

Abstract
Multiple molecular lesions in human cancers directly collaborate to deregulate proliferation and suppress apoptosis to promote tumorigenesis. The candidate tumor suppressor RASSF1A is commonly inactivated in a broad spectrum of human tumors and has been implicated as a pivotal gatekeeper of cell cycle progression. However, a mechanistic account of the role of RASSF1A gene inactivation in tumor initiation is lacking. Here we have employed loss-of-function analysis in human epithelial cells for a detailed investigation of the contribution of RASSF1 to cell cycle progression. We found that RASSF1A has dual opposing regulatory connections to G1/S phase cell cycle transit. RASSF1A associates with the Ewing sarcoma breakpoint protein, EWS, to limit accumulation of cyclin D1 and restrict exit from G1. Surprisingly, we found that RASSF1A is also required to restrict SCFβTrCP activity to allow G/S phase transition. This restriction is required for accumulation of the anaphase-promoting complex/cyclosome (APC/C) inhibitor Emi1 and the concomitant block of APC/C-dependent cyclin A turnover. The consequence of this relationship is inhibition of cell cycle progression in normal epithelial cells upon RASSF1A depletion despite elevated cyclin D1 concentrations. Progression to tumorigenicity upon RASSF1A gene inactivation should therefore require collaborating genetic aberrations that bypass the consequences of impaired APC/C regulation at the G1/S phase cell cycle transition.
Normal cellular proliferation proceeds through a regimented surveillance of proliferative and apoptotic checkpoints that integrate pro- and antigrowth signals. It is the responsibility of so called “tumor suppressor proteins” to regulate these checkpoints; their loss facilitates, and is likely required for, the development of the semiautonomous proliferative capacity of cancer cells. Recently, RASSF1A has emerged as a candidate tumor suppressor protein that may play a crucial role in mechanisms that curb aberrant, proliferative signals. RASSF1A is found in the 3p21.3 chromosomal region, which commonly exhibits loss of heterozygosity in lung, breast, ovarian, nasopharyngeal, and renal tumors (1). Although expressed in “normal” epithelial cells, RASSF1A is absent in many cancer cells due to a high level of methylation at the CpG sites in its promoter (6). A splice variant of RASSF1A regulated by an independent promoter, RASSF1C, is expressed in both normal and cancer cells and does not have a methylated promoter (1). RASSF1A inactivation is an extremely common event in many human cancers, including 80 to 100% of small-cell lung cancer cell lines and tumors, 30 to 40% of non-small-cell lung cancer cell lines and tumors, 49 to 62% of breast cancer cell lines and tumors, 67 to 70% of primary nasopharyngeal cancers, and 91% of primary renal cell carcinomas (1, 6, 10). Furthermore, evidence suggests that RASSF1A is silenced during early neoplastic changes in the breast, including intraductal papillomas and epithelial hyperplasia, indicating that its inactivation is an early event in cancer progression (8). Mice engineered to lack expression of RASSF1A are normal; however, they are more susceptible to spontaneous and radiation-induced tumorigenesis (25).
Together with the correlative observations described above, RASSF1A was implicated as a tumor suppressor gene through studies in which its reexpression in lung carcinoma cells reduced colony formation, suppressed anchorage-independent growth, and inhibited tumor formation in nude mice (6). Previously we have found that RASSF1A overexpression blocks proliferation and decreases the levels of cyclin D1, presumably preventing cells from passing through the Rb family cell cycle restriction point and entering S phase. Similarly, the reduction of RASSF1A protein levels by small interfering RNA (siRNA) increased cyclin D1 protein levels. Overexpression of viral oncoprotein E7, which inhibits the interaction between Rb and E2F, produced proliferative cells resistant to RASSF1A-induced cell cycle arrest, placing RASSF1A's antiproliferative effect prior to the Rb checkpoint (22). A supporting clinical correlation comes from studies of cervical cancer in which there is an inverse correlation between human papillomavirus infection (E7 expression) and RASSF1A methylation status, indicating that these two oncogenic changes disable similar tumorigenic pathways (5, 15).
A variety of interacting proteins have been characterized that may participate in RASSF1A-dependent regulatory events (3, 13, 23, 24). However, a mechanistic account of the consequences of RASSF1A loss for tumor progression remains elusive. Here we describe a detailed loss-of-function analysis to directly evaluate the impact of RASSF1A depletion on the molecular changes required for cell cycle progression. We found that RASSF1A inhibits cyclin D1 accumulation through an association with the Ewing sarcoma breakpoint protein (EWS). In addition, RASSF1A restricts APC/C activity in G1/S through a functional interaction with βTRCP. Together, these findings suggest that RASSF1A has both positive and negative inputs into cell cycle progression that may represent a fail-safe relationship. As a consequence, multiple genetic lesions would be required to overcome RASSF1A function during tumor progression. While loss of RASSF1A may not directly promote oncogenic transformation, it may provide a permissive environment for acquiring additional genetic lesions that lead to tumorigenesis.
MATERIALS AND METHODS
Yeast two-hybrid library screening and pairwise interaction assay.
RASSF1A (amino acids 1 to 210) lacking the Ras association domain was used as bait in a yeast two-hybrid screen of a HeLa cDNA library in yeast reporter strain L40 by standard methods (26).
Cell culture and transfection.
HeLa cells were maintained in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum (FBS). MCF10A cells were maintained in mammary epithelial cell basal medium supplemented with epidermal growth factor and bovine pituitary extract (Cambrex). A549 cells were maintained in RPMI medium plus 5% FBS. For siRNA transfections, cells were plated at 50 to 75% confluence and transfected with 100 nM total siRNA by using Oligofectamine (Invitrogen) according to manufacturer's protocol. MCF10A cells were transfected by a trypsin-mediated method in which cells are briefly trypsinzed for 30 s prior to addition of the lipid-siRNA complex (18).
siRNAs.
siRNA sequences were as follows: RASSF1A, GACCUCUGUGGCGACUUCATT and CACGUGGUGCGACCUCUGU; EWS, GACUCUGACAACAGUGCAATT and AAUGGCGUCCACGGAUUAC; Emi1, GAUGCUCAAACCAAGUUAU; CDH1, GAAGGGUCUGUUCACGUAU. For βTRCP silencing, the SMARTpool was obtained from Dharmacon (Lafayette, CO).
Immunoblotting and immunofluorescence.
Rabbit antibodies against RASSF1A and -C were generated as previously described (4). Polyclonal antibody 4169 was generated with a combination of genetic and peptide immunization procedures. cDNA encoding amino acids 1 to 119 of RASSF1A was inserted into mammalian expression vector pcDNA3.1 (Invitrogen, Carlsbad, CA). One milligram of the plasmid DNA was initially intrasplenically (i.s.) injected into a rabbit. Subsequently, 1 mg of the DNA was intramuscularly injected 30 and 45 days after the i.s. injection. The rabbit was subcutaneously injected with a keyhole limpet hemocyanin (KLH)-conjugated peptide (PAGRAGKGRTRLERANALRIA) corresponding to amino acids 15 to 35 of RASSF1A 14 days after the second intramuscular injection. Monoclonal antibody G5 was generated with a peptide specific to RASSF1A (SGEPELIELRELAPAGRAGKGR, corresponding to amino acids 2 to 22 of RASSF1A). BALB/c mice were injected i.s. with 100 μg of KLH-conjugated peptide. The mice were boosted four times with 60 μg of the KLH-conjugated peptide by subcutaneous injection during a 90-day period. Antibodies for cyclin A, cyclin B, EWS, FUS/TLS, and ERK2 were from Santa Cruz. Anti-Skp2 and -Emi1 antibodies were from Zymed, and anti-CDH1 antibody was from LabVision Corporation. MST2 antibody was obtained from Cell Signaling. A monoclonal antibody against bromodeoxyuridine (BrdU) was obtained from Becton Dickinson. Alexa 488 (Molecular Probes) was used as a secondary antibody for fluorescent labeling. Immunoblot assays were performed with proteins transferred to polyvinylidene difluoride membrane according to the manufacturer's protocol for each antibody. For BrdU visualization, cells were treated with 30 μM BrdU for 24 h and then fixed in 3.7% HCHO. Cells were permeabilized with methanol for 10 min at −20°C and then blocked in phosphate-buffered saline-5% bovine serum albumin-1% Tween for a minimum of 15 min. Anti-BrdU antibody was used at a dilution of 1:4. Cells were visualized on an Axiovert upright microscope (Zeiss) equipped with a Hamamatsu black and white camera.
Immunoprecipitation.
HeLa cells grown to confluence on 60-mm2 dishes were lysed in buffer containing 0.5% Triton X-100 and 150 mM NaCl and 0.5% deoxycholate. Soluble lysate was incubated with protein A beads for 30 min, followed by incubation overnight with anti-RASSF1A monoclonal antibody G5 or rabbit immunoglobulin G and protein A beads. Beads were washed three times in lysis buffer with 500 mM NaCl. Following washes, sample buffer was added and lysates were boiled and separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), followed by immunoblotting.
Fluorescence-activated cell sorter (FACS) analysis.
At 72 h posttransfection, cells were trypsinized and resuspended in a 50:50 mixture of ethanol and phosphate-buffered saline. Following fixation for 30 min, cells were washed and labeled with propidium iodide (Sigma) at 40 μg/ml for 30 min at 37°C. For each analysis, 10,000 cells were collected by FACScan and analyzed with the CellQuest program (Becton Dickinson).
Quantitative PCR (qPCR).
HeLa cells were transfected in 35-mm2 dishes with 100 nM siRNA. At 72 h posttransfection, RNA was extracted from cells with a High Pure RNA isolation kit (Roche Applied Science) according to the manufacturer's protocol. cDNA was synthesized with Super Script II reverse transcriptase (Invitrogen) according to the manufacturer's protocol. For cDNA synthesis, 1 μg of RNA and oligo(dT)12-18 primers were used. One-fifteenth of the cDNA reaction mixture was used with the Roche LightCycler System and LightCycler FastStart DNA Master Sybr green I (Roche Applied Systems). Primers were chosen to flank at least two siRNA target sequences and lie on separate exons. The primers used for cyclin D1 were 5′CCAGCTCCTGTGCTGCGAAG3′ (forward) and 5′GCGGCCAGGTTCCAC3′ (reverse). The primers used for βTRCP1 were 5′AGCTGTGCCAGACTCTGCTT3′ (forward) and 5′GCTGGCAGAGCAGTTATGAA3′ (reverse). The primers used for βTRCP2 were 5′TGCAGCGGGACTTTATTACC3′ (forward) and 5′TCTCGTAGGCCACTGATAATTT (reverse). Values were normalized to glyceraldehyde-3-phosphate dehydrogenase and analyzed with the relative quantification mathematical model (Pfaffl).
RESULTS
RASSF1A interacts with EWS.
Our previous observations had suggested that RASSF1A expression in tumor cells that lack RASSF1A inhibits cyclin D1 accumulation and cell cycle progression (22). To identify proteins that may participate in RASSF1A-dependent cell cycle modulation, we performed a yeast two-hybrid screen with an N-terminal fragment of RASSF1A (Fig. 1A). From this screen, we isolated the zinc finger domain of EWS as a specific RASSF1A binding partner that could be recapitulated by overexpression and coimmunoprecipitation (Fig. 1B). EWS has best been characterized for the fusion it forms with transcription factors such as Fli1 to cause Ewing's sarcoma (2). The full-length protein has both a transcriptional activation domain and a predicted zinc finger RNA binding domain. HeLa cells retain expression of RASSF1A presumably because E7, which directly inactivates Rb, allows G1 progression independently of a requirement for cyclin D1 accumulation. Therefore, we used these cells to examine native complexes of RASSF1A without potential complications from overexpression artifacts. Immunoprecipitates from whole-cell lysates with an antibody specific for the RASSF1A isoform coprecipitated endogenous EWS (Fig. 1C). We also identified the EWS family member FUS/TLS and a previously described RASSF1A-interacting protein, MST2, in the RASSF1A immunoprecipitates (13). Our previous studies had indicated that loss of RASSF1A induced an accumulation of cyclin D1 protein levels without a detectable change in cyclin D1 mRNA levels. Further observations found that neither cyclin D1 transcription nor protein degradation rates are altered in cells overexpressing RASSF1A, suggesting that cyclin D1 translation rates may be changing (22). While little is known about the function of wild-type EWS, there are a number of RNA binding motifs, indicating that EWS may be involved in translation. The identification of EWS in the polysome fraction of HEK293 cells further supports a role for EWS in active translation (12). Cyclin D1 has a complex promoter, and multiple regulatory elements have been implicated in its translational regulation. Given the role of RASSF1A in the regulation of cyclin D1 accumulation, we examined the contribution of EWS to this phenotype. We first compared cyclin D1 protein accumulation upon siRNA-mediated RASSF1A depletion with or without codepletion of EWS (Fig. 2A to C). As expected, cyclin D1 levels were elevated in cells depleted of RASSF1A alone. However, in either HeLa or MCF10A cells, codepletion of EWS and RASSF1A resulted in levels of cyclin D1 similar to those of the control, suggesting that EWS plays a positive role in cyclin D1 protein accumulation. To test this directly, HeLa cells depleted of RASSF1A and/or EWS were synchronized at G0 by overnight serum starvation, followed by a 6- to 8-h exposure to FBS. As shown in Fig. 2D, EWS depletion significantly impaired serum-induced cyclin D1 accumulation relative to the control. Previous reports have suggested that EWS can positively regulate c-fos transcription. As c-fos is an immediate-early gene product that can positively regulate cyclin D1 transcription, this could account for the effect of EWS depletion on cyclin D1 accumulation. However, we saw no effect of EWS depletion on c-fos expression following 45 min of serum stimulation (data not shown). To determine if the impact of EWS depletion on cyclin D1 was significant enough to affect cell cycle progression, we analyzed cells for BrdU incorporation. HeLa cell proliferation was only slightly affected by EWS knockdown, probably because HeLa cell cycle progression is independent of cyclin D1 regulation, as a consequence of Rb inactivation (Fig. 2E). Similarly, the A549 non-small-cell lung cancer cell line, which lacks RASSF1A and has bypassed Rb checkpoint control through p16 inactivation, was insensitive to EWS depletion. In contrast, proliferation in nontumorigenic MCF10A cells, which have an intact Rb checkpoint, is inhibited upon EWS depletion (Fig. 2D and E). This result suggests that in established tumor cell lines, which presumably have acquired multiple genetic alterations (including the loss of p16), EWS function is uncoupled from cell cycle progression (Fig. 2E). These phenotypes were also validated by a second, independent EWS siRNA (data not shown). Thus, it appears that EWS is a positive regulator of cyclin D1, perhaps at the level of translation, and is required for RASSF1A to modulate cyclin D1 accumulation.
FIG. 1.

RASSF1A interacts with EWS. (A) Schematic of RASSF1A protein. The region (amino acids 1 to 210) used in a yeast two-hybrid screen for interacting proteins is indicated. (B) 293 cells were transfected with Myc-tagged RASSF1A and the green fluorescent protein (GFP)-tagged EWS zinc finger domain (amino acids 175 to 210). After 48 h, cells were lysed and α-myc-coupled agarose was used to immunoprecipitate RASSF1A. Lysates and immunoprecipitates were resolved by SDS-PAGE and immunoblotted with myc and green fluorescent protein antibodies as indicated. (C) HeLa cell lysates were immunoprecipitated with a RASSF1A-specific monoclonal antibody, G5. Lysates and immunoprecipitates were resolved by SDS-PAGE, transferred to polyvinylidene difluoride membrane, and immunoblotted with a polyclonal RASSF1A-specific antibody, 4169, and an EWS antibody as indicated. WB, Western blot; I.P., immunoprecipitate; WCL, whole-cell lysate; IgG, immunoglobulin G.
FIG. 2.

EWS regulates cyclin D1 expression. (A) HeLa cells were transfected with the indicated siRNAs in the presence of serum. Seventy-two hours later, lysates were resolved by SDS-PAGE and immunoblotted for indicated proteins. (B) Cyclin D1 mRNA concentrations from cells treated as described for panel A were evaluated by quantitative reverse transcription-PCR. Error bars represent the standard deviation from the mean of three biological replicates. (C) MCF10A cells were transfected with the indicated siRNAs. Seventy-two hours later, whole-cell lysates were immunoblotted to visualize the indicated proteins. ERK1/2 was included as a loading control (Ctrl). (D) HeLa cells were transfected as described for panel A, with the exception that cells were incubated in serum-free medium for 24 h prior to lysate collection as indicated. ERK1/2 was included as a loading control. (E) The indicated cell lines were transfected as described for panel A. At 48 h following transfection, BrdU was added to the medium for an additional 24 h. BrdU incorporation was detected with an anti-BrdU antibody, nuclei were counterstained with 4′,6′-diamidino-2-phenylindole (DAPI), and the percentage of BrdU-positive cells was calculated by microscopic observation. At least 100 cells were analyzed for each condition in each experiment. Values were normalized to the BrdU incorporation frequency observed in untransfected (NT) cells, which was arbitrarily set to 100%. Error bars indicate the standard deviation from the mean of three biological replicates. Representative micrographs are shown for MCF10A cells at the bottom left. exp., expressed.
RASSF1A is required for cell cycle progression.
Surprisingly, BrdU labeling also demonstrated that depletion of RASSF1A from HeLa, MCF10A, or normal human bronchial epithelial cells impaired proliferation (Fig. 2E and data not shown). This observation is an apparent paradox, given that RASSF1A is a candidate tumor suppressor that limits cyclin D1 accumulation, and it suggests that RASSF1A may have multiple roles in the regulation of cell cycle progression.
To determine where in the cell cycle RASSF1A-depleted cells were arrested, we treated control siRNA-transfected and RASSF1A siRNA-transfected cells with nocodazole for 18 h. Nocodazole interferes with microtubule dynamics and through activation of the spindle assembly checkpoint. FACS analysis for DNA content indicated that while control transfected cells treated with nocodazole accumulated in G2/M as expected, cells depleted of RASSF1A and treated with nocodazole retained a significant population of cells in G1 (Fig. 3A). Similar results were obtained with MCF10A cells (data not shown). This analysis suggests that RASSF1A positively contributes to G1/S phase progression.
FIG. 3.

siRNA of RASSF1A induces G1 arrest. (A) HeLa cells were transfected with the indicated siRNAs. At 48 h posttransfection, 100 ng/ml nocodazole was added to the medium and the cells were incubated for an additional 18 to 20 h. Cells were then trypsinized, fixed, and stained with propidium iodide. FACS profiles of propidium iodide intensity are shown. M1 indicates 2N, M2 indicates >2N and <4N, and M3 indicates 4N DNA content. (B) Whole-cell lysates from cells treated as described for panel A were immunoblotted to visualize the indicated proteins. ERK1/2 was included as a loading control (Ctrl).
Progression through G1 requires accumulation of cyclin D1 to inactivate Rb, followed by activation of cyclin E and cyclin A. Despite an accumulation of cyclin D1 in RASSF1A-depleted cells, we found that cyclin A was dramatically downregulated and cyclin E levels remained unchanged (Fig. 3B and data not shown). In addition, cyclin B1, which is required for G2 progression, was also reduced in RASSF1A-depleted cells (Fig. 3B). We did not see an impact of EWS depletion on cyclin A or B accumulation (data not shown). Given that cyclins A and B are both substrates of APC/C while cyclin E is not, we examined an additional substrate of APC/C, SKP2 (19). This APC/C substrate was also dramatically reduced in RASSF1A-depleted cells compared to controls, and a SKP2 client protein, the cyclin-dependent kinase inhibitor p27, was concomitantly elevated (Fig. 3B and 4A). Given that APC/C inhibition is required at the G1/S transition to allow for stabilization of proteins required for S phase progression (20), RASSF1A depletion may result in unrestrained activation of APC/C at the G1/S transition, therefore arresting cells in late G1.
FIG. 4.

CDH1 depletion rescues G1 arrest induced by siRNA of RASSF1A. (A) HeLa cells were transfected with the indicated siRNAs, and whole-cell lysates were immunoblotted to visualize the indicated proteins. (B) Cells treated as described for panel A were analyzed for BrdU incorporation as described for Fig. 2D. (C) HeLa cells were transfected with the indicated siRNAs. At 48 h posttransfection, 100 ng/ml nocodazole was added to the medium and the cells were incubated for an additional 18 to 20 h. FACS analysis was performed as described for Fig. 3B. Population distributions are shown relative to DNA content. Results are representative of three independent experiments. NT, not transfected; Ctrl, control.
Inactivation of APCCDH1 restores proliferation in RASSF1A-depleted cells.
Inhibition of APC/C can occur through the cyclin A/CDK2 phosphorylation-induced inhibition of the APC coactivator CDH1 or by direct binding of the APC inhibitor Emi1 (19). To determine if unrestrained APC/C activity is responsible for the decreased cyclin A and Skp2 protein levels in RASSF1A-depleted cells, we inactivated APC by codepleting CDH1. As shown in Fig. 4, codepletion of CDH1 and RASSF1A restored cyclin A and Skp2 levels, reversed p27 accumulation, and restored BrdU incorporation, suggesting that RASSF1A contributes to APCCDH1 inactivation during the G1/S transition.
RASSF1A mediates cell cycle regulation through βTRCP.
Our observation that APC/C inactivation could reengage cell cycle progression in RASSF1A-depleted cells prompted us to examine the mechanism by which RASSF1A affects APC/C activity. Emi1 is an APC/C inhibitor that has been found to function at the G1/S transition and during G2 (21). Consistent with the hyperactive APC/C phenotype, we saw a significant decrease in Emi1 expression in RASSF1A knockdown cells (Fig. 5A). Transient overexpression of RASSF1A was also sufficient to induce a modest but reproducible increase in Emi1 accumulation (Fig. 5B). Surprisingly, we found that direct siRNA-mediated depletion of Emi1 resulted in apparent cell cycle arrest in S phase together with massive endoreduplication, a phenotype that was not altered by codepletion of RASSF1A (Fig. 5C and D). A very recent observation has demonstrated that Emi1 is required to allow cyclin A accumulation and S phase exit (16). To test the possibility that RASSF1A-dependent accumulation of Emi1 is required for the G1/S phase transition, we examined the consequence of depleting βTRCP, the F box protein required for SCF-mediated ubiquitination of Emi1 (17). We found that depletion of βTRCP1, -2, or both was sufficient to rescue Emi1 expression in the absence of RASSF1A expression (Fig. 6A and data not shown). Furthermore, siRNA of βTRCP1, -2, or both was sufficient to rescue cell cycle progression (Fig. 6B and data not shown). βTRCP knockdown was confirmed by qPCR (Fig. 6C). In combination with a recent report that RASSF1 proteins can bind directly to βTRCP, these observations indicate that RASSF1A plays a pivotal role in restricting βTRCP during the G1/S phase transition to allow APC/C inactivation by Emi1. In contrast to depletion of RASSF1A, depletion of EWS had no effect on Emi1 expression, suggesting that the βTRCP arm of RASSF1A regulation is distinct from effects on EWS function (Fig. 6D).
FIG. 5.

Emi1 mediates RASSF1A depletion-induced G1 arrest. (A) HeLa cells were transfected and immunoblotted for indicated proteins as described for Fig. 2A. (B) Cells were transfected with the indicated plasmids. At 48 h posttransfection, whole-cell lysates were immunoblotted for the indicated proteins. (C) BrdU incorporation was assayed as described for Fig. 2D. Representative micrographs are shown. (D) FACS analysis of HeLa cells transfected with the indicated siRNAs. Population distributions are shown relative to DNA content. Results are representative of three independent experiments. Ctrl, control; DAPI, 4′,6′-diamidino-2-phenylindole.
FIG. 6.

βTRCP siRNA restores Emi1 levels and rescues G1 arrest. (A) At 72 h posttransfection with the indicated siRNAs, HeLa cell lysates were immunoblotted to visualize the indicated proteins. (B) BrdU incorporation was assayed as described for Fig. 2D. (C) βTRCP1 and -2 mRNA concentrations were measured by qPCR from samples treated as described for panel A. (D) At 72 h posttransfection with the indicated siRNAs, HeLa cell lysates were immunoblotted to visualize the indicated proteins. Ctrl, control; DAPI, 4′,6′-diamidino-2-phenylindole.
DISCUSSION
RASSF1A gene inactivation has been established as a common event in many cancers. A number of studies have implicated RASSF1A function in the regulation of transcription, chromosome segregation, and apoptosis (3, 13, 22-24). However, a mechanistic account of the contribution of RASSF1A inactivation to cancer cell proliferation and survival has not been developed. Through a detailed loss-of-function analysis, we found that RASSF1A has both positive and negative inputs into cell cycle progression (Fig. 7). RASSF1A association with EWS restricts cyclin D1 accumulation, while interaction with βTRCP is necessary for the G1/S phase transition and cell cycle progression. These observations introduce an apparent paradox in which a tumor suppressor is both positively and negatively coupled to proliferation. This framework implies that the function of RASSF1A may be context dependent. In normal cells, this coupling may represent a tumor progression checkpoint that generates the necessity for multiple genetic lesions to release restraints on proliferation and survival. On the other hand, the loss of RASSF1A may result in a transient cell cycle arrest that provides a permissive environment for the accumulation of additional oncogenic insults that would otherwise engage an apoptotic response in cycling cells. Future studies that determine which genetic alterations occur after RASSF1A inactivation are required to delineate the order of specific genetic changes that must occur to cause tumorigenesis.
FIG. 7.
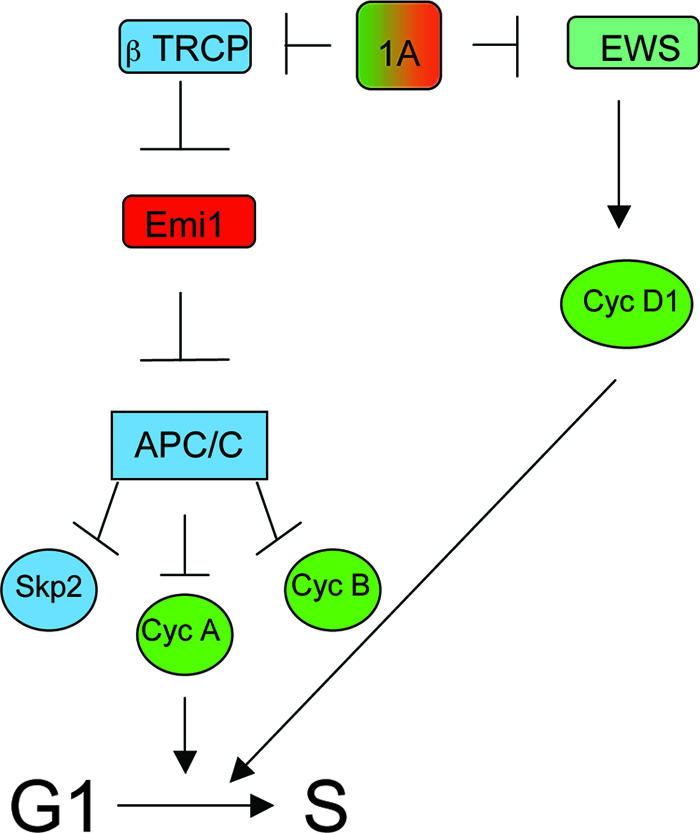
Model of dual opposing regulatory connections of RASSF1A to cell cycle progression. The presence of RASSF1A provides a regulatory input for the restriction of EWS-dependent cyclin (Cyc) D1 accumulation (antiproliferative) and for the restriction of βTRCP-dependent Emi1 inactivation (proproliferative). Loss of this regulatory input allows aberrant cyclin D1 accumulation but also restricts cyclin A accumulation as a consequence of aberrant APC/C activity during the G1/S phase transition.
Mechanistically, a molecular coupler between RASSF1A and cell cycle progression appears to be regulation of Emi1 accumulation in late G1 through restriction of βTRCP activity. While we found that loss of RASSF1A results in βTRCP-dependent Emi1 loss and G1 arrest, when Emi1 is silenced directly an S phase arrest occurs concomitant with endoreduplication (9, 16). This observation further supports the notion that Emi1 is important in S phase, not only to restrict APC/C as UBCH10 levels begin to increase but also to ensure proper regulation of DNA replication (16, 20). This highlights the critical role of Emi1 in multiple cell cycle control points and suggests that distinct cell cycle transitions require different stoichiometric ratios between APC/C and Emi1. These ratios are likely dependent on the availability of additional APC regulatory molecules such as UBCH10, cyclin A, and CDK1/2.
SCFβTRCP has been implicated as a regulator of Emi1 during prophase (17). Here, we show that inhibition of SCFβTRCP during G1 is an additional control point for Emi1 regulation. Recently, RASSF1A family member RASSF1C has been shown to interact directly with SCFβTRCP and regulate the degradation of β-catenin and IκB (11). Given the observation that RASSF1A family members, including RASSF1C, can function as a complex (14; our unpublished observations), SCFβTRCP likely represents the proximal molecular entry point for RASSF1A control of G1/S phase transitions, perhaps through deflection of the capacity of RASSF1C to activate SCFβTRCP (11). This scenario would predict that any protumorigenic consequences of unrestrained SCFβTRCP activity, bestowed upon the loss of the RASSF1A tumor suppressor, would require the continued expression of RASSF1C. In fact, RASSF1C is rarely, if ever, lost in human cancers despite frequent loss of heterozygosity at this locus coupled with selective inactivation of the RASSF1A splice form (7, 25).
Acknowledgments
This work was supported by the National Institutes of Health (CA71443), the Robert E. Welch Foundation (I-1414), and the Department of Defense (PC040054). J.D.M. is supported by CA71618 and SPORE P50CA70907. B.G. is supported by the Komen Foundation.
We thank Hongtao Yu and members of our laboratories for helpful discussions.
Footnotes
Published ahead of print on 17 March 2008.
REFERENCES
- 1.Agathanggelou, A., S. Honorio, D. P. Macartney, A. Martinez, A. Dallol, J. Rader, P. Fullwood, A. Chauhan, R. Walker, J. A. Shaw, S. Hosoe, M. I. Lerman, J. D. Minna, E. R. Maher, and F. Latif. 2001. Methylation associated inactivation of RASSF1A from region 3p21.3 in lung, breast and ovarian tumours. Oncogene 201509-1518. [DOI] [PubMed] [Google Scholar]
- 2.Arvand, A., and C. T. Denny. 2001. Biology of EWS/ETS fusions in Ewing's family tumors. Oncogene 205747-5754. [DOI] [PubMed] [Google Scholar]
- 3.Baksh, S., S. Tommasi, S. Fenton, V. C. Yu, L. M. Martins, G. P. Pfeifer, F. Latif, J. Downward, and B. G. Neel. 2005. The tumor suppressor RASSF1A and MAP-1 link death receptor signaling to Bax conformational change and cell death. Mol. Cell 18637-650. [DOI] [PubMed] [Google Scholar]
- 4.Burbee, D. G., E. Forgacs, S. Zochbauer-Muller, L. Shivakumar, K. Fong, B. Gao, D. Randle, M. Kondo, A. Virmani, S. Bader, Y. Sekido, F. Latif, S. Milchgrub, S. Toyooka, A. F. Gazdar, M. I. Lerman, E. Zabarovsky, M. White, and J. D. Minna. 2001. Epigenetic inactivation of RASSF1A in lung and breast cancers and malignant phenotype suppression. J. Natl. Cancer Inst. 93691-699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cohen, Y., G. Singer, O. Lavie, S. M. Dong, U. Beller, and D. Sidransky. 2003. The RASSF1A tumor suppressor gene is commonly inactivated in adenocarcinoma of the uterine cervix. Clin. Cancer Res. 92981-2984. [PubMed] [Google Scholar]
- 6.Dammann, R., C. Li, J. H. Yoon, P. L. Chin, S. Bates, and G. P. Pfeifer. 2000. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat. Genet. 25315-319. [DOI] [PubMed] [Google Scholar]
- 7.Dammann, R., U. Schagdarsurengin, C. Seidel, M. Strunnikova, M. Rastetter, K. Baier, and G. P. Pfeifer. 2005. The tumor suppressor RASSF1A in human carcinogenesis: an update. Histol. Histopathol. 20645-663. [DOI] [PubMed] [Google Scholar]
- 8.Dammann, R., G. Yang, and G. P. Pfeifer. 2001. Hypermethylation of the cpG island of Ras association domain family 1A (RASSF1A), a putative tumor suppressor gene from the 3p21.3 locus, occurs in a large percentage of human breast cancers. Cancer Res. 613105-3109. [PubMed] [Google Scholar]
- 9.Di Fiore, B., and J. Pines. 2007. Emi1 is needed to couple DNA replication with mitosis but does not regulate activation of the mitotic APC/C. J. Cell Biol. 177425-437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dreijerink, K., E. Braga, I. Kuzmin, L. Geil, F. M. Duh, D. Angeloni, B. Zbar, M. I. Lerman, E. J. Stanbridge, J. D. Minna, A. Protopopov, J. Li, V. Kashuba, G. Klein, and E. R. Zabarovsky. 2001. The candidate tumor suppressor gene, RASSF1A, from human chromosome 3p21.3 is involved in kidney tumorigenesis. Proc. Natl. Acad. Sci. USA 987504-7509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Estrabaud, E., I. Lassot, G. Blot, E. Le Rouzic, V. Tanchou, E. Quemeneur, L. Daviet, F. Margottin-Goguet, and R. Benarous. 2007. RASSF1C, an isoform of the tumor suppressor RASSF1A, promotes the accumulation of β-catenin by interacting with βTrCP. Cancer Res. 671054-1061. [DOI] [PubMed] [Google Scholar]
- 12.Felsch, J. S., W. S. Lane, and E. G. Peralta. 1999. Tyrosine kinase Pyk2 mediates G-protein-coupled receptor regulation of the Ewing sarcoma RNA-binding protein EWS. Curr. Biol. 9485-488. [DOI] [PubMed] [Google Scholar]
- 13.Guo, C., S. Tommasi, L. Liu, J. K. Yee, R. Dammann, and G. P. Pfeifer. 2007. RASSF1A is part of a complex similar to the Drosophila Hippo/Salvador/Lats tumor-suppressor network. Curr. Biol. 17700-705. [DOI] [PubMed] [Google Scholar]
- 14.Khokhlatchev, A., S. Rabizadeh, R. Xavier, M. Nedwidek, T. Chen, X. F. Zhang, B. Seed, and J. Avruch. 2002. Identification of a novel Ras-regulated proapoptotic pathway. Curr. Biol. 12253-265. [DOI] [PubMed] [Google Scholar]
- 15.Kuzmin, I., L. Liu, R. Dammann, L. Geil, E. J. Stanbridge, S. P. Wilczynski, M. I. Lerman, and G. P. Pfeifer. 2003. Inactivation of RAS association domain family 1A gene in cervical carcinomas and the role of human papillomavirus infection. Cancer Res. 631888-1893. [PubMed] [Google Scholar]
- 16.Machida, Y. J., and A. Dutta. 2007. The APC/C inhibitor, Emi1, is essential for prevention of rereplication. Genes Dev. 21184-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Margottin-Goguet, F., J. Y. Hsu, A. Loktev, H. M. Hsieh, J. D. Reimann, and P. K. Jackson. 2003. Prophase destruction of Emi1 by the SCF(βTrCP/Slimb) ubiquitin ligase activates the anaphase promoting complex to allow progression beyond prometaphase. Dev. Cell 4813-826. [DOI] [PubMed] [Google Scholar]
- 18.Matheny, S. A., C. Chen, R. L. Kortum, G. L. Razidlo, R. E. Lewis, and M. A. White. 2004. Ras regulates assembly of mitogenic signalling complexes through the effector protein IMP. Nature 427256-260. [DOI] [PubMed] [Google Scholar]
- 19.Peters, J. M. 2006. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat. Rev. Mol. Cell Biol. 7644-656. [DOI] [PubMed] [Google Scholar]
- 20.Rape, M., and M. W. Kirschner. 2004. Autonomous regulation of the anaphase-promoting complex couples mitosis to S-phase entry. Nature 432588-595. [DOI] [PubMed] [Google Scholar]
- 21.Reimann, J. D., E. Freed, J. Y. Hsu, E. R. Kramer, J. M. Peters, and P. K. Jackson. 2001. Emi1 is a mitotic regulator that interacts with Cdc20 and inhibits the anaphase promoting complex. Cell 105645-655. [DOI] [PubMed] [Google Scholar]
- 22.Shivakumar, L., J. Minna, T. Sakamaki, R. Pestell, and M. A. White. 2002. The RASSF1A tumor suppressor blocks cell cycle progression and inhibits cyclin D1 accumulation. Mol. Cell. Biol. 224309-4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song, M. S., J. S. Chang, S. J. Song, T. H. Yang, H. Lee, and D. S. Lim. 2005. The centrosomal protein RAS association domain family protein 1A (RASSF1A)-binding protein 1 regulates mitotic progression by recruiting RASSF1A to spindle poles. J. Biol. Chem. 2803920-3927. [DOI] [PubMed] [Google Scholar]
- 24.Song, M. S., S. J. Song, N. G. Ayad, J. S. Chang, J. H. Lee, H. K. Hong, H. Lee, N. Choi, J. Kim, H. Kim, J. W. Kim, E. J. Choi, M. W. Kirschner, and D. S. Lim. 2004. The tumour suppressor RASSF1A regulates mitosis by inhibiting the APC-Cdc20 complex. Nat. Cell Biol. 6129-137. [DOI] [PubMed] [Google Scholar]
- 25.Tommasi, S., R. Dammann, Z. Zhang, Y. Wang, L. Liu, W. M. Tsark, S. P. Wilczynski, J. Li, M. You, and G. P. Pfeifer. 2005. Tumor susceptibility of Rassf1a knockout mice. Cancer Res. 6592-98. [PubMed] [Google Scholar]
- 26.Vojtek, A. B., and S. M. Hollenberg. 1995. Ras-Raf interaction: two-hybrid analysis. Methods Enzymol. 255331-342. [DOI] [PubMed] [Google Scholar]