

Abstract
Unlike responses to acute stressful events that are protective and adaptive in nature, chronic stress elicits neurochemical, neuroanatomical and cellular changes that may have deleterious consequences upon higher brain functioning. For example, while exposure to acute stress facilitates memory formation and consolidation, chronic stress or chronic exposure to stress levels of glucocorticoids impairs cognitive performance. Chronic stress or glucocorticoid exposure, as well as impairments in hypothalamic-pituitary-adrenal (HPA) axis function are proposed to participate in the etiology and progression of neurological disorders such as depressive illness, anxiety disorders and post-traumatic stress disorder (PTSD). HPA axis dysfunction, impaired stress responses and elevated basal levels of Glucocorticoids are also hallmark features of experimental models of type 1 and type 2 diabetes, as well as diabetic subjects in poor glycemic control. Such results suggest that stress and glucocorticoids contribute to the neurological complications observed in diabetes patients. Interestingly, many of the hyperglycemia mediated changes in the brain are similar to those observed in depressive illness patients and in experimental models of chronic stress. Such results suggest that common mechanisms may be involved in the development of the neurological complications associated with Anxiety, Depressive illness and Diabetes: the As and Ds of stress. The aim of the current review will be to discuss the mechanisms through which limbic structures such as the hippocampus and amygdala respond and adapt to the deleterious consequences of chronic stress and hyperglycemia.
Keywords: diabetes, glucocorticoid, glutamate, insulin, depressive illness, hippocampus
1. Introduction
Acute exposure to stress leads to the activation of the hypothalamic-pituitary-adrenal (HPA) axis, leading to the release of epinephrine and glucocorticoids from the adrenal gland (Jacobson and Sapolsky, 1991). Once released, epinephrine and glucocorticoids may produce a wide variety of effects in the periphery ranging from increases in cardiovascular activities, decreases in gastrointestinal and immune function, and increasing energy mobilization. Stress hormones also play critical functional roles in the central nervous system (CNS), including the facilitation and consolidation of strong emotional memories that involves glucocorticoid binding and activation of glucocorticoid receptors in limbic regions such as the hippocampus and amygdala (Reul and de Kloet, 1985). Unlike these physiological responses to acute stressful events, chronic stress, exposure to stress levels of glucocorticoids and HPA axis dysfunction are critical mediators in disease states in the periphery and the CNS. For example, chronic stress is proposed to contribute to the etiology and progression of cardiovascular disease, hypertension, cancer metastasis, immune impairments, among other peripheral disorders. Impairments in HPA axis function and elevated basal glucocorticoids are also implicated in the peripheral complications of type 1 and type 2 diabetes. In the CNS, the consequences of chronic stress or glucocorticoid exposure have been extensively studied and are proposed to contribute to the etiology and progression of Cushing’s syndrome, major depressive disorder and post-traumatic stress disorder (PTSD) (Reagan and McEwen, 1997). Interestingly, many of the neurological consequences observed in experimental models of type 1 and type 2 diabetes are strikingly similar those observed following chronic stress, suggesting that glucocorticoids may be common mechanistic mediators in the pathophysiological consequences of diabetes and stress-related disorders.
1.1. Structural and functional deficits in major depression
One of the hallmark clinical features of stress-related disorders is atrophy of the hippocampus. Functional imaging studies have determined that hippocampal formation volume is decreased in patients with Cushing’s syndrome (Starkman et al., 1992), PTSD (Bremner et al., 1995; Gurvits et al., 1996), and in aging populations (Lupien et al., 1998). The volume of the hippocampus is also decreased in depressive illness patients. Major depressive illness is one of the most common psychiatric disorders, affecting an estimated 12–15% of the general population (Kessler et al., 1994). The symptoms of depression include alterations in mood and perception, as well as physiological changes such as loss of appetite, changes in body weight and disruption in sleep patterns. The aim of antidepressant treatments is the improvement of these depressive symptoms; it should also address all the potential complications resulting from major depression including structural changes in the central nervous system (CNS). For example, volumetric magnetic resonance imaging (MRI) studies of depressive illness patients have revealed decreases in left hippocampal volumes (Vythilingam et al., 2002; Frodl et al., 2002b; Bremner et al., 2000; Mervaala et al., 2000), right hippocampal volumes (Steffens et al., 2000) and total hippocampal volumes (MacQueen et al., 2003; Sheline et al., 1999). These studies have also found that depressive illness patients with decreases in hippocampal formation volume exhibit deficits in hippocampal-dependent measures of cognitive function.
More recent analyses have suggested an important role for the amygdala in depressive illness (McEwen, 2003), including imaging studies that have revealed that amygdala volumes may be increased (Bremner et al, 2000; Frodl et al., 2002a) or decreased (Sheline et al., 1998; Sheline et al, 1999; von Gunten et al., 2000) in major depression patients. While these disparities may be related to illness duration and/or therapeutic interventions (Campbell et al., 2004), the results demonstrate that the amygdala is also a site for neuroanatomical alterations in depressive illness and suggest that the amygdala may exhibit time and treatment dependent changes. Since the hippocampus and the amygdala are major sites of glucocorticoid action in the CNS, these results lead to the suggestion that stress may be responsible for neuroanatomical alterations observed in recurrent depressive illness patients (Sheline et al., 1996).
1.2. Chronic stress as an experimental model of depressive illness: advantages and limitations
Clinical studies provide invaluable information regarding structural and functional integrity of the hippocampus and amygdala in pathological settings. Unfortunately, these studies are limited in their ability to identify the underlying molecular, neurochemical and neuropharmacological deficits that contribute to structural and functional deficits in patients with stress related disorders such as depressive illness. For these reasons, many investigators rely upon stress paradigms as experimental models of depressive illness. While there are limitations associated with experimental stress paradigms as preclinical models of mood disorders (Nestler et al., 2002), such studies provide important information regarding stress related effects upon brain neurochemistry and the mechanisms of antidepressant drugs. Animals subjected to stress exhibit many similarities to those observed in depressive illness patients, including changes in body weight, disrupted sleep cycles, impairments in HPA axis function, as well as morphological changes in the hippocampus and amygdala. The validity of chronic stress paradigms as a preclinical model of depression gains support from studies that demonstrated that several clinically-relevant treatments, including tianeptine, clomipramine and lithium effectively inhibit stress-mediated changes in the hippocampus (Kole et al., 2002; Czeh et al., 2001; Lucassen et al., 2004; Wood et al., 2004; Reagan et al., 2004; Watanabe et al., 1992; Magariños et al., 1999; McEwen et al., 2002a; van der Hart et al., 2002; Campbell et al., 2007) and the amygdala (Vouimba et al., 2006; Reznikov et al., 2007; McEwen and Chattarji, 2004; Reagan et al., 2007).
1.3. Regulation of the glutamatergic system in chronic stress models: relation to limbic system plasticity
The role of glucocorticoids in the stress-related disorders might involve steroid modulation of neurotransmitter activity, including the serotonergic, GABAergic and glutamatergic systems. The dynamic interactions of these neurotransmitter systems have previously been established in stress effects upon HPA axis function and activity (Herman and Cullinan, 1997; Van de Kar and Blair, 1999). Similarly, these neurotransmitter systems are known to participate in stress-induced changes in the hippocampus [For reviews, see (McEwen, 1997; Reagan and McEwen, 1997)] More specifically, an intimate relationship exists between the glutamatergic system and the neurological consequences of experimental models of stress (McEwen and Sapolsky, 1995; Reagan and McEwen, 1997). For example, stress affects the electrophysiological properties of ionotropic glutamate receptors in the hippocampus (Kole et al, 2002) and stress-induced atrophy of CA3 pyramidal neurons is inhibited by NMDA receptor antagonists (Magariños and McEwen, 1995). Acute stress also elicits increases in extracellular glutamate levels that quickly return to baseline in the rat hippocampus and amygdala, as measured by in vivo microdialysis (Reznikov et al, 2007; Lowy et al., 1993; Bagley and Moghaddam, 1997). Conversely, acute stress-induced increases in extracellular glutamate efflux remain elevated in the hippocampus of rats subjected to prior chronic stress (Yamamoto and Reagan, 2006). This dysregulation of glutamatergic tone that results in sustained increases in extracellular glutamate levels may be the stimulus for stress mediated morphological changes in the hippocampus, as well as increases in glial glutamate transporter expression in the CA3 region of the hippocampus (Reagan et al, 2004). As noted above, tianeptine effectively blocks these glutamate-dependent changes in the hippocampus and amygdala, thereby providing a potential mechanism of action through which this antidepressant inhibits stress-induced effects in the hippocampus and amygdala.
1.4. The glutamate system in depressive illness
Clinical studies also support a role for the glutamatergic system in the pathology of major depressive illness. For instance, plasma and cerebrospinal fluid (CSF) glutamate levels and glutamate/glutamine ratios are modulated in affective disorders (Levine et al., 2000; Kim et al., 1982; Altamura et al., 1995; Altamura et al., 1993). More recent clinical studies support these findings and identified specific neuronal populations where these changes in glutamatergic neurotransmission may take place. For example, proton magnetic resonance spectroscopic analyses have identified decreases in glutamate levels the anterior cingulate cortex of depressive illness patients (Mirza et al., 2004; Auer et al., 2000). Conversely, occipital cortex glutamate levels were significantly increased in depressed subjects (Sanacora et al., 2004). Decreased GABA levels were also observed in these subjects, suggesting that an imbalance between excitatory and inhibitory tone in depressive illness patients (Sanacora et al., 2000). Decreases in glial cell densities observed in depressive illness patients (Rajkowska et al., 1999; Hamidi et al., 2004; Cotter et al., 2001; Ongur et al., 1998) may result in decreased glial glutamate transporter expression, thereby reducing the capacity to regulate synaptic concentrations of glutamate. Indeed, a recent microarray study suggested that glial glutamate transporter expression is decreased in the anterior cingulate cortex and dorsolateral prefrontal cortex of depressive illness patients (Choudary et al., 2005). When combined with results from experimental models of stress, these clinical observations support the hypothesis that modulation of glutamatergic neurochemistry participates in the pathophysiology of major depressive disorder.
2. Neurological complications in diabetes phenotypes: adaptive plasticity or increased vulnerability?
It is interesting to note that many of the neurological consequences observed following chronic stress are strikingly similar to the observed in experimental models of type 1 and type 2 diabetes (Reagan, 2002; Gispen and Biessels, 2000). The accumulated data from experimental models of diabetes suggest that a consequence of chronic hyperglycemia is accelerated brain aging, a hypothesis originally proposed by Gispen, Biessels and co-workers (Kamal et al., 2000). For example, streptozotocin diabetic rats, an experimental model of type 1 diabetes, rapidly exhibit dendritic remodeling in the CA3 region of the rat hippocampus (Magariños and McEwen, 2000). Subsequent studies revealed that dendritic changes may be more widespread in the hippocampus of streptozotocin rats and may also include synaptic reorganization (Grillo et al., 2005). These neuroanatomical changes may represent the initiation of irreversible neuronal damage in the hippocampus of diabetic subjects since neuronal apoptosis has been observed in the hippocampus of type 1 diabetic rats (Li et al., 2002b). However, it is important to note that neuronal loss, decreases in neuronal density and increases in apoptosis are only observed following several months of uncontrolled hyperglycemia, suggesting that short-term exposure to hyperglycemia/insulinopenia is associated with reversible, not irreversible neuronal changes. Indeed, our previous studies illustrated that diabetes-induced morphological changes in the CA3 region of the rat hippocampus are reversed with insulin replacement (Magariños et al., 2001) and are not associated with irreversible neuronal loss (Reagan, 2002; Grillo et al, 2005). Nonetheless, in view the consequences of hippocampal atrophy associated with other clinical disorders (McEwen, 1997), diabetes-induced neuronal atrophy and redistribution of synaptic proteins would undoubtedly have functional consequences and may also increase neuronal vulnerability to subsequent insults or pathologies (Brands et al., 2004; Reagan, 2002)
2.1. Glucocorticoids elicit insulin resistance in the hippocampus
Impairments in HPA axis function and elevated basal glucocorticoids are implicated in the peripheral complications of type 1 and type 2 diabetes. Diabetic patients in poor glycemic control exhibit elevated plasma cortisol levels (Couch, 1992); neuroendocrine dysfunction, including greater sensitivity to both acute and chronic stress is also observed in experimental models of diabetes (Leedom et al., 1987; Scribner et al., 1991; Oster et al., 1988; Magariños and McEwen, 2000; Winocur et al., 2005). One of the major metabolic effects of chronic increases in glucocorticoid levels is insulin resistance, a condition characterized by increases in serum glucose levels which are not effectively regulated by the elevations in serum insulin (Amatruda et al., 1985; McMahon et al., 1988). Under physiological conditions, increases in plasma insulin levels stimulate the translocation of the insulin sensitive glucose transporter GLUT4 from intracellular stores to the plasma membrane in organs such as muscle and adipose tissue, hereby increasing glucose uptake and utilization [For review see (Saltiel and Pessin, 2002)]. Elevated levels of circulating glucocorticoid lead to impairment in the functionality of GLUT4, an effect that may contribute to peripheral insulin resistance (Garvey et al., 1989; Dimitriadis et al., 1997). Based upon these peripheral findings, we have recently analyzed the effects of short-term corticosterone treatment on insulin receptor expression and signaling in the rat hippocampus (Piroli et al., 2007a). Our results indicate that insulin receptor expression was unchanged in corticosterone-treated rats compared to vehicle-treated rats. However, insulin stimulated phosphorylation of insulin receptor, as well as total Akt levels, were reduced in corticosterone-treated rats when compared to vehicle-treated controls. Furthermore, total expression of GLUT4 and insulin-stimulated translocation of GLUT4 to the plasma membrane was reduced by corticosterone administration. These results demonstrate short-term corticosterone administration impairs insulin signaling in rat hippocampus, thereby providing a potential mechanism thorough which glucocorticoids modulate hippocampal glucose utilization in rats (Kadekaro et al., 1988; Landgraf et al., 1978) and humans (de Leon et al., 1997). Moreover, hippocampal insulin resistance elicited by corticosterone may contribute to the deleterious consequences of hypercortisolinemic/hyperglycemic states observed in type 2 diabetes.
2.2. Diabetes mediated disruption of pro-oxidant and anti-oxidant cascades
Hyperglycemia-induced shifts in the balance of pro-oxidant and anti-oxidant cascades may contribute to increased neuronal vulnerability in diabetic subjects. Oxidative stress, lipid peroxidation and increased production of reactive oxygen species reduce the activity of a variety of proteins that are critical to neuronal homeostasis (Mattson, 1998). Oxidative stress and reactive oxygen species are increased in diabetes (Baynes, 1991; Wolff, 1993) and are proposed to contribute to the development of diabetic encephalopathy (Gispen and Biessels, 2000). In this regard, superoxide production is increased in the serum of type 1 diabetic patients, increases that are reduced with improved glycemic control (Ceriello et al., 1991). Lipid peroxidation products such as 4-hydroxynonenal (HNE) and malondialdehyde (MDA) are increased in the brains of type 2 diabetic mice, and more specifically in the hippocampus of streptozotocin rats (Grillo et al., 2003; Reagan et al., 2000; Reagan, 2002; Tuzcu and Baydas, 2006). HNE has been shown to mediate β-amyloid toxicity (Mark et al., 1997) and oxidative stress-induced apoptosis in hippocampal primary cultures (Kruman et al., 1997). The glial glutamate transporter GLT-1 is a protein target of HNE protein conjugation, which may contribute to HNE mediated decreases in glutamate transport in primary astrocytic cultures (Blanc et al., 1998) and in rat cortical synaptosomal fractions (Keller et al., 1997a). HNE treatment also impairs glucose uptake (Keller et al., 1997b) and our previous studies have identified the neuron-specific glucose transporter GLUT3 as a target of HNE protein conjugation in the hippocampus of diabetic rats (Reagan et al, 2000). Collectively, these results suggest that lipid peroxidation products such as HNE adversely affect neuronal metabolism and neurochemistry. Moreover, since oxidative stress and lipid peroxidation are hallmark features of Alzheimer’s disease pathology (Mattson, 1998), hyperglycemia-induced increases in oxidative stress provide another example of accelerated brain aging in diabetic subjects.
In addition to increases in pro-oxidant pathways, hyperglycemia also decreases ant-oxidant pathways in the diabetic brain. For example, glutathione levels (Tuzcu and Baydas, 2006), as well as the expression and activity of glyceraldehyde-3-phosphate dehydrogenase (Aragno et al., 2005), are reduced in the hippocampus of streptozotocin rats. Interestingly, anti-oxidant treatments such as melatonin, vitamin E (Tuzcu and Baydas, 2006) and dehydroepiandrosterone (DHEA) (Aragno et al., 2000; Aragno et al, 2005) reverse the imbalances in anti-oxidant/pro-oxidant ratios by increasing anti-oxidant expression and activity. In view of these observations, we examined the ability of DHEA to reduce diabetes/stress mediated increases in lipid peroxidation in the hippocampus. Diabetic rats subjected to chronic stress were provided access to normal rat chow or rat chow supplemented with 0.4% DHEA; control rats were provided control chow. With the exception of increased plasma levels of DHEA, diabetic rats provided the DHEA-supplemented chow exhibited similar plasma endocrine measures as diabetic rats provided standard chow (Table 1). Diabetic rats subjected to stress and provided standard chow exhibited the expected increases in lipid peroxidation in the Ammon’s Horn, increases that were attenuated in DHEA-treated rats. However, HNE radioimmunoreactive levels were reduced in CA1, CA2 and CA3 of Ammon’s Horn in diabetic rats subjected to chronic stress provided the DHEA-supplemented diet (Figure 1. Panel A). Similarly, DHEA attenuated diabetes/stress-induced increases in malondialdehyde (MDA) immunoreactive levels in CA1, CA2 and CA3 compared with diabetes/stress group provided normal chow (Figure 1, Panel B). These results suggest that anti-oxidant treatments provide neuroprotection against hyperglycemia-induced increases in oxidative stress. In addition to supplementation with anti-oxidants, the hippocampus itself may activate compensatory mechanisms in diabetic rats. In this regard, our previous studies revealed that superoxide dismutase (SOD) isoform expression increases during prolonged periods of hyperglycemia in streptozotocin diabetic rats, which may account for decreases in lipid peroxidation products observed in these rats (Grillo et al, 2003). As such, both endogenous and exogenous mechanisms may help to restore the appropriate balance between anti-oxidant and pro-oxidant activities in the diabetic brain.
Table 1.
Plasma endocrine analysis following DHEA administration
| Plasma endocrine measure | Control | STZ + Chow | STZ + DHEA |
|---|---|---|---|
| Glucose (mg/dl) | 124.2 ± 3.2 | 489.3 ± 20.6a | 444.3 ± 21.7a |
| Insulin (ng/ml) | 0.437 ± 0.16 | 0.034 ± 0.018a | 0.037 ± 0.03a |
| DHEA-SO4 (ug/dl) | 0.00 | 0.00 | 75.8 ± 19.5a/b |
| Corticosterone (ug/dl) | 3.39 ± 0.43 | 2.19 ± 0.42 | 3.89 ± 0.63 |
Rats were provided standard chow (control and STZ + Chow) or standard chow supplemented with 0.4% DHEA (STZ + DHEA). Fasting glucose levels were measured by glucose-trinder assay; insulin, DHEA-SO4 and corticosterone were measured by radioimmunoassay.
= significantly different from control;
= significantly different from STZ + Chow.
/# = p = 0.001.
Figure 1.
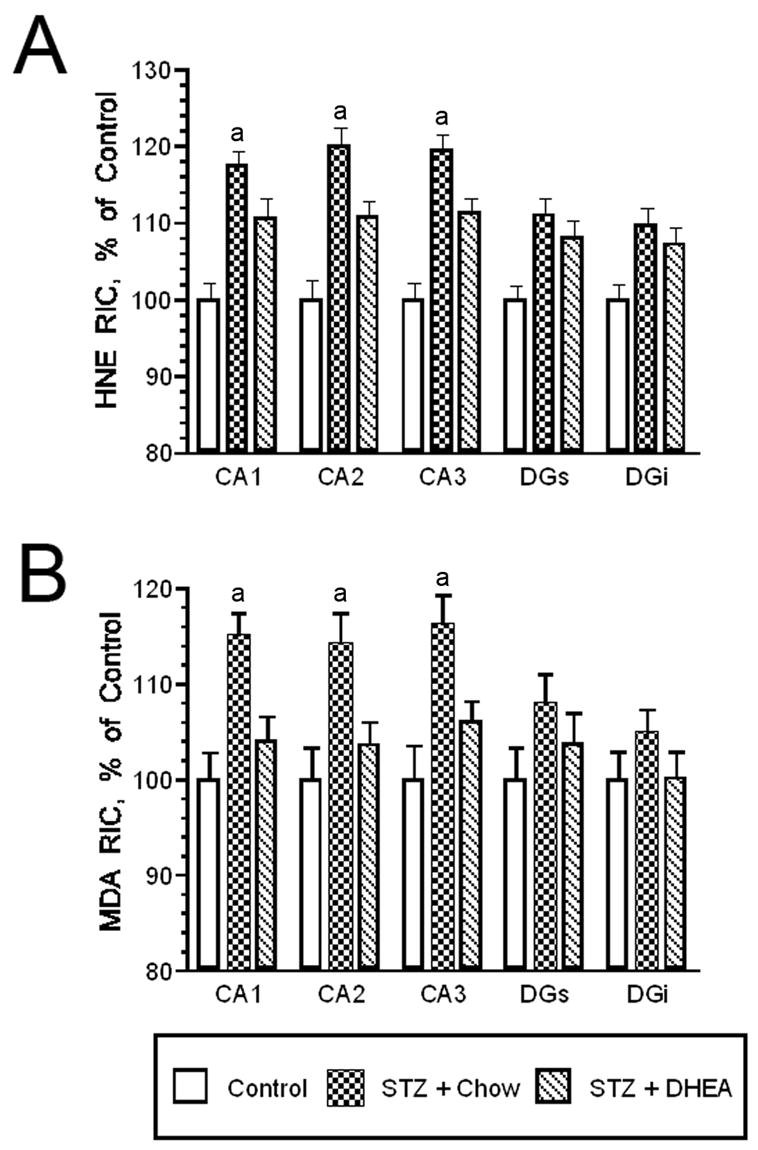
DHEA administration attenuates diabetes/stress mediated increases in oxidative stress. Streptozotocin diabetic rats subjected to chronic (i.e. 21 day) stress were provided access to normal chow or chow supplemented with 0.4% DHEA. Panel A. Radioimmunocytochemistry determined that diabetic rats subjected to stress provided normal chow exhibited the expected increases in HNE radioimmunolabeling the Ammon’s Horn of the hippocampus, increases that were significantly reduced in diabetic/stress rats provided the DHEA supplemented diet. Panel B. Similarly, the DHEA supplemented diet significantly reduced MDA radioimmunoreactive levels in Ammon’s Horn in diabetic rats subjected to chronic stress. See text for details. [a = P < 0.05]
2.3. Diabetes mediated changes in hippocampal synaptic plasticity
As seen in experimental models of chronic stress, neurochemical and cellular deficits in hippocampal synaptic plasticity have been identified as long-term consequences of hyperglycemia. For example, cell proliferation and/or neurogenesis is a form of synaptic plasticity that is observed in the dentate gyrus region of the hippocampus (Gould and Gross, 2002; Gage, 2002) and glucocorticoids have been shown to reduce the birth of new neurons in this region (Pham et al., 2003; Gould et al., 1992). Cell proliferation and neurogenesis are also reduced in the dentate gyrus in experimental models of type 1 diabetes (Kim et al., 2003; Beauquis et al., 2006; Saravia et al., 2006). Interestingly, diabetes-induced decreases in neurogenesis/cell proliferation were reversed by treatments that have previously been shown to increase cell proliferation in the dentate gyrus, including exercise (Kim et al, 2003), estrogen treatment (Saravia et al, 2006) and antidepressant treatment (Beauquis et al, 2006). Electrophysiological deficits have also been identified in experimental models of diabetes. For example, type 1 diabetic rodents exhibit impairments in hippocampal long-term potentiation and enhancement of long-term depression (Biessels et al., 1996; Kamal et al., 1999; Artola et al., 2005; Izumi et al., 2003; Valastro et al., 2002), electrophysiological deficits that are inhibited by insulin replacement. Electrophysiological studies in experimental models of type 2 diabetes have failed to reach a consensus, with some studies stating that type 2 animals exhibit deficits in long-term potentiation (Gerges et al., 2003; Li et al., 2002a), while others have failed to observe electrophysiological changes (Belanger et al., 2004). A potential explanation for these discrepancies is that the physiological characteristics of the type 2 diabetic animals used in these studies may be dissimilar; such considerations may also be particularly relevant to the equivocal findings from clinical studies (see below).
2.4. Glutamatergic activity in diabetes phenotypes
Alterations in glutamate neuropharmacology may contribute to these diabetes mediated deficits in hippocampal synaptic plasticity. For example, deficits in long-term potentiation are associated with increased expression of NMDA and AMPA receptors in the hippocampus of NOD mice, as determined by quantitative autoradiographic receptor binding studies (Valastro et al, 2002). Conversely, these same investigators reported that AMPA receptor binding activity is decreased in the hippocampus of streptozotocin-treated rats (Gagne et al., 1997), suggesting that hyperglycemia may mediate species-specific changes in glutamate receptor pharmacology. In support of this hypothesis, unlike the observations in NOD mice, NR2B mRNA and protein are decreased in the hippocampus of streptozotocin rats (Di Luca et al., 1999). Moreover, Ca+/CaM stimulated phosphorylation of hippocampal NR2A and NR2B subunits expressed in the post-synaptic density were reduced in streptozotocin rats. As described above, redistribution and reorganization of synaptic proteins such as post-synaptic density-95 may be indicative of ongoing synaptogenesis in the hippocampus of streptozotocin rats (Grillo et al, 2005) and thereby modulate glutamatergic tone. Collectively, these results suggest that transcriptional, translational and post-translational modification of glutamate receptors may adversely affect synaptic transmission and the electrophysiological properties of hippocampal neurons in diabetic rodents.
Other components of glutamatergic synapses may also be modulated by diabetes phenotypes. As noted above, glutamate transporters are regulated by stress and antidepressant treatment; glutamate transporter expression has also been examined in experimental models of diabetes. Glial glutamate transporter expression was not modulated in the hippocampus and cortex of streptozotocin rats compared to control rats, as determined by immunoblot analysis (Coleman et al., 2004). In agreement with these findings, we examined GLT-1 expression using radioimmunocytochemical approaches and determined that total GLT-1 expression is unchanged in the hippocampus of streptozotocin rats or diabetic rats subjected to acute stress. However, more region-specific analysis revealed that GLT-1 protein expression is significantly increased in the CA3 region of streptozotocin rats subjected to acute stress compared to non-stressed streptozotocin rats or control rats (Figure 2). Since increases in GLT-1 expression in the CA3 region are associated with elevations in extracellular levels of glutamate in the hippocampus of rats subjected to chronic stress (see above), these results suggest that basal glutamatergic tone may also be increased in the hippocampus of diabetic rats.
Figure 2.
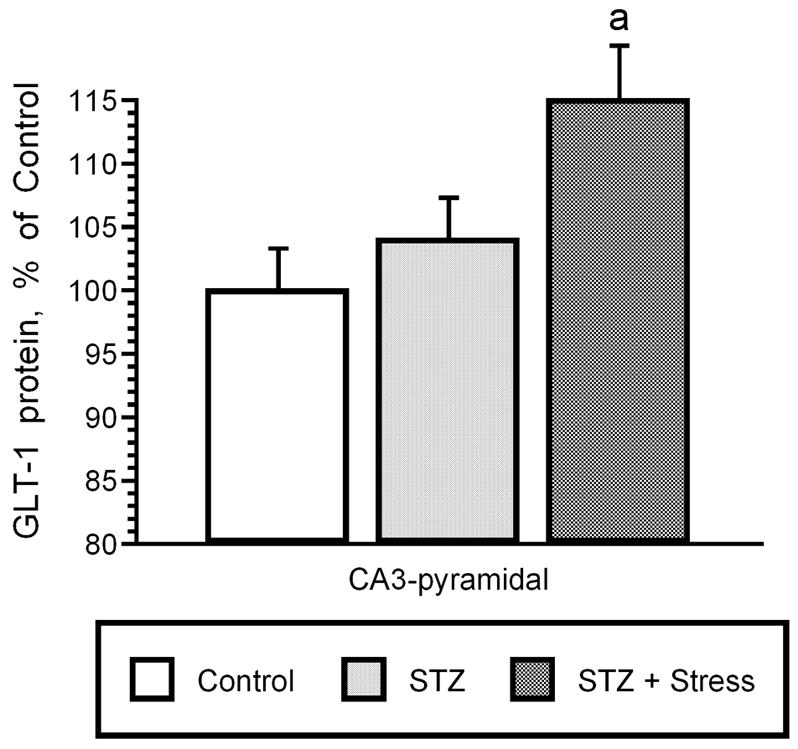
Glial glutamate transporter expression is increased in the hippocampus of diabetic rats subjected to acute stress. Radioimmunocytochemistry revealed that diabetic rats subjected to acute (i.e. 7 day) stress exhibit region-specific increases in GLT-1 protein expression in the CA3 region of the hippocampus. Since chronic stress increases GLT-1 expression in the CA3 region of the rat hippocampus that are associated with increases in basal glutamatergic tone, these results suggest that basal glutamatergic tone may also be elevated in diabetic rats subjected to stress. See text for details. [a = P < 0.05]
2.5. Insulin receptor expression and signaling in diabetes phenotypes
The insulin receptor is expressed in discrete neuronal populations in the CNS, including the cerebellum, hypothalamus and the hippocampus (Marks et al., 1991; Doré et al., 1997; Kar et al., 1993). The insulin receptor is proposed to participate in a variety of functional activities of the CNS, including cognition (Park, 2001). For example, insulin improves cognitive performance in humans and animals in a wide variety of settings, including healthy subjects (Kopf and Baratti, 1994; Parkes and White, 2000; Park et al., 2000), aged subjects (Winocur and Gagnon, 1998; Messier et al., 1997; Manning et al., 1998), Alzheimer's disease patients (Messier and Gagnon, 1996; Manning et al., 1993) and in experimental models of insulin resistance (Greenwood and Winocur, 2001). Additionally, insulin receptor expression and signaling is increased following spatial learning of a hippocampal-dependent task, the Morris water maze, (Zhao et al., 1999). Our recent studies have focused upon insulin signaling deficits in diabetic rodents, especially as how they may contribute to impairments in hippocampal synaptic plasticity. To this end, we have examined the expression, localization and translocation of the insulin-sensitive glucose transporters GLUT4 and GLUT8 in the hippocampus. GLUT4 is expressed in principle and non-principle neurons in the rat hippocampus (Aplet et al., 1999; LeLoup et al., 1996; McEwen and Reagan, 2004) and physiologically relevant increases in plasma insulin levels stimulates the translocation of GLUT4 to the plasma membrane (McEwen and Reagan, 2004; Piroli et al, 2007a). GLUT8 is localized to the cytosol and the rough endoplasmic reticulum (RER) in the rat hippocampus (Piroli et al., 2002) and cortex (Piroli et al., 2007b). Unlike GLUT4 translocation to the plasma membrane, insulin stimulates GLUT8 translocation from the cytosol to the RER in the rat hippocampus (Piroli et al, 2002; Piroli et al., 2004). Under pathological conditions such as diabetes phenotypes, plasma membrane association of GLUT4 (McEwen and Reagan, 2004; Winocur et al, 2005) and RER association of GLUT8 is reduced (Piroli et al, 2004), further supporting a role for insulin the in the translocation of these GLUTs.
One of the lingering questions that remains to be determined is the functional significance of insulin-stimulated trafficking of GLUTs in the hippocampus. Given the abundant expression of GLUT1 and GLUT3, it is unlikely that GLUT4 makes a significant contribution to glucose uptake and metabolism in the hippocampus. The functional role of GLUT8 remains to be determined, although we have previously speculated that GLUT8 participates in protein glycosylation events that occur in the RER (Piroli et al, 2002; Piroli et al., 2005). Nonetheless, it is interesting to note that GLUT4 and GLUT8 translocation events are correlated with behavioral performance of hippocampal-dependent tasks. Moreover, the rapid time course of increased hippocampal glucose uptake during learning and memory tasks (McNay et al., 2000; McNay and Gold, 1999) suggests that a readily mobilizable pool of GLUTs quickly enhances metabolic capacity during increased neuronal activity. Conversely, decreases in plasma membrane association of GLUT4 are observed in streptozotocin diabetic rats (McEwen and Reagan, 2004) that exhibit deficits in hippocampal dependent tasks (Biessels et al, 1996). More recently we have shown that deficits in hippocampal-dependent behavioral tasks in type 2 Zucker diabetic rats is associated with decreases in plasma membrane levels of GLUT4 (Winocur et al, 2005). Since total GLUT4 levels were not affected in diabetic rats compared to control rats in these studies, these data strengthen the link between insulin signaling, GLUT4 trafficking events and behavioral performance.
2.6. Neurological consequences of diabetes and stress: common etiological mechanisms?
As detailed above, there are striking similarities when comparing the neurological consequences of chronic exposure to stress levels of glucocorticoids and those observed in diabetes phenotypes. These include deficits in long-term potentiation, changes in neuronal morphology and synapse formation, elevations in oxidative stress, all of which may contribute to the development of cognitive deficits. One of the major metabolic effects of chronic increases in glucocorticoids levels is insulin resistance (see above) and our recent studies suggest that these effects of glucocorticoid administration extend to the CNS in that insulin receptor signaling and GLUT4 translocation is decreased in the hippocampus of corticosterone-treated rats (Piroli et al, 2007a). In view of the proposed role of insulin in cognition, it is interesting to speculate that glucocorticoid mediated deficits in insulin receptor signaling contribute to memory impairments in Cushing’s syndrome patients (Starkman et al, 1992) Common endocrine profiles are present in diabetic subjects and following chronic exposure to stress levels of glucocorticoids, including hyperglycemia, deficits in insulin receptor signaling, and HPA axis dysfunction, in particular hypercorticosteronemia. As a result, while insulin receptor signaling deficits occur via different mechanisms, the similar endocrine profiles in diabetic subjects and chronic stress paradigms make it difficult to assess the pathophysiological etiology of the morphological, electrophysiological and cellular deficits observed in these paradigms.
2.7. Molecular approaches to examine insulin receptor activity in the CNS
Another important consideration regarding insulin CNS signaling deficits in diabetes and stress paradigms is that insulin may mediates its effects through the insulin receptor or the insulin-like growth factor receptor family (Adamo et al., 1989). In the absence of receptor-specific ligands, recent studies have utilized molecular approaches to more selectively examine the functional activities of central insulin receptors. For example, disruption of insulin receptor throughout the brain results in increased body adiposity and plasma insulin levels (Bruning et al., 2000). More selective knockdown of insulin receptor expression in the hypothalamus using antisense oligodeoxynucleotides leads to greater adiposity specifically in the subcutaneous depot (Obici et al., 2002). Nevertheless, there are several caveats and limitations associated with these molecular approaches that have examined insulin receptor function in the CNS. For example, one potential complication associated with the use of knock-out mice is that compensatory changes may occur during development due to elimination of the gene of interest. As described above, this may be particularly relevant to insulin receptor expression in the CNS since insulin also exhibits high affinity for insulin-like growth factor I (IGF-I) receptor and IGF-I receptors are expressed in the hypothalamus (Kar et al, 1993). Administration of antisense oligonucleotides avoids this potential limitation associated with knock-out mice, but requires constant infusion of antisense sequences that may produce short-lived effects.
An emerging technology that provides an alternative to these approaches is virus-mediated gene transfer (Wilson and Yeomans, 2002). Virus-mediated gene transfer induces long lasting changes in gene expression in targeted brain regions in adult animals, thereby allowing for examination of the role of a particular gene in neuronal function from the cellular to the behavioral levels. In view of the advantages of this approach, we developed a lentivirus vector packaged with an antisense sequence selective for the insulin receptor (LV-IRAS) and injected this construct into the third ventricle to target insulin receptors expressed in the hypothalamus (Grillo et al., 2007). Insulin receptor expression and insulin-stimulated translocation of GLUT4 to the plasma membrane was decreased in the hypothalamus of rats treated with the LV-IRAS construct, decreases not observed in the hippocampus. LV-IRAS-treated rats also exhibited the expected increases in body weight gain, subcutaneous fat and plasma leptin levels. As such, the LV-IRAS construct is an experimental tool that will allow for the more selective examination of insulin receptor activities in the CNS, including the potential role of insulin as a neurotrophic factor in regions like the hippocampus. Indeed, our recent studies suggest that downregulation of insulin receptor expression and signaling adversely affects hippocampal synaptic plasticity (Grillo et al, unpublished observations). Such experimental approaches may identify common mechanisms and pathologies that provide etiological links between co-morbidities like diabetes and depressive illness.
3. Consequences of stress and diabetes: clinical correlates to animal studies
The cumulative neurological consequences of stress paradigms and experimental models of diabetes ultimately contribute to behavioral deficits. As such, one of the most critical questions that remains to be determined is whether these pre-clinical data translate to the clinical setting. There is strong evidence to suggest that the data provided by animal models of stress may accurately reflect and predict some of the structural and functional deficits observed in stress-related disorders such as depressive illness, PTSD and anxiety disorders, as well as in normal aging and in Cushing’s syndrome. Interestingly, as seen in experimental models (Luine et al., 1994; Conrad et al., 1999), the structural and functional changes observed in the hippocampus of Cushing’s syndrome patients are at least partially reversible (Starkman et al., 1999). These data suggest that pharmacological interventions that restore hippocampal and amygdalar synaptic plasticity in pre-clinical studies may produce similar beneficial effects in patients with mood-related disorders.
The clinical literature that has described structural and functional changes in diabetic patients is somewhat more equivocal in that the magnitude and significance of cognitive deficits in diabetic patients is a subject of debate. The complexity of the pathophysiological causes and consequences of diabetes may contribute to the dissimilar findings in clinical studies. For example, a variety of factors have been proposed to negatively influence the structural and functional integrity of the brain in diabetes patients, including the degree of glycemic control, the number and severity of hypoglycemic episodes, the age of onset and the duration of diabetes [For reviews, see (Ryan, 2006; Ryan, 1999)]. A consequence of these metabolic deficits is impairments in HPA axis function, which may further exacerbate the neurological complications of diabetes (Bruehl et al., 2007). Neuroanatomical abnormalities have been reported in type 1 and type 2 patients [For review see (Reagan, 2002)] and the advent of imaging technologies has confirmed and extended these previous observations. For example, while MRI techniques have not identified cerebral or hippocampal atrophy in type 1 patients (Lobnig et al., 2006; Ferguson et al., 2003), voxel-based morphometry (VBM) revealed decreases in grey matter density in type 1 patients (Musen et al., 2006). Imaging studies in type 2 patients have yielded more consistent findings suggestive of cerebral atrophy, in that MRI analyses have identified structural atrophy (Manschot et al., 2006), particularly in the limbic structures such as the hippocampus and amygdala (den Heijer et al., 2003). Decreases in hippocampal formation volume in type 2 patients have also been identified using a combined MRI/VBM approach (Gold et al., 2007). Importantly, these structural changes are often associated with neuropsychological deficits in type 2 pateints (Gold et al, 2007; Manschot et al, 2006)
Some investigators suggest that CNS structural and functional deficits in diabetic patients are subtle and do not represent a significant cognitive burden in diabetic individuals compared to the general population. Irrespective of the ‘significance’ of cognitive impairments identified in diabetes, the life-long complications of hyperglycemia may make diabetic patients more vulnerable to develop co-morbidities such as recurrent depressive illness, dementia and Alzheimer’s disease (Ott et al., 1996; Ott et al., 1999; Brown et al., 2004; McEwen et al., 2002b). Therefore, while the magnitude of cognitive deficits observed in diabetic subjects may remain a subject of debate, the significance of the underlying molecular, cellular, neurophysiological and neuroanatomical changes that may ultimately produce cognitive deficits and co-morbidities in diabetes subjects should not be overlooked or marginalized. Indeed, the long-term consequences of diabetes upon the CNS will become an important health care issue over the next decade in view of the growing diabetes patient population. As a result, diabetic encephalopathy could surpass renal failure, heart disease and retinopathy as the major complication of diabetes.
Acknowledgments
The authors’ work is supported in part by Juvenile Diabetes Research Foundation grant number 2-03-675 (LPR), NIH grant number NS047728 (LPR) and the University of South Carolina Research Foundation (LPR). The authors would like to acknowledge the efforts of their collaborators, including: Dr. Bruce McEwen (The Rockefeller University); Dr. Maureen Charron (Albert Einstein College of Medicine); Dr. Carol Greenwood and Dr. Gordon Winocur (University of Toronto); Dr. Randall Sakai (University of Cincinnati); and Dr. Steven Wilson, Dr. Jim Fadel and Leah Reznikov (University of South Carolina School of Medicine).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adamo M, Raizada MK, LeRoith D. Insulin and insulin-like growth factor receptors in the nervous system. Mol Neurobiol. 1989;3:71–100. doi: 10.1007/BF02935589. [DOI] [PubMed] [Google Scholar]
- Altamura C, Maes M, Dai J, Meltzer HY. Plasma concentrations of excitatory amino acids, serine, glycine, taurine and histidine in major depression. Eur Neuropsychopharmacol. 1995;5(Suppl):71–75. doi: 10.1016/0924-977x(95)00033-l. [DOI] [PubMed] [Google Scholar]
- Altamura CA, Mauri MC, Ferrara A, Moro AR, D’Andrea G, Zamberlan F. Plasma and platelet excitatory amino acids in psychiatric disorders. Am J Psychiatry. 1993;150:1731–1733. doi: 10.1176/ajp.150.11.1731. [DOI] [PubMed] [Google Scholar]
- Amatruda JM, Livingston JN, Lockwood DH. Cellular mechanisms in selected states of insulin resistance: human obesity, glucocorticoid excess, and chronic renal failure. Diabetes Metab Rev. 1985;1:293–317. doi: 10.1002/dmr.5610010304. [DOI] [PubMed] [Google Scholar]
- Aplet J, Melhorn G, Schliebs R. Insulin-sensitive GLUT4 glucose transporters are colocalized with GLUT3-expressing cells and demonstrate a chemically distinct neuron-specific localization in rat brain. J Neurosci Res. 1999;57:693–705. [PubMed] [Google Scholar]
- Aragno M, Mastrocola R, Medana C, Restivo F, Catalano MG, Pons N, Danni O, Boccuzzi G. Up-regulation of advanced glycated products receptors in the brain of diabetic rats is prevented by antioxidant treatment. Endocrinology. 2005;146:5561–5567. doi: 10.1210/en.2005-0712. [DOI] [PubMed] [Google Scholar]
- Aragno M, Parola S, Tamagno E, Brignardello E, Manti R, Danni O, Boccuzzi G. Oxidative derangement in rat synaptosomes induced by hyperglycemia: restorative effect of dehydroepiandrosterone treatment. Biochem Pharmacol. 2000;60:389–395. doi: 10.1016/s0006-2952(00)00327-0. [DOI] [PubMed] [Google Scholar]
- Artola A, Kamal A, Ramakers GM, Biessels GJ, Gispen WH. Diabetes mellitus concomitantly facilitates the induction of long-term depression and inhibits that of long-term potentiation in hippocampus. Eur J Neurosci. 2005;22:169–178. doi: 10.1111/j.1460-9568.2005.04205.x. [DOI] [PubMed] [Google Scholar]
- Auer DP, Putz B, Kraft E, Lipinski B, Schill J, Holsboer F. Reduced glutamate in the anterior cingulate cortex in depression: an in vivo proton magnetic resonance spectroscopy study. Biol Psychiatry. 2000;47:305–313. doi: 10.1016/s0006-3223(99)00159-6. [DOI] [PubMed] [Google Scholar]
- Bagley J, Moghaddam B. Temporal dynamics of glutamate efflux in the prefrontal cortex and in the hippocampus following repeated stress: effects of pretreatment with saline or diazepam. Neuroscience. 1997;77:65–73. doi: 10.1016/s0306-4522(96)00435-6. [DOI] [PubMed] [Google Scholar]
- Baynes JW. Role of oxidative stress in development of complications in diabetes. Diabetes. 1991;40:405–412. doi: 10.2337/diab.40.4.405. [DOI] [PubMed] [Google Scholar]
- Beauquis J, Roig P, Homo-Delarche F, De Nicola A, Saravia F. Reduced hippocampal neurogenesis and number of hilar neurones in streptozotocin-induced diabetic mice: reversion by antidepressant treatment. Eur J Neurosci. 2006;23:1539–1546. doi: 10.1111/j.1460-9568.2006.04691.x. [DOI] [PubMed] [Google Scholar]
- Belanger A, Lavoie N, Trudeau F, Massicotte G, Gagnon S. Preserved LTP and water maze learning in hyperglycaemic-hyperinsulinemic ZDF rats. Physiol Behav. 2004;83:483–494. doi: 10.1016/j.physbeh.2004.08.031. [DOI] [PubMed] [Google Scholar]
- Biessels GJ, Kamal A, Ramakers GM, Urban IJ, Spruijt BM, Erkelens DW, Gispen WH. Place learning and hippocampal synaptic plasticity in streptozotocin-induced diabteic rats. Diabetes. 1996;45:1259–1266. doi: 10.2337/diab.45.9.1259. [DOI] [PubMed] [Google Scholar]
- Blanc EM, Keller JN, Fernandez S, Mattson MP. 4-hydroxynonenal, a lipid peroxidation product, impairs glutamate transport in cortical astrocytes. Glia. 1998;22:149–160. doi: 10.1002/(sici)1098-1136(199802)22:2<149::aid-glia6>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Brands AM, Kessels RP, de Haan EH, Kappelle LJ, Biessels GJ. Cerebral dysfunction in type 1 diabetes: effects of insulin, vascular risk factors and blood-glucose levels. Eur J Pharmacol. 2004;490:159–168. doi: 10.1016/j.ejphar.2004.02.053. [DOI] [PubMed] [Google Scholar]
- Bremner JD, Narayan M, Anderson ER, Staib LH, Miller HL, Charney DS. Hippocampal volume reduction in major depression. Am J Psychiatry. 2000;157:115–118. doi: 10.1176/ajp.157.1.115. [DOI] [PubMed] [Google Scholar]
- Bremner JD, Randall P, Scott TM, Bronen RA, Seibyl JP, Southwick SM, Delaney RC, McCarthy G, Charney DS, Innis RB. MRI-based measurement of hippocampal volume of patients with combat-related posttraumatic stress disorder. Am J Psychiatry. 1995;152:973–981. doi: 10.1176/ajp.152.7.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown ES, Varghese FP, McEwen BS. Association of depression with medical illness: does cortisol play a role? Biol Psychiatry. 2004;55:1–9. doi: 10.1016/s0006-3223(03)00473-6. [DOI] [PubMed] [Google Scholar]
- Bruehl H, Rueger M, Dziobek I, Sweat V, Tirsi A, Javier E, Arentoft A, Wolf OT, Convit A. Hypothalamic-pituitary-adrenal axis dysregulation and memory impairments in type 2 diabetes. J Clin Endocrinol Metab. 2007;92:2439–2445. doi: 10.1210/jc.2006-2540. [DOI] [PubMed] [Google Scholar]
- Bruning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, Klein R, Krone W, Muller-Wieland D, Kahn CR. Role of brain insulin receptor in control of body weight and reproduction. Science. 2000;289:2122–2125. doi: 10.1126/science.289.5487.2122. [DOI] [PubMed] [Google Scholar]
- Campbell AM, Park CR, Zoladz PR, Munoz C, Fleshner M, Diamond DM. Pre-training administration of tianeptine, but not propranolol, protects hippocampus-dependent memory from being impaired by predator stress. Eur Neuropsychopharmacol. 2007 doi: 10.1016/j.euroneuro.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Campbell S, Marriott M, Nahmias C, MacQueen GM. Lower hippocampal volume in patients suffering from depression: a meta-analysis. Am J Psychiatry. 2004;161:598–607. doi: 10.1176/appi.ajp.161.4.598. [DOI] [PubMed] [Google Scholar]
- Ceriello A, Giugliano D, Quatraro A, Dello Russo P, Lefèbvre PJ. Metabolic control may influence the increased superoxide generation in diabetic serum. Diabetic Med. 1991;8:540–542. doi: 10.1111/j.1464-5491.1991.tb01647.x. [DOI] [PubMed] [Google Scholar]
- Choudary PV, Molnar M, Evans SJ, Tomita H, Li JZ, Vawter MP, Myers RM, Bunney WE, Jr, Akil H, Watson SJ, Jones EG. Altered cortical glutamatergic and GABAergic signal transmission with glial involvement in depression. Proc Natl Acad Sci U S A. 2005;102:15653–15658. doi: 10.1073/pnas.0507901102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman E, Judd R, Hoe L, Dennis J, Posner P. Effects of diabetes mellitus on astrocyte GFAP and glutamate transporters in the CNS. Glia. 2004;48:166–178. doi: 10.1002/glia.20068. [DOI] [PubMed] [Google Scholar]
- Conrad CD, LeDoux JE, Magarinos AM, McEwen BS. Repeated restraint stress facilitates fear conditioning independently of causing hippocampal CA3 dendritic atrophy. Behav Neurosci. 1999;113:902–913. doi: 10.1037//0735-7044.113.5.902. [DOI] [PubMed] [Google Scholar]
- Cotter D, Mackay D, Landau S, Kerwin R, Everall I. Reduced glial cell density and neuronal size in the anterior cingulate cortex in major depressive disorder. Arch Gen Psychiatry. 2001;58:545–553. doi: 10.1001/archpsyc.58.6.545. [DOI] [PubMed] [Google Scholar]
- Couch RM. Dissociation of cortisol and adrenal androgen secretion in poorly controlled insulin-dependent diabetes mellitus. Acta Endocrinol. 1992;127:115–117. doi: 10.1530/acta.0.1270115. [DOI] [PubMed] [Google Scholar]
- Czeh B, Michaelis T, Watanabe T, Frahm J, de Biurrun G, van Kampen M, Bartolomucci A, Fuchs E. Stress-induced changes in cerebral metabolites, hippocampal volume, and cell proliferation are prevented by antidepressant treatment with tianeptine. Proc Natl Acad Sci USA. 2001;98:12796–12801. doi: 10.1073/pnas.211427898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leon MJ, McRae T, Rusinek H, Convit A, De Santi S, Tarshish C, Golomb J, Volkow N, Daisley K, Orentreich N, McEwen BS. Cortisol reduces hippocampal glucose metabolism in normal elderly, but not in Alzheimer’s disease. J Clin Endocrinol Metab. 1997;82:3251–3259. doi: 10.1210/jcem.82.10.4305. [DOI] [PubMed] [Google Scholar]
- den Heijer T, Vermeer SE, van Dijk EJ, Prins ND, Koudstaal PJ, Hofman A, Breteler MM. Type 2 diabetes and atrophy of medial temporal lobe structures on brain MRI. Diabetologia. 2003;46:1604–1610. doi: 10.1007/s00125-003-1235-0. [DOI] [PubMed] [Google Scholar]
- Di Luca M, Ruts L, Gardoni F, Cattabeni F, Biessels GJ, Gispen WH. NMDA receptor subunits are modified transcriptionally and post-translationally in the brain of streptozotocin-diabetic rats. Diabetologia. 1999;42:693–701. doi: 10.1007/s001250051217. [DOI] [PubMed] [Google Scholar]
- Dimitriadis G, Leighton B, Parry-Billings M, Sasson S, Young M, Krause U, Bevan S, Piva T, Wegener G, Newsholme EA. Effects of glucocorticoid excess on the sensitivity of glucose transport and metabolism to insulin in rat skeletal muscle. Biochem J. 1997;321(Pt 3):707–712. doi: 10.1042/bj3210707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doré S, Kar S, Rowe W, Quirion R. Distribution and levels of [125I]IGF-I, [125I]IGF-II and [125I]Insulin receptor binding sites in the hippocampus of aged memory-unimpaired and -impaired rats. Neuroscience. 1997;80:1033–1040. doi: 10.1016/s0306-4522(97)00154-1. [DOI] [PubMed] [Google Scholar]
- Ferguson SC, Blane A, Perros P, McCrimmon RJ, Best JJ, Wardlaw J, Deary IJ, Frier BM. Cognitive ability and brain structure in type 1 diabetes: relation to microangiopathy and preceding severe hypoglycemia. Diabetes. 2003;52:149–156. doi: 10.2337/diabetes.52.1.149. [DOI] [PubMed] [Google Scholar]
- Frodl T, Meisenzahl E, Zetzsche T, Bottlender R, Born C, Groll C, Jager M, Leinsinger G, Hahn K, Moller HJ. Enlargement of the amygdala in patients with a first episode of major depression. Biol Psychiatry. 2002a;51:708–714. doi: 10.1016/s0006-3223(01)01359-2. [DOI] [PubMed] [Google Scholar]
- Frodl T, Meisenzahl EM, Zetzsche T, Born C, Groll C, Jager M, Leinsinger G, Bottlender R, Hahn K, Moller HJ. Hippocampal changes in patients with a first episode of major depression. Am J Psychiatry. 2002b;159:1112–1118. doi: 10.1176/appi.ajp.159.7.1112. [DOI] [PubMed] [Google Scholar]
- Gage FH. Neurogenesis in the adult brain. J Neurosci. 2002;22:612–613. doi: 10.1523/JNEUROSCI.22-03-00612.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagne J, Milot M, Gelinas S, Lahsaini A, Trudeau F, Martinoli MG, Massicotte G. Binding properties of glutamate receptors in streptozotocin-induced diabetes in rats. Diabetes. 1997;46:841–846. doi: 10.2337/diabetes.46.5.841. [DOI] [PubMed] [Google Scholar]
- Garvey WT, Huecksteadt TP, Lima FB, Birnbaum MJ. Expression of a glucose transporter gene cloned from brain in cellular models of insulin resistance: dexamethasone decreases transporter mRNA in primary cultured adipocytes. Mol Endocrinol. 1989;3:1132–1141. doi: 10.1210/mend-3-7-1132. [DOI] [PubMed] [Google Scholar]
- Gerges NZ, Aleisa AM, Alkadhi KA. Impaired long-term potentiation in obese zucker rats: possible involvement of presynaptic mechanism. Neuroscience. 2003;120:535–539. doi: 10.1016/s0306-4522(03)00297-5. [DOI] [PubMed] [Google Scholar]
- Gispen WH, Biessels G-J. Cognition and synaptic plasticity in diabetes mellitus. Trends Neurosci. 2000;23:542–549. doi: 10.1016/s0166-2236(00)01656-8. [DOI] [PubMed] [Google Scholar]
- Gold SM, Dziobek I, Sweat V, Tirsi A, Rogers K, Bruehl H, Tsui W, Richardson S, Javier E, Convit A. Hippocampal damage and memory impairments as possible early brain complications of type 2 diabetes. Diabetologia. 2007;50:711–719. doi: 10.1007/s00125-007-0602-7. [DOI] [PubMed] [Google Scholar]
- Gould E, Cameron HA, Daniels DC, Woolley CS, McEwen BS. Adrenal hormones suppress cell division in the adult rat dentate gyrus. J Neurosci. 1992;12:3642–3650. doi: 10.1523/JNEUROSCI.12-09-03642.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould E, Gross CG. Neurogenesis in adult mammals: some progress and problems. J Neurosci. 2002;22:619–623. doi: 10.1523/JNEUROSCI.22-03-00619.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood CE, Winocur G. Glucose treatment reduces memory deficits in young adult rats fed high-fed diets. Neurobiol Learn Mem. 2001;75:179–189. doi: 10.1006/nlme.2000.3964. [DOI] [PubMed] [Google Scholar]
- Grillo CA, Piroli GG, Rosell DR, Hoskin EK, McEwen BS, Reagan LP. Region specific increases in oxidative stress and superoxide dismutase in the hippocampus of diabetic rats subjected to stress. Neuroscience. 2003;121:133–140. doi: 10.1016/s0306-4522(03)00343-9. [DOI] [PubMed] [Google Scholar]
- Grillo CA, Piroli GG, Wood GE, Reznikov LR, McEwen BS, Reagan LP. Immunocytochemical analysis of synaptic proteins provides new insights into diabetes-mediated plasticity in the rat hippocampus. Neuroscience. 2005;136:477–486. doi: 10.1016/j.neuroscience.2005.08.019. [DOI] [PubMed] [Google Scholar]
- Grillo CA, Tamashiro KL, Piroli GG, Melhorn S, Gass JT, Newsom RJ, Reznikov LR, Smith A, Wilson SP, Sakai RR, Reagan LP. Lentivirus-mediated downregulation of hypothalamic insulin receptor expression. Physiol Behav. 2007 doi: 10.1016/j.physbeh.2007.05.043. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurvits TV, Shenton ME, Hokama H, Ohta H, Lasko NB, Gilbertson MW, Orr SP, Kikinis R, Jolesz FA, McCarley RW, Pitman RK. Magnetic resonance imaging study of hippocampal volume in chronic, combat-related posttraumatic stress disorder. Biol Psychiatry. 1996;40:1091–1099. doi: 10.1016/S0006-3223(96)00229-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamidi M, Drevets WC, Price JL. Glial reduction in amygdala in major depressive disorder is due to oligodendrocytes. Biol Psychiatry. 2004;55:563–569. doi: 10.1016/j.biopsych.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Herman JP, Cullinan WE. Neurocircuitry of stress: central control of the hypothalamo-pituitary-adrenocortical axis. Trends Neurosci. 1997;20:78–84. doi: 10.1016/s0166-2236(96)10069-2. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Yamada KA, Matsukawa M, Zorumski CF. Effects of insulin on long-term potentiation in hippocampal slices from diabetic rats. Diabetologia. 2003;46:1007–1012. doi: 10.1007/s00125-003-1144-2. [DOI] [PubMed] [Google Scholar]
- Jacobson L, Sapolsky R. The role of the hippocampus in feedback regulation of the hypothalamic-pituitary-adrenocortical axis. Endocrin Reviews. 1991;12:118–134. doi: 10.1210/edrv-12-2-118. [DOI] [PubMed] [Google Scholar]
- Kadekaro M, Ito M, Gross PM. Local cerebral glucose utilization is increased in acutely adrenalectomized rats. Neuroendocrinology. 1988;47:329–334. doi: 10.1159/000124933. [DOI] [PubMed] [Google Scholar]
- Kamal A, Biessels GJ, Duis SEJ, Gispen WH. Learning and hippocampal synaptic plasticity in streptozotocin-diabetic rats: interaction of diabetes and ageing. Diabetologia. 2000;43:500–506. doi: 10.1007/s001250051335. [DOI] [PubMed] [Google Scholar]
- Kamal A, Biessels GJ, Urban IJA, Gispen WH. Hippocampal synaptic plasticity in streptozotocin-diabetic rats: impairment of long-term potentiation and facilitation of long-term depression. Neuroscience. 1999;90:737–745. doi: 10.1016/s0306-4522(98)00485-0. [DOI] [PubMed] [Google Scholar]
- Kar S, Chabot J-G, Quirion R. Quantitative autoradiographic localization of [125I]Insulin-like growth factor I, [125I]Insulin-like growth factor II and [125I]Insulin binding sites in developing and adult rat brain. J Comp Neurol. 1993;333:375–397. doi: 10.1002/cne.903330306. [DOI] [PubMed] [Google Scholar]
- Keller JN, Mark RJ, Bruce AJ, Blanc E, Rothstein JD, Uchida K, Waeg G, Mattson MP. 4-hydroxynonenal, an aldehyde product of membrane lipid peroxidation, impairs glutamate transport and mitochondrial function in synaptosomes. Neuroscience. 1997a;80:685–696. doi: 10.1016/s0306-4522(97)00065-1. [DOI] [PubMed] [Google Scholar]
- Keller JN, Pang Z, Geddes JW, Begley JG, Germeyer A, Waeg G, Mattson MP. Impairment of glucose and glutamate transport and induction of mitochondrial oxidative stress and dysfunction in synaptosomes by amyloid β-peptide: role of the lipid peroxidation product 4-hydroxynonenal. J Neurochem. 1997b;69:273–284. doi: 10.1046/j.1471-4159.1997.69010273.x. [DOI] [PubMed] [Google Scholar]
- Kessler RC, McGonagle KA, Zhao S, Nelson CB, Hughes M, Eshleman S, Wittchen HU, Kendler KS. Lifetime and 12-month prevalence of DSM-III-R psychiatric disorders in the United States. Results from the National Comorbidity Survey. Arch Gen Psychiatry. 1994;51:8–19. doi: 10.1001/archpsyc.1994.03950010008002. [DOI] [PubMed] [Google Scholar]
- Kim HB, Jang MH, Shin MC, Lim BV, Kim YP, Kim KJ, Kim EH, Kim CJ. Treadmill exercise increases cell proliferation in dentate gyrus of rats with streptozotocin-induced diabetes. J Diabetes Complications. 2003;17:29–33. doi: 10.1016/s1056-8727(02)00186-1. [DOI] [PubMed] [Google Scholar]
- Kim JS, Schmid-Burgk W, Claus D, Kornhuber HH. Increased serum glutamate in depressed patients. Arch Psychiatr Nervenkr. 1982;232:299–304. doi: 10.1007/BF00345492. [DOI] [PubMed] [Google Scholar]
- Kole MH, Swan L, Fuchs E. The antidepressant tianeptine persistently modulates glutamate receptor currents of the hippocampal CA3 commissural associational synapse in chronically stressed rats. Eur J Neurosci. 2002;16:807–816. doi: 10.1046/j.1460-9568.2002.02136.x. [DOI] [PubMed] [Google Scholar]
- Kopf SR, Baratti CM. Memory-improving actions of glucose: involvement of a central cholinergic muscarinic mechanism. Behav Neur Biol. 1994;62:237–243. doi: 10.1016/s0163-1047(05)80022-6. [DOI] [PubMed] [Google Scholar]
- Kruman I, Bruce-Keller AJ, Bredesen D, Waeg G, Mattson MP. Evidence that 4-hydroxynonenal mediates oxidative stress-induced neuronal apoptosis. J Neurosci. 1997;17:5089–5100. doi: 10.1523/JNEUROSCI.17-13-05089.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landgraf R, Mitro A, Hess J. Regional net uptake of 14C-glucose by rat brain under the influence of corticosterone. Endocrinol Exp. 1978;12:119–129. [PubMed] [Google Scholar]
- Leedom LJ, Meehan WP, Zeidler A. Avoidance responding in mice with diabetes mellitus. Physiol Behav. 1987;40:447–451. doi: 10.1016/0031-9384(87)90029-1. [DOI] [PubMed] [Google Scholar]
- LeLoup C, Arluison M, Kassis N, Lepetit N, Cartier N, Ferré P, Pénicaud L. Discrete brain areas express the insulin-responsive glucose transporter GLUT4. Mol Brain Res. 1996;38:45–53. doi: 10.1016/0169-328x(95)00306-d. [DOI] [PubMed] [Google Scholar]
- Levine J, Panchalingam K, Rapoport A, Gershon S, McClure RJ, Pettegrew JW. Increased cerebrospinal fluid glutamine levels in depressed patients. Biol Psychiatry. 2000;47:586–593. doi: 10.1016/s0006-3223(99)00284-x. [DOI] [PubMed] [Google Scholar]
- Li XL, Aou S, Oomura Y, Hori N, Fukunaga K, Hori T. Impairment of long-term potentiation and spatial memory in leptin receptor-deficient rodents. Neuroscience. 2002a;113:607–615. doi: 10.1016/s0306-4522(02)00162-8. [DOI] [PubMed] [Google Scholar]
- Li ZG, Zhang W, Grunberger G, Sima AA. Hippocampal neuronal apoptosis in type 1 diabetes. Brain Res. 2002b;946:221–231. doi: 10.1016/s0006-8993(02)02887-1. [DOI] [PubMed] [Google Scholar]
- Lobnig BM, Kromeke O, Optenhostert-Porst C, Wolf OT. Hippocampal volume and cognitive performance in long-standing Type 1 diabetic patients without macrovascular complications. Diabet Med. 2006;23:32–39. doi: 10.1111/j.1464-5491.2005.01716.x. [DOI] [PubMed] [Google Scholar]
- Lowy MT, Gault L, Yamamoto BK. Adrenalectomy attenuates stress-induced elavations in extracellular glutamate concentrations in the hippocampus. J Neurochem. 1993;61:1957–1960. doi: 10.1111/j.1471-4159.1993.tb09839.x. [DOI] [PubMed] [Google Scholar]
- Lucassen PJ, Fuchs E, Czeh B. Antidepressant treatment with tianeptine reduces apoptosis in the hippocampal dentate gyrus and temporal cortex. Biol Psychiatry. 2004;55:789–796. doi: 10.1016/j.biopsych.2003.12.014. [DOI] [PubMed] [Google Scholar]
- Luine V, Villegas M, Martinez C, McEwen BS. Repeated stress causes reversible impairments of spatial memory impairments. Brain Res. 1994;639:167–170. doi: 10.1016/0006-8993(94)91778-7. [DOI] [PubMed] [Google Scholar]
- Lupien SJ, de Leon MJ, De Santi S, Convit A, Tarshish C, Nair NPV, McEwen BS, Hauger RL, Meaney MJ. Cortisol levels during human aging predict hippocampal atrophy and memory deficits. Nat Neurosci. 1998;1:69–73. doi: 10.1038/271. [DOI] [PubMed] [Google Scholar]
- MacQueen GM, Campbell S, McEwen BS, Macdonald K, Amano S, Joffe RT, Nahmais C, Young LT. Course of illness, hippocampal function, and hippocampal volume in major depression. Proc Natl Acad Sci USA. 2003;100:1387–1392. doi: 10.1073/pnas.0337481100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magariños AM, Deslandes A, McEwen BS. Effects of antidepressants and benzodiazepine treatments on the dendritic structure of CA3 pyramidal neurons after chronic stress. Eur J Pharmacol. 1999;371:113–122. doi: 10.1016/s0014-2999(99)00163-6. [DOI] [PubMed] [Google Scholar]
- Magariños AM, Jain K, Blount ED, Reagan L, Smith BH, McEwen BS. Peritoneal implantation of microencapsulated porcine pancreatic islets in diabetic rats ameliorates severe hyperglycemia and prevents retraction and simplification of hippocampal dendrites. Brain Res. 2001;902:282–287. doi: 10.1016/s0006-8993(01)02400-3. [DOI] [PubMed] [Google Scholar]
- Magariños AM, McEwen BS. Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: involvement of glucocorticoid secretion and excitatory amino acid receptors. Neuroscience. 1995;69:89–98. doi: 10.1016/0306-4522(95)00259-l. [DOI] [PubMed] [Google Scholar]
- Magariños AM, McEwen BS. Experimental diabetes in rats causes hippocampal dendritic and synaptic reorganization and increased glucocorticoid reactivity to stress. Proc Natl Acad Sci USA. 2000;97:11056–11061. doi: 10.1073/pnas.97.20.11056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning CA, Ragozzino ME, Gold PE. Glucose enhancement of memory in patients with probable senile dementia of the Alzheimer’s type. Neurobiol Aging. 1993;14:523–528. doi: 10.1016/0197-4580(93)90034-9. [DOI] [PubMed] [Google Scholar]
- Manning CA, Stone WS, Korol DL, Gold PE. Glucose enhancement of 24-h memory retrival in healthy elderly humans. Behav Brain Res. 1998;93:71–76. doi: 10.1016/s0166-4328(97)00136-8. [DOI] [PubMed] [Google Scholar]
- Manschot SM, Brands AM, van der GJ, Kessels RP, Algra A, Kappelle LJ, Biessels GJ. Brain magnetic resonance imaging correlates of impaired cognition in patients with type 2 diabetes. Diabetes. 2006;55:1106–1113. doi: 10.2337/diabetes.55.04.06.db05-1323. [DOI] [PubMed] [Google Scholar]
- Mark RJ, Lovell MA, Markesbery WR, Uchida K, Mattson MP. A role for 4-hydroxynonenal, an aldehyde product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid β-peptide. J Neurochem. 1997;68:255–264. doi: 10.1046/j.1471-4159.1997.68010255.x. [DOI] [PubMed] [Google Scholar]
- Marks JL, Porte D, Jr, Stahl WL, Baskin DG. Localization of insulin receptor mRNA in rat brain by in situ hybridization. Endocrinology. 1991;127:3234–3236. doi: 10.1210/endo-127-6-3234. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Modification of ion homeostasis by lipid peroxidation: roles in neuronal degeneration and adaptive plasticity. Trends Neurosci. 1998;21:53–57. doi: 10.1016/s0166-2236(97)01188-0. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Possible mechanisms for atrophy of the human hippocampus. Mol Psychiatry. 1997;2:255–262. doi: 10.1038/sj.mp.4000254. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Mood disorders and allostatic load. Biol Psychiatry. 2003;54:200–207. doi: 10.1016/s0006-3223(03)00177-x. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Chattarji S. Molecular mechanisms of neuroplasticity and pharmacological implications: the example of tianeptine. Eur Neuropsychopharmacol. 2004;14(Suppl 5):S497–S502. doi: 10.1016/j.euroneuro.2004.09.008. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Magariños AM, Reagan LP. Structural plasticity and Stablon: cellular and molecular targets. Eur Psychiatry. 2002a;17:1–13. doi: 10.1016/s0924-9338(02)00650-8. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Magarinos AM, Reagan LP. Studies of hormone action in the hippocampal formation: possible relevance to depression and diabetes. J Psychosom Res. 2002b;53:883–890. doi: 10.1016/s0022-3999(02)00307-0. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Reagan LP. Glucose transporter expression in the central nervous system: relationship to synaptic function. Eur J Pharmacol. 2004;490:13–24. doi: 10.1016/j.ejphar.2004.02.041. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Sapolsky RM. Stress and cognitive function. Curr Opin Neurobiol. 1995;5:205–216. doi: 10.1016/0959-4388(95)80028-x. [DOI] [PubMed] [Google Scholar]
- McMahon M, Gerich J, Rizza R. Effects of glucocorticoids on carbohydrate metabolism. Diabetes Metab Rev. 1988;4:17–30. doi: 10.1002/dmr.5610040105. [DOI] [PubMed] [Google Scholar]
- McNay EC, Fries TM, Gold PE. Decreases in rat extracellular hippocampal glucose concentration associated with cognitive demand during a spatial task. Proc Natl Acad Sci USA. 2000;97:2881–2885. doi: 10.1073/pnas.050583697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNay EC, Gold PE. Extracellular glucose concentrations in the rat hippocampus measured by zero-net-flux: effects of microdialysis flow rate, strain, and age. J Neurochem. 1999;72:785–790. doi: 10.1046/j.1471-4159.1999.720785.x. [DOI] [PubMed] [Google Scholar]
- Mervaala E, Fohr J, Kononen M, Valkonen-Korhonen M, Vainio P, Partanen K, Partanen J, Tiihonen J, Viinamaki H, Karjalainen AK, Lehtonen J. Quantitative MRI of the hippocampus and amygdala in severe depression. Psychol Med. 2000;30:117–125. doi: 10.1017/s0033291799001567. [DOI] [PubMed] [Google Scholar]
- Messier C, Gagnon M. Glucose regulation and cognitive functions: relation to Alzheimer’s disease and diabetes. Behav Brain Res. 1996;75:1–11. doi: 10.1016/0166-4328(95)00153-0. [DOI] [PubMed] [Google Scholar]
- Messier C, Gagnon M, Knott V. Effect of glucose and peripheral glucose regulation on memory in the ederly. Neurobiol Aging. 1997;18:297–304. doi: 10.1016/s0197-4580(97)80311-9. [DOI] [PubMed] [Google Scholar]
- Mirza Y, Tang J, Russell A, Banerjee SP, Bhandari R, Ivey J, Rose M, Moore GJ, Rosenberg DR. Reduced anterior cingulate cortex glutamatergic concentrations in childhood major depression. J Am Acad Child Adolesc Psychiatry. 2004;43:341–348. doi: 10.1097/00004583-200403000-00017. [DOI] [PubMed] [Google Scholar]
- Musen G, Lyoo IK, Sparks CR, Weinger K, Hwang J, Ryan CM, Jimerson DC, Hennen J, Renshaw PF, Jacobson AM. Effects of type 1 diabetes on gray matter density as measured by voxel-based morphometry. Diabetes. 2006;55:326–333. doi: 10.2337/diabetes.55.02.06.db05-0520. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Gould E, Manji H, Buncan M, Duman RS, Greshenfeld HK, Hen R, Koester S, Lederhendler I, Meaney M, Robbins T, Winsky L, Zalcman S. Preclinical models: status of basic research in depression. Biol Psychiatry. 2002;52:503–528. doi: 10.1016/s0006-3223(02)01405-1. [DOI] [PubMed] [Google Scholar]
- Obici S, Feng Z, Karkanias G, Baskin DG, Rossetti L. Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat Neurosci. 2002;5:566–572. doi: 10.1038/nn0602-861. [DOI] [PubMed] [Google Scholar]
- Ongur D, Drevets WC, Price JL. Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc Natl Acad Sci U S A. 1998;95:13290–13295. doi: 10.1073/pnas.95.22.13290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oster MH, Castonguay TM, Keen CL, Stern JS. Circadian rhythm of corticosterone in diabetic rats. Life Sci. 1988;43:1643–1645. doi: 10.1016/0024-3205(88)90536-x. [DOI] [PubMed] [Google Scholar]
- Ott A, Stolk RP, Hofman A, van Harskamp F, Grobbee DE, Breteler MM. Association of diabetes mellitus and dementia: the Rotterdam Study. Diabetologia. 1996;39:1392–1397. doi: 10.1007/s001250050588. [DOI] [PubMed] [Google Scholar]
- Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology. 1999;53:1937–1942. doi: 10.1212/wnl.53.9.1937. [DOI] [PubMed] [Google Scholar]
- Park CR. Cognitive effects of insulin in the central nervous system. Neurosci Biobehav Rev. 2001;25:311–323. doi: 10.1016/s0149-7634(01)00016-1. [DOI] [PubMed] [Google Scholar]
- Park CR, Seely RJ, Craft S, Woods SC. Intracerebroventricular insulin enhances memory in a passive-avoidance task. Physiol Behav. 2000;68:509–514. doi: 10.1016/s0031-9384(99)00220-6. [DOI] [PubMed] [Google Scholar]
- Parkes M, White KG. Glucose attenuation of memory impairments. Behav Neurosci. 2000;114:307–319. [PubMed] [Google Scholar]
- Pham K, Nacher J, Hof PR, McEwen BS. Repeated restraint stress suppresses neurogenesis and induces biphasic PSA-NCAM expression in the adult rat dentate gyrus. Eur J Neurosci. 2003;17:879–886. doi: 10.1046/j.1460-9568.2003.02513.x. [DOI] [PubMed] [Google Scholar]
- Piroli GG, Grillo CA, Charron MJ, McEwen BS, Reagan LP. Biphasic effects of stress upon GLUT8 glucose transporter expression and trafficking in the diabetic rat hippocampus. Brain Res. 2004;1006:28–35. doi: 10.1016/j.brainres.2004.01.044. [DOI] [PubMed] [Google Scholar]
- Piroli GG, Grillo CA, Hoskin EK, Znamensky V, Katz EB, Milner TA, McEwen BS, Charron MJ, Reagan LP. Peripheral glucose administration stimulates the translocation of GLUT8 glucose transporter to the endoplasmic reticulum in the rat hippocampus. J Comp Neurol. 2002;452:103–114. doi: 10.1002/cne.10368. [DOI] [PubMed] [Google Scholar]
- Piroli GG, Grillo CA, Reznikov LR, Adams S, McEwen BS, Charron MJ, Reagan LP. Corticosterone impairs insulin-stimulated translocation of GLUT4 in the rat hippocampus. Neuroendocrinology. 2007a;85:71–80. doi: 10.1159/000101694. [DOI] [PubMed] [Google Scholar]
- Piroli GG, Grillo CA, Reznikov LR, Reagan LP. Expression and Functional Activities of Glucose Transporters in the Central Nervous System. In: Lajtha A, editor. Handbook of Neurochemistry and Molecular Neurobiology. Springer; New York: 2005. in press. [Google Scholar]
- Piroli GG, Grillo CA, Reznikov LR, Reagan LP. Expression and Functional Activities of Glucose Transporters in the Central Nervous System. In: Lajtha A, editor. Handbook of Neurochemistry and Molecular Neurobiology. Springer; New York: 2007b. pp. 387–404. [Google Scholar]
- Rajkowska G, Miguel-Hidalgo JJ, Wei J, Dilley G, Pittman SD, Meltzer HY, Overholser JC, Roth BL, Stockmeier CA. Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol Psychiatry. 1999;45:1085–1098. doi: 10.1016/s0006-3223(99)00041-4. [DOI] [PubMed] [Google Scholar]
- Reagan LP. Glucose, stress and hippocampal neuronal vulnerability. Int Rev Neurobiol. 2002;51:289–324. doi: 10.1016/s0074-7742(02)51009-6. [DOI] [PubMed] [Google Scholar]
- Reagan LP, Hendry RM, Reznikov LR, Piroli GG, Wood GE, McEwen BS, Grillo CA. Tianeptine increases brain-derived neurotrophic factor expression in the rat amygdala. Eur J Pharmacol. 2007;565:68–75. doi: 10.1016/j.ejphar.2007.02.023. [DOI] [PubMed] [Google Scholar]
- Reagan LP, Magariños AM, Yee DK, Szweda LI, Van Bueren A, McCall AL, McEwen BS. Oxidative stress and HNE conjugation of GLUT3 are increased in the hippocampus of diabetic rats subjected to stress. Brain Res. 2000;862:292–300. doi: 10.1016/s0006-8993(00)02212-5. [DOI] [PubMed] [Google Scholar]
- Reagan LP, McEwen BS. Controversies surrounding glucocorticoid-mediated cell death in the hippocampus. J Chem Neuroanat. 1997;13:149–167. doi: 10.1016/s0891-0618(97)00031-8. [DOI] [PubMed] [Google Scholar]
- Reagan LP, Rosell DR, Wood GE, Spedding M, Munoz C, Rothstein J, McEwen BS. Chronic restraint stress up-regulates GLT-1 mRNA and protein expression in the rat hippocampus: reversal by tianeptine. Proc Natl Acad Sci U S A. 2004;101:2179–2184. doi: 10.1073/pnas.0307294101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reul JMH, de Kloet ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology. 1985;117:2505–2511. doi: 10.1210/endo-117-6-2505. [DOI] [PubMed] [Google Scholar]
- Reznikov LR, Grillo CA, Piroli GG, Pasumarthi RK, Reagan LP, Fadel J. Acute stress-mediated increases in extracellular glutamate levels in the rat amygdala: differential effects of antidepressant treatment. Eur J Neurosci. 2007;25:3109–3114. doi: 10.1111/j.1460-9568.2007.05560.x. [DOI] [PubMed] [Google Scholar]
- Ryan CM. Memory and metabolic control in children. Diabetes Care. 1999;22:1239–1241. doi: 10.2337/diacare.22.8.1239. [DOI] [PubMed] [Google Scholar]
- Ryan CM. Why is cognitive dysfunction associated with the development of diabetes early in life? The diathesis hypothesis. Pediatr Diabetes. 2006;7:289–297. doi: 10.1111/j.1399-5448.2006.00206.x. [DOI] [PubMed] [Google Scholar]
- Saltiel AR, Pessin JE. Insulin signaling pathways in time and space. Trends Cell Biol. 2002;12:65–71. doi: 10.1016/s0962-8924(01)02207-3. [DOI] [PubMed] [Google Scholar]
- Sanacora G, Gueorguieva R, Epperson CN, Wu YT, Appel M, Rothman DL, Krystal JH, Mason GF. Subtype-specific alterations of gamma-aminobutyric acid and glutamate in patients with major depression. Arch Gen Psychiatry. 2004;61:705–713. doi: 10.1001/archpsyc.61.7.705. [DOI] [PubMed] [Google Scholar]
- Sanacora G, Mason GF, Krystal JH. Impairment of GABAergic transmission in depression: new insights from neuroimaging studies. Crit Rev Neurobiol. 2000;14:23–45. doi: 10.1615/critrevneurobiol.v14.i1.20. [DOI] [PubMed] [Google Scholar]
- Saravia FE, Beauquis J, Revsin Y, Homo-Delarche F, de Kloet ER, De Nicola AF. Hippocampal neuropathology of diabetes mellitus is relieved by estrogen treatment. Cell Mol Neurobiol. 2006;26:943–957. doi: 10.1007/s10571-006-9096-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scribner KA, Walker CD, Cascio CS, Dallman MF. Chronic streptozotocin diabetes in rats facilitates the acute stress response without altering pituitary or adrenal responsiveness to secretagogues. Endocrinology. 1991;129:99–108. doi: 10.1210/endo-129-1-99. [DOI] [PubMed] [Google Scholar]
- Sheline YI, Gado MH, Price JL. Amygdala core nuclei volumes are decreased in recurrent major depression. NeuroReport. 1998;9:2023–2028. doi: 10.1097/00001756-199806220-00021. [DOI] [PubMed] [Google Scholar]
- Sheline YI, Sanghavi M, Mintun MA, Gado MH. Depression duration but not age predicts hippocampal volume loss in medically healthy women with recurrent major depression. J Neurosci. 1999;19:5034–5043. doi: 10.1523/JNEUROSCI.19-12-05034.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheline YI, Wang PW, Gado MH, Csernansky JG, Vannier MW. Hippocampal atrophy in recurrent major depression. Proc Natl Acad Sci USA. 1996;93:3908–3913. doi: 10.1073/pnas.93.9.3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkman MN, Gebarski SS, Berent S, Schteingart DE. Hippocampal formation volume, memory dysfunction, and cortisol levels in patients with Cushing’s syndrome. Biol Psychiatry. 1992;32:756–765. doi: 10.1016/0006-3223(92)90079-f. [DOI] [PubMed] [Google Scholar]
- Starkman MN, Giordani B, Gebarski SS, Berent S, Schork MA, Schteingart DE. Decrease in cortisol reverses human hippocampal atrophy following treatment of Cushing’s Disease. Biol Psychiatry. 1999;46:1595–1602. doi: 10.1016/s0006-3223(99)00203-6. [DOI] [PubMed] [Google Scholar]
- Steffens DC, Byrum CE, McQuoid DR, Greenberg DL, Payne ME, Blitchington TF, MacFall JR, Krishnan KR. Hippocampal volume in geriatric depression. Biol Psychiatry. 2000;48:301–309. doi: 10.1016/s0006-3223(00)00829-5. [DOI] [PubMed] [Google Scholar]
- Tuzcu M, Baydas G. Effect of melatonin and vitamin E on diabetes-induced learning and memory impairment in rats. Eur J Pharmacol. 2006;537:106–110. doi: 10.1016/j.ejphar.2006.03.024. [DOI] [PubMed] [Google Scholar]
- Valastro B, Cossette J, Lavoie N, Gagnon S, Trudeau F, Massicotte G. Up-regulation of glutamate receptors is associated with LTP defects in the early stages of diabetes mellitus. Diabetologia. 2002;45:642–650. doi: 10.1007/s00125-002-0818-5. [DOI] [PubMed] [Google Scholar]
- Van de Kar LD, Blair ML. Forebrain pathways mediating stress-induced hormone secretion. Front Neuroendocrinol. 1999;20:1–48. doi: 10.1006/frne.1998.0172. [DOI] [PubMed] [Google Scholar]
- van der Hart MG, Czeh B, de Biurrun G, Michaelis T, Watanabe T, Natt O, Frahm J, Fuchs E. Substance P receptor antagonist and clomipramine prevent stress-induced alterations in cerebral metabolites, cytogenesis in the dentate gyrus and hippocampal volume. Mol Psychiatry. 2002;7:933–941. doi: 10.1038/sj.mp.4001130. [DOI] [PubMed] [Google Scholar]
- von Gunten A, Fox NC, Cipolotti L, Ron MA. A volumetric study of hippocampus and amygdala in depressed patients with subjective memory problems. J Neuropsychiatry Clin Neurosci. 2000;12:493–498. doi: 10.1176/jnp.12.4.493. [DOI] [PubMed] [Google Scholar]
- Vouimba RM, Munoz C, Diamond DM. Differential effects of predator stress and the antidepressant tianeptine on physiological plasticity in the hippocampus and basolateral amygdala. Stress. 2006;9:29–40. doi: 10.1080/10253890600610973. [DOI] [PubMed] [Google Scholar]
- Vythilingam M, Heim C, Newport J, Miller AH, Anderson E, Bronen R, Brummer M, Staib L, Vermetten E, Charney DS, Nemeroff CB, Bremner JD. Childhood trauma associated with smaller hippocampal volume in women with major depression. Am J Psychiatry. 2002;159:2072–2080. doi: 10.1176/appi.ajp.159.12.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Gould E, Daniels DC, Cameron H, McEwen BS. Tianeptine attenuates stress-induced morphological changes in the hippocampus. Eur J Pharmacol. 1992;222:157–162. doi: 10.1016/0014-2999(92)90830-w. [DOI] [PubMed] [Google Scholar]
- Wilson SP, Yeomans DC. Virally mediated delivery of enkephalin and other neuropeptide transgenes in experimental pain models. Ann N Y Acad Sci. 2002;971:521. doi: 10.1111/j.1749-6632.2002.tb04516.x. [DOI] [PubMed] [Google Scholar]
- Winocur G, Gagnon S. Glucose treatment attenuates spatial learning and memory deficts of aged rats on tests of hippocampal function. Neurobiol Aging. 1998;19:233–241. doi: 10.1016/s0197-4580(98)00057-8. [DOI] [PubMed] [Google Scholar]
- Winocur G, Greenwood CE, Piroli GG, Grillo CA, Reznikov LR, Reagan LP, McEwen BS. Memory Impairment in Obese Zucker Rats: An Investigation of Cognitive Function in an Animal Model of Insulin Resistance and Obesity. Behav Neurosci. 2005;119:1389–1395. doi: 10.1037/0735-7044.119.5.1389. [DOI] [PubMed] [Google Scholar]
- Wolff SP. Diabetes mellitus and free radicals. Brit Med Bull. 1993;49:642–652. doi: 10.1093/oxfordjournals.bmb.a072637. [DOI] [PubMed] [Google Scholar]
- Wood GE, Young LT, Reagan LP, Chen B, McEwen BS. Stress-induced structural remodeling in hippocampus: prevention by lithium treatment. Proc Natl Acad Sci U S A. 2004;101:3973–3978. doi: 10.1073/pnas.0400208101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto BK, Reagan LP. The glutamatergic system in neuronal plasticity and vulnerability in mood disorders. Neuropsychiatric Disease and Treatment 2006 [Google Scholar]
- Zhao W, Chen H, Moore E, Meiri N, Quon MJ, Alkon DL. Brain insulin receptors and spatial memory. J Biol Chem. 1999;274:34893–34902. doi: 10.1074/jbc.274.49.34893. [DOI] [PubMed] [Google Scholar]