize lipoprotein and lipoteichoic acid (LTA) from diverse species of bacteria (26, 35, 43). TLR6 in combination with TLR2 recognizes zymosan and peptidoglycan (29, 39). TLR9 and TLR5 have been shown to recognize bacterially derived CpG DNA (14) and flagellin (12), respectively. The interaction of TLRs with their target motifs initiates a cascade of intracellular signaling events that culminate in the activation of NF-κB and the production of proinflammatory cytokines, chemokines, and costimulatory molecules that are important for host defenses (1, 4, 36). Upon recognition of microbial components, these TLRs associate with several adaptor molecules, such as myeloid differentiation factor 88 (MyD88), which are critical for downstream signaling events (25, 28).
Based on previous studies reporting that several constituents of S. pyogenes can be recognized by the TLR family of receptors (18, 29), we hypothesized that this receptor family might be involved in the recognition of the whole microorganism. To investigate this, we evaluated the responses of DCs isolated from animals that lack the common adapter molecule MyD88, which is required for almost all TLR activation cascades, to stimulation with S. pyogenes. Our results show that the MyD88 signaling pathway is crucial for S. pyogenes induction of maturation and secretion of proinflammatory cytokines by DCs, suggesting the participation of TLRs in bacterial sensing. However, using DCs from mice deficient in the expression of TLR1, TLR2, TLR4, or TLR2/6 or DCs pretreated with an inhibitor of TLR9, we have excluded a major involvement of each of these specific TLRs in DC sensing of S. pyogenes. Taken together, these observations probably reflect the involvement of multiple TLR pathways, triggered by different bacterial components, in the activation of DCs by S. pyogenes. This multimodal recognition may be essential for a rapid and effective response to the invading pathogen.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The S. pyogenes strain KTL3 (M1 type), initially isolated from the blood of a patient with streptococcal bacteremia, has been described previously (32). The S. pyogenes strain 90-226, a serotype M1 strain cultured from the blood of a patient with sepsis, and the M protein-deficient emm1::Km isogenic mutant, constructed by insertional inactivation of emm1 with aphA-3 (8), were kindly provided by P. Cleary (University of Minnesota Medical School, Minneapolis, MN). Stock cultures were maintained at −70°C and were cultured at 37°C in Todd-Hewitt broth (Oxoid, Basingstoke, United Kingdom) supplemented with 1% yeast extract. Bacteria were collected in mid-log phase, washed twice with sterile phate-buffered saline (PBS), and diluted to the required inoculum, and the number of viable bacteria was determined by counting CFU after dilution and plating of cells on blood agar plates (Gibco, Paisley, United Kingdom) containing 5% sheep blood.
Mice.
C57BL/6 and TLR2−/− mice were obtained from The Jackson Laboratory (Bar Harbor, ME). MyD88−/− and TLR4−/− mice were obtained from the Max Planck Institute for Infection Biology (Berlin, Germany). TLR1−/− and TLR2/6−/− mice were originally from the Centre National de la Recherche Scientifique (Paris, France) and were kindly provided by T. Ebensen (Helmholtz Center for Infection Research [HZI]). These mice were housed in a pathogen-free animal facility at the HZI. Mice were maintained under standard conditions and according to institutional guidelines. All experiments were approved by the appropriate national ethical board (Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit, Oldenburg, Germany).
Isolation and culture of bone marrow-derived DCs.
For the generation of bone marrow-derived DCs, bone marrow was removed from the tibias and femurs of 5- to 10-week-old mice. Following red blood cell lysis and washing, progenitor cells (106/ml) were resuspended and plated in RPMI 1640 containing 10 ng/ml of recombinant mouse granulocyte-macrophage colony-stimulating factor (Sigma, St. Louis, MO) and 10 ng/ml of IL-4 (R&D Systems, Minneapolis, MN). DCs were cultured for 6 days at 37°C in 5% CO2 and were gently washed and fed with fresh medium and cytokines on days 2 and 4. On day 6, the DCs were used for infection experiments.
Experimental infection of DCs.
DCs were infected with the corresponding S. pyogenes strain at a multiplicity of infection (MOI) of 10 bacteria per DC and incubated for 2 h in antibiotic-free medium. DCs were washed twice with PBS to remove unbound bacteria, and pure fractions of DCs were obtained by labeling cells with anti-CD11c MACS beads (Miltenyi Biotec Inc., Bergisch-Gladbach, Germany) and passing them twice through a MACS+ selection column (Miltenyi Biotec Inc.). The cells purified from the column were routinely >95% CD11c+, as determined by flow cytometry. Cells were further incubated in the presence of antibiotics for an additional 24 or 48 h.
For inhibition of TLR9, DCs were pretreated with ODN2088 (5 μM) (Invivogen, San Diego, CA) for 2 h prior to infection or stimulation. The TLR9 activator ODN1826 (0.5 μM) (Invivogen, San Diego, CA) was used to stimulate DCs through TLR9. DCs stimulated with 2 μg/ml of ultrapure LPS from Escherichia coli (Invivogen, San Diego, CA) were used as a control.
Fluorescence-activated cell sorter analysis of DCs.
DCs were harvested 48 h after infection, washed, incubated for 5 min with anti-CD16/32 antibodies to block the Fc receptor, and double stained with phycoerythrin (PE)-conjugated anti-CD11c and fluorescein isothiocyanate (FITC)-conjugated anti-CD40, anti-CD80, and anti-CD86 antibodies along with their isotype controls (BD Pharmingen) for 30 min. Labeled DCs were washed and analyzed on a FACSCalibur flow cytometer (Becton Dickinson, Erembodegem-Aalst, Belgium).
Cytokine assay.
The levels of IL-12, IL-6, and tumor necrosis factor alpha (TNF-α) in the supernatants of S. pyogenes-stimulated, LPS-stimulated, and unstimulated DCs collected at 48 h of culture were determined by ELISA. Briefly, polystyrene microtiter plates (Costar, Fernwald, Germany) were incubated overnight at 4°C with purified rat anti-mouse IL-12 or IL-6 antibody (BD Pharmingen) diluted in coating buffer. Wells were blocked with 3% bovine serum albumin-PBS, and sample supernatants, serum samples, and standards (serial concentrations of recombinant murine IL-12 or IL-6) were added and incubated overnight at 4°C. Bound cytokine was detected with either biotinylated rat anti-mouse IL-12 or IL-6 antibody (BD Pharmingen), followed by streptavidin-peroxidase conjugate, and color was developed with 2,2′-azinobis(3-ethylbenzthiazolinesulfonic acid) (ABTS). The optical densities of samples and standards were measured at 405 nm, with a correction wavelength of 650 nm. The levels of TNF-α were determined by using a BD OptEIA mouse TNF-ELISA set (mono/poly) (BD Biosciences, San Diego, CA) according to the manufacturer's instructions.
RNA isolation, RT-PCR, and quantitative real-time PCR.
Total RNAs were isolated from purified S. pyogenes-infected, LPS-stimulated, and unstimulated DCs at 24 h of culture. DCs were resuspended in RLT buffer and homogenized by using QIAshedder columns (Qiagen). Total RNA was isolated with an RNeasy Mini kit (Qiagen) according to the manufacturer's protocol, and cDNA synthesis was performed using a Gibco reverse transcription-PCR (RT-PCR) kit following the manufacturer's instructions. The β-actin gene was used as an internal reference gene for RT-PCR. rsp-9 was used as a housekeeping gene for quantitative real-time PCR. The following oligonucleotides were generated: for β-actin, 5′-TGG AAT CCT GTG GCA TCC ATG AAA C-3′ and 5′-TAA AAC GCA GCT CAG TAA CAG TCC G-3′; for rsp-9, 5′-CTG GAC GAG GGC AAG ATG AAG C-3′ and 5′-TGA CGT TGG CGG ATG AGC ACA-3′; and for IL-12p40, 5′-CGT GCT CAT GGC TGG TGC AAA G-3′ and 5′-CTT CAT CTG CAA GTT CTT GGG C-3′. Cycling conditions for PCR amplification were 15 s at 94°C, 30 s at 58°C, and 45 s at 72°C for 30 cycles.
For quantitative real-time PCR, amplification of rsp-9 and IL-12p40 cDNAs was performed using a rotor gene cycler (Corbett Research, Cambridgeshire, United Kingdom) and SYBR green PCR master mix (Applied Biosystems). Templates used to generate standard curves for rsp-9 and IL-12p40 real-time PCRs were produced by amplification of full-length cDNAs, which were then subcloned into the TOPO PCR2.1 cloning vector (Invitrogen). The resulting plasmids were prepared using a QIAprep Spin miniprep kit (Qiagen) following the manufacturer's instructions and then used to prepare a 10-fold dilution series of each amplification standard. For the amplification standards, the cycle threshold (CT) was plotted against the log10 copy number to obtain the standard curve used to calculate cDNA copy numbers. Cycle threshold values for IL-12p40 were normalized to the value for β-actin expression. The relative expression levels for stimulated DCs were compared to the expression levels for unstimulated DCs. Data are presented as changes in mRNA level in infected or stimulated DCs compared to the mRNA level in unstimulated control DCs.
Immunofluorescence microscopy.
For double-immunofluorescence staining of extracellular and intracellular bacteria, DCs were applied to sterile coverslips and infected with S. pyogenes for 2 h. Coverslips were then rinsed to remove unbound cells, and adherent cells were fixed with 4% formaldehyde. For double-immunofluorescence staining, extracellular bacteria were stained with polyclonal rabbit anti-S. pyogenes antibodies, followed by Alexa green-conjugated goat anti-rabbit antibodies (Sigma-Aldrich, Germany). After several washes, cells were permeabilized by 0.025% Triton X-100 in PBS and washed again, and intracellular bacteria were stained by anti-S. pyogenes antibodies, followed by Alexa red-conjugated goat anti-rabbit antibodies (Sigma-Aldrich). The fluorescence images were obtained with an Axiophot microscope (Zeiss, Oberkochen, Germany) and analyzed using AxioVision, release 4.5, software (Zeiss). Intracellular bacteria thus appear red, while extracellular S. pyogenes cells appear yellow-green.
Gentamicin protection assay.
DCs were infected with S. pyogenes (MOI, 10:1) for 2 h. DCs were then washed to remove unbound bacteria and were further incubated in the presence of gentamicin (100 μg/ml) at 37°C in 5% CO2 to kill extracellular bacteria. After different times, DCs were washed and disrupted with distilled H2O to release intracellular bacteria. The resulting suspension was serially diluted, and bacterial numbers were determined after plating of cells onto blood agar.
Preparation of S. pyogenes DNA.
Overnight cultures of S. pyogenes were centrifuged, and the obtained pellet was resuspended in sucrose-TES [sucrose-N-tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid] buffer, followed by the addition of lysozyme and mutanolysin and 1 h of incubation at 37°C. RNase and proteinase K were added, and the mixture was incubated for 15 min at 37°C. After addition of 500 μl of 10% sodium dodecyl sulfate solution and incubation for 1 h, the DNA was isolated by phenol-chloroform extraction. The DNA fraction was then precipitated overnight with sodium acetate and ethanol at −20°C. After being washed with ethanol, the DNA was resuspended in Tris-EDTA buffer. Quantification was performed by photometric measurement at 260 nm. Bacterial DNA was stored at 4°C.
RESULTS
Effect of MyD88 deficiency on DC maturation.
To determine the role of the MyD88 signaling pathway in S. pyogenes-induced maturation of DCs, the expression of CD40, CD80, and CD86 on the surfaces of DCs differentiated from bone marrows obtained from either C57BL/6 (wild-type DCs) or MyD88−/− (MyD88−/− DCs) mice was determined 48 h after S. pyogenes infection by flow cytometry analysis. While wild-type DCs had significantly increased expression of CD40, CD80, and CD86 upon infection (Fig. 1, left histograms), S. pyogenes-stimulated MyD88−/− DCs completely failed to up-regulate the surface expression of these molecules (Fig. 1, right histograms). These results indicate that S. pyogenes induces DC maturation through a MyD88-dependent pathway.
FIG. 1.

S. pyogenes-induced maturation of DCs is MyD88 dependent. Bone marrow-derived DCs from C57BL/6 (left histograms) and MyD88−/− (right histograms) mice were infected for 2 h with S. pyogenes and further incubated for 48 h. Uninfected DCs were used as a control. DCs were collected, stained with PE-labeled monoclonal anti-mouse CD11c antibody and FITC-labeled monoclonal anti-mouse CD40, CD80, or CD86 antibody, and subjected to flow cytometry analysis. CD11c+ cell populations were gated and analyzed for CD40, CD80, and CD86 expression. Thin lines represent uninfected DCs; thick lines represent DCs infected with S. pyogenes. Results are representative of four independent experiments.
MyD88 is essential for S. pyogenes-induced IL-12 production from DCs.
We have recently shown that DCs are the major source of IL-12 during S. pyogenes infection (22). Therefore, we investigated the role of MyD88 signaling in the production of IL-12 by DCs in response to S. pyogenes. Wild-type and MyD88−/− DCs were infected for 2 h with S. pyogenes and further cultured in the presence of antibiotics. The supernatant of DCs stimulated with LPS was used as a control. The levels of IL-12 in the supernatants were measured 48 h after infection by ELISA determination of IL-12p40 levels. As shown in Fig. 2A, secretion of IL-12p40 was completely abolished in MyD88−/− DCs infected with S. pyogenes. To test whether the impaired capacity of MyD88−/− DCs to release IL-12 in response to S. pyogenes was caused by a lack of induction of IL-12 gene expression, we compared the levels of mRNA for the IL-12p40 subunit, which is induced in DCs after activation (40), in S. pyogenes-infected and uninfected wild-type and MyD88−/− DCs. The absence of MyD88 resulted in suppression of IL-12p40 transcription, as measured by RT-PCR (Fig. 2B) as well as real-time PCR (Fig. 2C). These results indicate that S. pyogenes induction of the IL-12p40 gene is completely MyD88 dependent.
FIG. 2.

DCs from MyD88−/− mice show impaired S. pyogenes-induced IL-12 responses in vitro. Purified bone marrow-derived DCs from C57BL/6 or MyD88−/− mice were infected in vitro with S. pyogenes for 2 h and further incubated in the presence of antibiotics. (A) IL-12p40 was measured in the supernatant of S. pyogenes-infected (hatched bars) or uninfected (white bars) DCs by ELISA 48 h after infection. DCs stimulated with 2 μg/ml of LPS were used as a control (black bars). Bars show means plus standard deviations (SD) for triplicates. Similar experiments were performed at least three times. (B) RT-PCR amplification of IL-12p40 cDNA from unstimulated, LPS-stimulated, and S. pyogenes-infected DCs isolated from wild-type C57BL/6 (left) or MyD88−/− (right) mice. β-actin expression served as a loading control. Plasmids containing the sequences encoding IL-12p40 or β-actin were used as positive controls (CTR), and reactions in the absence of cDNA were used as negative controls. (C) Changes (x-fold) in the ratio of IL-12p40 mRNA to β-actin mRNA in S. pyogenes-infected or LPS-stimulated DCs compared to those for unstimulated control DCs isolated from either wild-type C57BL/6 mice (black bars) or MyD88−/− mice (white bars).
Secretion of TNF-α and IL-6 by DCs in response to S. pyogenes is MyD88 dependent.
We next examined if MyD88 was involved in the production of other inflammatory cytokines, such as TNF-α and IL-6. These cytokines were detected by ELISA in the supernatants of uninfected and S. pyogenes-infected wild-type or MyD88−/− DCs 48 h after in vitro infection. Wild-type DCs and MyD88−/− DCs stimulated with LPS were used as controls. The production of both TNF-α (Fig. 3A) and IL-6 (Fig. 3B) in response to S. pyogenes infection was drastically reduced in MyD88−/− DCs compared to that in wild-type cells. These data indicate that S. pyogenes-induced production of proinflammatory cytokines in DCs is TLR mediated and dependent on MyD88.
FIG. 3.
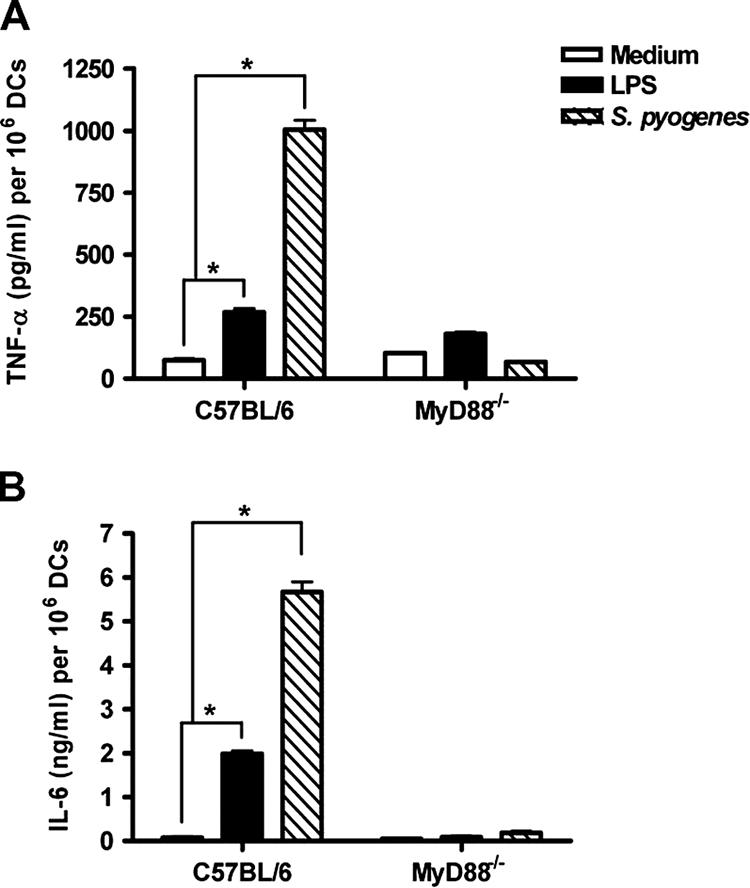
Production of TNF-α (A) and IL-6 (B) in response to S. pyogenes in wild-type and MyD88−/− DCs. DCs from wild-type C57BL/6 or MyD88−/− mice were infected with S. pyogenes or stimulated with 2 μg/ml LPS and further cultured for 48 h. Concentrations of TNF-α and IL-6 in the culture supernatants were measured by ELISA. The results are shown as means plus SD for three experiments. *, P < 0.05 (by F test).
Uptake and killing of internalized S. pyogenes bacteria by DCs are independent on MyD88 expression.
It has been reported that activation of the TLR signaling pathway by bacteria regulates phagocytosis at multiple steps, including internalization and phagosome maturation (6). Therefore, we assessed whether DCs that lacked MyD88 expression differed from normal DCs with respect to the internalization of S. pyogenes by using double-immunofluorescence microscopy to discriminate between extracellular and intracellular bacteria. In contrast to the impressive phenotype of these cells with respect to the induced activation of inflammatory cytokines, MyD88 expression did not affect the ability of DCs to internalize S. pyogenes, since the uptake of S. pyogenes by MyD88−/− DCs (Fig. 4B) was comparable to that of wild-type DCs (Fig. 4A). Similarly, wild-type and MyD88−/− DCs did not differ in the ability to kill internalized S. pyogenes cells, as shown by the kinetics of bacterial killing displayed in Fig. 4C.
FIG. 4.

In vitro phagocytosis and killing of S. pyogenes by DCs isolated from wild-type C57BL/6 or MyD88−/− mice. (A and B) Double-immunofluorescence staining of S. pyogenes-infected DCs isolated from wild-type C57BL/6 (A) or MyD88−/− (B) mice showing the intracellular or extracellular location of S. pyogenes within the DCs. Extracellular bacteria are shown in yellow-green, intracellular bacteria are shown in red, and DNA in the nucleus is stained in blue. (C) Kinetics of S. pyogenes killing by DCs isolated from wild-type C57BL/6 (squares) or MyD88−/− (triangles) mice. DCs from the indicated mice were infected with S. pyogenes at an MOI of 1:10 for 2 h and further incubated in the presence of gentamicin (100 μg/ml) at 37°C in 5% CO2 to kill extracellular bacteria. The numbers of viable intracellular S. pyogenes cells were determined after different periods by disrupting infected DCs with distilled H2O and plating them onto blood agar. Each point represents the mean value ± SD for four independent experiments.
Signaling through TLR2 is not absolutely required for S. pyogenes-induced activation of DCs.
The finding that the DC response to S. pyogenes was MyD88 dependent prompted an effort to define which TLRs were involved in S. pyogenes-induced DC activation. Since gram-positive cell wall products, such as LTA and peptidoglycans, have been implicated in the activation of DCs through TLR2 (26), we first investigated the participation of TLR2 in DC sensing of S. pyogenes. DCs from TLR2−/− mice as well as wild-type C57BL/6 control mice were infected with S. pyogenes for 2 h and further incubated after removal of unbound bacteria. Maturation markers in S. pyogenes-stimulated and nonstimulated DCs were determined after 48 h of culture. The obtained results show that both TLR2−/− and wild-type DCs up-regulated the expression of maturation markers upon exposure to S. pyogenes, indicating that a functional TLR2 was not absolutely required for this process. Representative histograms showing up-regulation of CD40 in DCs from wild-type (left histogram) and TLR2−/− (right histogram) mice are depicted in Fig. 5A. Similar results were observed when the expression of other surface markers (CD80 and CD86) was examined (data not shown).
FIG. 5.

S. pyogenes-induced maturation and production of cytokines in DCs deficient in expression of TLR2. (A) Flow cytometry analysis for detection of CD40 expression on DCs derived from bone marrows of wild-type C57BL/6 (left histograms) or TLR2−/− (right histograms) mice (uninfected [thin lines] or after infection with S. pyogenes [thick lines]). DCs were infected with S. pyogenes for 2 h and further cultured in the presence of antibiotics for 48 h. The DCs were then stained with PE-conjugated anti-CD11c and FITC-conjugated anti-CD40 antibodies and analyzed by fluorescence-activated cell sorting. (B and C) Induction of IL-12p40 (B) and IL-6 (C) in DCs derived from bone marrows of C57BL/6 or TLR2−/− mice by wild-type S. pyogenes (black bars) or the isogenic S. pyogenes mutant strain deficient in the expression of M1 protein (hatched bars). Cytokines in the supernatants of uninfected and infected DCs were determined after 48 h of culture. Bars represent the means plus SD for three experiments. *, P < 0.05 (by F test).
In addition, examination of cytokine secretion revealed that wild-type and TLR2−/− DCs produced comparable amounts of IL-12p40 in response to stimulation with S. pyogenes (Fig. 5B). Thus, S. pyogenes is clearly able to induced IL-12p40 production by DCs by signaling though TLRs other than TLR2.
It was reported recently (30) that the streptococcal M protein, a classical virulence determinant of S. pyogenes, interacts with TLR2 on human blood monocytes, thereby inducing the production of large amounts of proinflammatory cytokines. To investigate the relative contribution of M protein-TLR2 receptor interaction to the overall production of IL-12p40 by DCs, we compared the levels of IL-12p40 induced in wild-type and TLR2−/− DCs by an S. pyogenes mutant strain deficient in the expression of M protein and by the S. pyogenes parental strain. The IL-12p40 response induced by the M protein-deficient strain was roughly equivalent to that of the corresponding wild-type strain, irrespective of the integrity of TLR2 expression by the DCs (Fig. 5B). Additionally, the pattern of secretion of other inflammatory cytokines, such as IL-6, did not differ between wild-type and TLR2−/− DCs after stimulation with the parental or M protein-deficient strain of S. pyogenes (Fig. 5C).
It has been reported that TLR2 possesses the ability to colocalize with TLR6 and may function as a heterodimeric receptor in response to ligation with the appropriate microbial product (29). Accordingly, we compared maturation and cytokine secretion between DCs derived from bone marrows of mice deficient in the expression of both TLR2 and TLR6 after in vitro exposure to S. pyogenes. Again, no significant differences in the expression of maturation markers (data not shown) were seen. IL-12p40 production was also equivalent between wild-type and TLR2/6−/− DCs (Fig. 6).
FIG. 6.
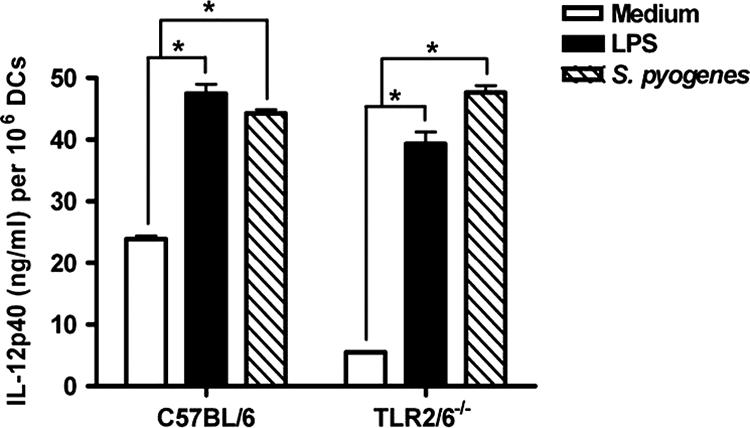
Secretion of IL-12p40 by DCs derived from bone marrows of wild-type C57BL/6 or TLR2/6−/− mice after infection with S. pyogenes (hatched bars) or stimulation with LPS (black bars). DCs were infected with S. pyogenes for 2 h and further cultured in the presence of antibiotics for 48 h. IL-12p40 levels in the supernatants of untreated and treated DCs were determined at 48 h of culture. Bars represent means plus SD for three experiments. *, P < 0.05 (by F test).
Inhibition of TLR9 does not affect the ability of S. pyogenes to induce activation of DCs.
Bacterially derived DNA containing CpG motifs can also induce cytokine production and DC maturation through TLR9 (14). DNA from S. pyogenes has been reported to contain stimulatory motifs that induce the activation of human lymphocytes (7). To determine whether DNA isolated from S. pyogenes could stimulate IL-12 production through the stimulation of TLR9 signaling, DCs were stimulated with S. pyogenes DNA in the absence or presence of the TLR9 inhibitor ODN 2088, and the secretion of IL-12p40 was determined at 48 h of culture. The production of appreciable levels of IL-12p40 by DCs stimulated with S. pyogenes DNA compared with those in unstimulated DCs demonstrated that bone marrow DCs actively responded to this agent (Fig. 7A). S. pyogenes DNA-induced IL-12p40 production by DCs is dependent on TLR9, since the pretreatment of DCs with the TLR9 inhibitor ODN 2088 significantly attenuated IL-12p40 production by DNA-stimulated DCs (Fig. 7A).
FIG. 7.
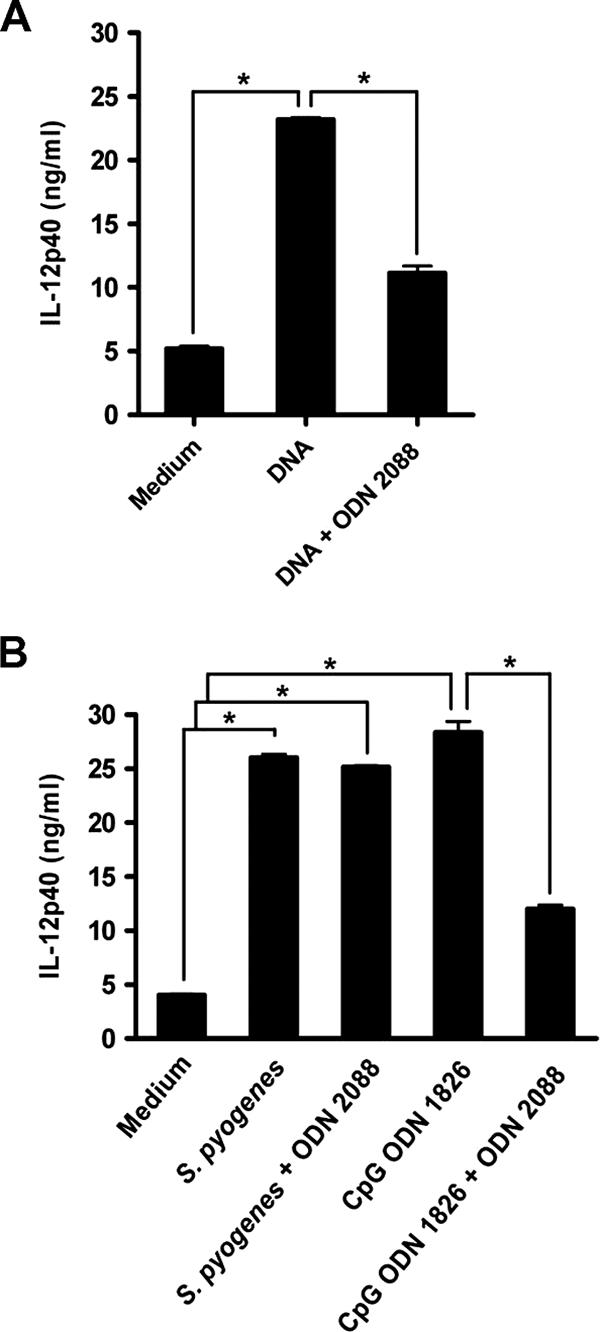
S. pyogenes DNA stimulation of IL-12p40 secretion by DCs is dependent on TLR9, but stimulation of DC IL-12p40 secretion by the whole S. pyogenes microorganism is TLR9 independent. (A) DCs isolated from bone marrows of C57BL/6 mice were pretreated or not with the TLR9 inhibitor ODN 2088 for 2 h, followed by stimulation with 50 μg/ml of purified S. pyogenes DNA for 48 h and analysis of IL-12p40 by ELISA. Data are means plus SD for triplicates. *, P < 0.05 (F test). (B) DCs were pretreated or not with the TLR9 inhibitor ODN 2088 for 2 h prior to stimulation with the TLR9 agonist CpG ODN 1826 or infection with S. pyogenes and were further cultured for 48 h. IL-12p40 levels in the supernatants were determined by ELISA. Data are means plus SD for triplicates. *, P < 0.05 (F test).
To determine the relative contribution of TLR9 to IL-12p40 production by whole S. pyogenes microorganisms, DCs were stimulated either with S. pyogenes or with ODN 1826 as a positive control, in either the presence or absence of the TLR9 inhibitor ODN 2088. The results show that in contrast to what was observed with purified DNA, inhibition of TLR9 did not affect the production of IL-12p40 by DCs in response to S. pyogenes stimulation. Therefore, these data ruled out a primary role of TLR9 signaling in DC activation by S. pyogenes.
A major contribution of TLR1 or TLR4 in immune recognition of S. pyogenes by DCs was also discarded after evaluating the response of DCs isolated from mice deficient in the expression of either TLR1 or TLR4 (data not shown).
DISCUSSION
DCs play an important role in host defense against S. pyogenes (22). In this report, we attempted to identify the receptors involved in innate immune recognition of S. pyogenes by DCs. To determine the possible contribution of TLRs to this process, we tested the requirement for the TLR-associated adapter molecule MyD88 in maturation and production of cytokines by DCs after exposure to S. pyogenes. MyD88 has been shown to be a critical signaling molecule for most TLRs as well as for IL-1 receptor family responses (25). After recognition of a pathogen-associated molecule, TLRs associate with MyD88 and induce a cascade of signaling events, leading to NF-κB activation and subsequent transcription of genes encoding proinflammatory cytokines, such as TNF-α, IL-6, and IL-12 (36). Using DCs deficient in the expression of MyD88, we show here that the induction of DC maturation and production of cytokines in response to S. pyogenes infection is entirely dependent on the MyD88-mediated signaling pathway. Our results are in agreement with previously published reports suggesting that the production of inflammatory cytokines, in particular IL-12, by DCs is completely abolished in the absence of MyD88 (19, 21). The observation that S. pyogenes-mediated activation of DCs was MyD88 dependent suggested that one or multiple TLRs were involved. However, in contrast to our expectations, DCs lacking either TLR1, TLR2, TLR4, or TLR2/TLR6 or inhibited in TLR9 signaling retained the ability to mature and to produce inflammatory cytokines in response to S. pyogenes infection. Therefore, despite the indications that TLRs mediate the response of DCs to S. pyogenes, we failed to identify a specific TLR with a predominant impact on this response.
These observations indicate that immune recognition of S. pyogenes is not likely to occur through recognition of a single microbial target molecule by a single TLR. Instead, innate immune cells are exposed to multiple bacterial components capable of engaging different TLR family members. Thus, we hypothesize that TLRs 1, 2, 4, 6, and 9 may all be involved in the immune sensing of S. pyogenes through the recognition of lipoproteins, peptidoglycans, LTA (29, 38), and bacterial DNA (14). Deletion of an individual TLR can therefore be compensated for by signaling triggered through the others. To cope with the complexity of sensing pathogens, the innate immune system probably evolved to respond via multiple interactions to maximize activation. Simultaneous or sequential activation of multiple TLRs can also be important for tailoring inflammatory responses to be effective against specific microbial pathogens.
In accordance with our observations with S. pyogenes and DCs, Henneke and colleagues (15, 16) reported that macrophages deficient in MyD88 expression are significantly impaired in the immune response to group B streptococcus. However, macrophages isolated from mutant mice with targeted deletions of the individual TLRs 1, 2, 4, 6, and 9 did not show a significantly decreased cytokine response to this pathogen, thus excluding all of the TLRs that have been reported as potential receptors for cell components of gram-positive bacteria (15, 16).
It is noteworthy that, in general, the impact of TLR deletion on microbial infection phenotypes is often not severe. Thus, although LPS from E. coli is recognized by TLR4, mice deficient in TLR4 do not display enhanced susceptibility to infection with this pathogen (10, 13). A similar situation has been observed with Listeria monocytogenes, which can induce inflammatory signaling through TLR2, but mice deficient in TLR2 are not more susceptible to infection (9). These observations further demonstrate the complex interactions between a pathogen and innate immune cells, in which a single receptor cannot be assigned a sole recognition molecule and the induction of all components of the inflammatory response.
Another conclusion from this study is that the uptake and killing of S. pyogenes by DCs were unaffected in the absence of MyD88, suggesting that in these settings, phagocytic and killing mechanisms of DCs do not require TLR-mediated signaling for their induction.
In summary, our results indicate that immune recognition of S. pyogenes by DCs is highly complex and requires a coordinated interplay between MyD88-independent events involving phagocytosis and antigen processing as well as MyD88-dependent signaling pathways involved in the induction of DC maturation and cytokine production. Adding an additional level of complexity, the data presented here argue for multimodal detection of S. pyogenes by DCs, involving several TLR signaling pathways, resulting in a complex triggering of proinflammatory responses that impact the nature and intensity of the ensuing immune response. The evolution of such a multireceptor, integrated system for sensing pathogens is crucial for innate immune defenses to prevent pathogens from easily subverting recognition by mutating or deleting critical TLR ligands.
Acknowledgments
This work was supported in part by the Nationales Genomforschungsnetz II (grant 01GS0404) and by the Impuls und Vernetzungsfond, Helmholtz-Gemeinschaft deutscher Forschungszentren (grant in immunogenetics of group A streptococci to E.M.).
We thank P. Cleary for providing the M1-deficient S. pyogenes mutant strain.
Editor: B. A. McCormick
Footnotes
Published ahead of print on 7 April 2008.
REFERENCES
- 1.Akira, S., and K. Takeda. 2004. Toll-like receptor signaling. Nat. Rev. Immunol. 4499-511. [DOI] [PubMed] [Google Scholar]
- 2.Akira, S., K. Takeda, and T. Kaisho. 2001. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat. Immunol. 2675-680. [DOI] [PubMed] [Google Scholar]
- 3.Banchereau, J., and R. M. Steinman. 1998. Dendritic cells and the control of immunity. Nature 392245-252. [DOI] [PubMed] [Google Scholar]
- 4.Barton, G. M., and R. Medzhitov. 2003. Toll-like receptor signaling pathways. Science 3001524-1525. [DOI] [PubMed] [Google Scholar]
- 5.Beutler, B., K. Hoebe, X. Du, and R. J. Ulevitch. 2003. How we detect microbes and respond to them: the Toll-like receptors and their transducers. J. Leukoc. Biol. 74479-485. [DOI] [PubMed] [Google Scholar]
- 6.Blander, J. M., and R. Medzhitov. 2004. Regulation of phagosome maturation by signals from Toll-like receptors. Science 3041014-1018. [DOI] [PubMed] [Google Scholar]
- 7.Chatellier, S., and M. Kotb. 2000. Preferential stimulation of human lymphocytes by oligodeoxynucleotides that copy DNA CpG motifs present in virulent genes of group A streptococci. Eur. J. Immunol. 30993-1001. [DOI] [PubMed] [Google Scholar]
- 8.Cue, D., P. E. Dombek, H. Lam, and P. P. Cleary. 1998. Streptococcus pyogenes serotype M1 encodes multiple pathways for entry into human epithelial cells. Infect. Immun. 664593-4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Edelson, B. T., and E. R. Unanue. 2002. MyD88-dependent but Toll-like receptor 2-independent innate immunity to Listeria: no role for either in macrophage listericidal activity. J. Immunol. 1693869-3875. [DOI] [PubMed] [Google Scholar]
- 10.Evans, T. J., E. Strivens, A. Carpenter, and J. Cohen. 1991. Differences in cytokine response and induction of nitric oxide synthase in endotoxin-resistant and endotoxin-sensitive mice after intravenous gram-negative infection. J. Immunol. 1505033-5040. [PubMed] [Google Scholar]
- 11.Goldman, O., M. Rohde, G. S. Chhatwal, and E. Medina. 2004. Role of macrophages in host resistance to group A streptococci. Infect. Immun. 722956-2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hayashi, F., K. D. Smith, A. Ozinsky, T. R. Hawn, E. C. Yi, D. R. Goodlett, J. K. Eng, S. Akira, D. M. Underhill, and A. Aderem. 2001. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 4101099-1103. [DOI] [PubMed] [Google Scholar]
- 13.Haziot, A., N. Hijiya, S. C. Gangloff, J. Silver, and S. M. Goyert. 2001. Induction of a novel mechanism of accelerated bacterial clearance by lipopolysaccharide in CD14-deficient and Toll-like receptor 4-deficient mice. J. Immunol. 1661075-1078. [DOI] [PubMed] [Google Scholar]
- 14.Hemmi, H., O. Takeuchi, T. Kawai, T. Kaisho, S. Sato, H. Sanjo, M. Matsumoto, K. Hoshino, H. Wagner, K. Takeda, and S. Akira. 2000. A Toll-like receptor recognizes bacterial DNA. Nature 408740-745. [DOI] [PubMed] [Google Scholar]
- 15.Henneke, P., O. Takeuchi, R. Malley, E. Lien, R. R. Ingalls, M. W. Freeman, T. Mayadas, V. Nizet, S. Akira, D. L. Kasper, and D. T. Golenbock. 2002. Cellular activation, phagocytosis, and bactericidal activity against group B streptococcus involve parallel myeloid differentiation factor 88-dependent and independent signaling pathways. J. Immunol. 1693970-3977. [DOI] [PubMed] [Google Scholar]
- 16.Henneke, P., O. Takeuchi, J. A. van Strijp, H.-K. Guttormsen, J. A. Smith, A. B. Schromm, T. A. Espevik, S. Akira, V. Nizet, D. L. Kasper, and D. T. Golenbock. 2001. Novel engagement of CD14 and multiple Toll-like receptors by group B streptococci. J. Immunol. 1677069-7076. [DOI] [PubMed] [Google Scholar]
- 17.Reference deleted.
- 18.Joosten, L. A., M. I. Koenders, R. L. Smeets, M. Heuvelmans-Jacobs, M. M. Helsen, K. Takeda, S. Akira, E. Lubberts, F. A. van de Loo, and W. B. van den Berg. 2003. Toll-like receptor 2 pathway drives streptococcal cell wall-induced joint inflammation: critical role of myeloid differentiation factor 88. J. Immunol. 1716145-6153. [DOI] [PubMed] [Google Scholar]
- 19.Kaisho, T., and S. Akira. 2001. Dendritic-cell function in Toll-like receptor- and MyD88-knockout mice. Trends Immunol. 2278-83. [DOI] [PubMed] [Google Scholar]
- 20.Kaisho, T., and S. Akira. 2003. Regulation of dendritic cell function through Toll-like receptors. Curr. Mol. Med. 3759-771. [DOI] [PubMed] [Google Scholar]
- 21.Kawai, T., O. Adachi, T. Ogawa, K. Takeda, and S. Akira. 1999. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity 11115-122. [DOI] [PubMed] [Google Scholar]
- 22.Loof, T. G., M. Rohde, G. S. Chhatwal, S. Jung, and E. Medina. 2007. The contribution of dendritic cells to host defenses against Streptococcus pyogenes. J. Infect. Dis. 1961794-1803. [DOI] [PubMed] [Google Scholar]
- 23.Martin, J. M., and M. Green. 2006. Group A streptococcus. Semin. Pediatr. Infect. Dis. 17140-148. [DOI] [PubMed] [Google Scholar]
- 24.Medina, E., O. Goldmann, M. Rohde, A. Lengeling, and G. S. Chhatwal. 2001. Genetic control of susceptibility to group A streptococcal infection in mice. J. Infect. Dis. 184846-852. [DOI] [PubMed] [Google Scholar]
- 25.Medzhitov, R., P. Preston-Hurlburt, E. Kopp, A. Stadlen, C. Chen, S. Ghosh, and C. A. Janeway, Jr. 1998. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol. Cell 2253-258. [DOI] [PubMed] [Google Scholar]
- 26.Michelsen, K. S., A. Aicher, M. Mohaupt, T. Hartung, S. Dimmeler, C. J. Kirschning, and R. R. Schumann. 2001. The role of Toll-like receptors (TLRs) in bacteria-induced maturation of murine dendritic cells (DCs). Peptidoglycan and lipoteichoic acid are inducers of DC maturation and require TLR2. J. Biol. Chem. 27625680-25686. [DOI] [PubMed] [Google Scholar]
- 27.O'Loughlin, R. E., A. Roberson, P. R. Cieslak, R. Lynfield, K. Gershman, A. Craig, B. A. Albanese, M. M. Farley, N. L. Barrett, N. L. Spina, B. Beall, L. H. Harrison, A. Reingold, C. Van Beneden, et al. 2007. The epidemiology of invasive group A streptococcal infection and potential vaccine implications: United States, 2000-2004. Clin. Infect. Dis. 45853-862. [DOI] [PubMed] [Google Scholar]
- 28.O'Neill, L. A., K. A. Fitzgerald, and A. G. Bowie. 2003. The Toll-IL-1 receptor adaptor family grows to five members. Trends Immunol. 24286-290. [DOI] [PubMed] [Google Scholar]
- 29.Ozinsky, A., D. M. Underhill, J. D. Fontenot, A. M. Hajjar, K. D. Smith, C. B. Wilson, L. Schroeder, and A. Aderem. 2000. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between Toll-like receptors. Proc. Natl. Acad. Sci. USA 9713766-13771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pahlman, L. I., M. Morgelin, J. Eckert, L. Johansson, W. Russell, K. Riesbeck, O. Soehnlein, L. Lindbom, A. Norrby-Teglund, R. R. Schumann, L. Bjorck, and H. Herwald. 2006. Streptococcal M protein: a multipotent and powerful inducer of inflammation. J. Immunol. 1771221-1228. [DOI] [PubMed] [Google Scholar]
- 31.Poltorak, A., X. He, I. Smirnova, M. Y. Liu, C. Van Huffel, X. Du, D. Birdwell, E. Alejos, M. Silva, C. Galanos, M. Freudenberg, P. Ricciardi-Castagnoli, B. Layton, and B. Beutler. 1998. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 2822085-2088. [DOI] [PubMed] [Google Scholar]
- 32.Rasmussen, M., H. P. Muller, and L. Bjorck. 1999. Protein GRAB of Streptococcus pyogenes regulates proteolysis at the bacterial surface by binding alpha2-macroglobulin. J. Biol. Chem. 27415336-15344. [DOI] [PubMed] [Google Scholar]
- 33.Reis e Sousa, C. 2001. Dendritic cells as sensors of infection. Immunity 14495-498. [DOI] [PubMed] [Google Scholar]
- 34.Reis e Sousa, C. 2004. Toll-like receptors and dendritic cells: for whom the bug tolls. Semin. Immunol. 1627-34. [DOI] [PubMed] [Google Scholar]
- 35.Schwandner, R., R. Dziarski, H. Wesche, M. Rothe, and C. J. Kirschning. 1999. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by Toll-like receptor 2. J. Biol. Chem. 27417406-17409. [DOI] [PubMed] [Google Scholar]
- 36.Takeda, K., and S. Akira. 2005. Toll-like receptors in innate immunity. Int. Immunol. 171-14. [DOI] [PubMed] [Google Scholar]
- 37.Takeda, K., T. Kaisho, and S. Akira. 2003. Toll-like receptors. Annu. Rev. Immunol. 21335-376. [DOI] [PubMed] [Google Scholar]
- 38.Takeuchi, O., K. Hoshino, T. Kawai, H. Sanjo, H. Takada, T. Ogawa, K. Takeda, and S. Akira. 1999. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 11443-451. [DOI] [PubMed] [Google Scholar]
- 39.Takeuchi, O., T. Kawai, P. F. Muhlradt, M. Morr, J. D. Radolf, A. Zychlinsky, K. Takeda, and S. Akira. 2001. Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int. Immunol. 13933-940. [DOI] [PubMed] [Google Scholar]
- 40.Trinchieri, G. 2003. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 3133-146. [DOI] [PubMed] [Google Scholar]
- 41.Veckman, V., and I. Julkunen. 2008. Streptococcus pyogenes activates human plasmacytoid and myeloid dendritic cells. J. Leukoc. Biol. 83296-304. [DOI] [PubMed] [Google Scholar]
- 42.Veckman, V., M. Miettinen, J. Pirhonen, J. Siren, S. Matikainen, and I. Julkunen. 2004. Streptococcus pyogenes and Lactobacillus rhamnosus differentially induce maturation and production of Th1-type cytokines and chemokines in human monocyte-derived dendritic cells. J. Leukoc. Biol. 75764-771. [DOI] [PubMed] [Google Scholar]
- 43.Yoshimura, A., E. Lien, R. R. Ingalls, E. Tuomanen, R. Dziarski, and D. Golenbock. 1999. Cutting edge: recognition of gram-positive bacterial cell wall components by the innate immune system occurs via Toll-like receptor 2. J. Immunol. 1631-5. [PubMed] [Google Scholar]