

Summary
Culturing and subcultivation of normal human diploid fibroblasts have advanced our understanding of the molecular events involved in aging. This progress is leading to the development of therapies that slow or ablate the adverse physiological and pathological changes associated with aging. It has been established that normal human diploid fibro-blasts can proliferate in culture for only finite periods of time. Hayflick and Moorhead and others have described numerous types of cells, ranging from fetal to adult, that were incapable of indefinite proliferation. There are many ways to study aging in vitro, and this chapter summarizes some laboratory procedures.
Keywords: Senescence, aging, bio-marker, fibroblasts, cell culture
1. Introduction
Normal somatic cells that are serially cultured are a well studied model system that mimics the cellular and molecular changes associated with development and aging. Aging is a complex process that may consist of both environmentally stimulated and inherently programmed components. The cell culture system could be better likened to a model of cell proliferation regulatory mechanisms that shows age-associated changes. It is interesting to note that the two major age-associated diseases, cancer and atherosclerosis, demonstrate failures in cell proliferation regulation. Rubin (1) proposes that the limit on replicative life span is a result of the inability of cells to adapt to the trauma of explantation and foreign conditions of cell culture. Rubin also addresses the doubts as to whether normal somatic cells divide only approx 50 times in an average life span in vivo. Extrapolating from data garnered from radiolabeled mouse cells and comparing it to the 70-yr life span of humans, studies have projected that human cells are able to divide between 500 and 5000 times during a lifetime (depending on cell type), and these numbers differ greatly from the 20–70 population doublings observed in vitro (reviewed in ref. 1).
Much information has been gained in the area of therapeutic strategies, centering on the prevention and treatment of the ailments known to increase with age. Many studies have concentrated on specific age-related conditions that augment morbidity and mortality, as aging itself is more difficult to address and mimic in an in vitro culture condition (2). To this end, research approaches either focus on the age-associated changes with unfavorable consequences or the processes underlying many age-related dysfunctions (3). Questions arise as to whether such studies accurately represent what occurs inside of an organism. Cultivation requires that cells continuously undergo proliferation, which does not occur in an organism and which stresses cells unnaturally (4). Moreover, cells in culture and those in an intact organism differ drastically in metabolic needs, growth conditions, etc. The conventional 10-fold dilution of serum in culture media contains a lower concentration of protein than is normally found in extracellular tissue fluids (1). To strengthen the argument that the needs of cultured cells differ from those of cells in intact tissue, Rubin cites the study illustrating that freshly isolated cells require 13 amino acids for stable growth, whereas cells in an organism require 8–10. Many cells fail to multiply unless seeded at a relatively high population density. This fact is reiterated by the great effect on proliferative capacity of conditioned media, especially on small numbers of cells (5).
The limited growth potential observed in cultured somatic cells has been labeled an in vitro artifact of genetic damage caused by the synthetic environment, but a normal human cell population with infinite proliferative capacity has not been reported. Moreover, in light of the extremely low incidence of spontaneous immortalization, it is indeed probable that the replicative potential of human cells is inherently limited. Although the decrease in cell growth potential during serial cultivation has been implicated in causing aging, there is no evidence that correlates human aging with somatic cells losing their potential to divide (reviewed in ref. 6). Furthermore, studies have failed to consistently correlate decreased telomere length, increased lysosomal enzyme function (i.e. β-galactosidase staining), or genes involved in cell cycling with senescence rather than a reversible, prolonged postmitotic state. Yeast use recombination and Drosophila utilize specific retrotransposons to lengthen the ends of chromosomes (7). The limited proliferative capacity of somatic cells appears an appropriate system by which to study human aging, as long as correct correlations are made between observed phenomena and the physiology of the entire organism. The culture system exhibits the gradual, stepwise changes that permanently alter the organism, not so much in discrete phases as in a continuum of events that mirror the time from the fusion of the gametes to the demise of the organism.
2. Culturing Aging Fibroblasts
2.1. Procedures for Culturing Aging Fibroblasts
2.1.1. Materials
The following materials are for mammalian cells grown in a monolayer in Petri plates or flasks. Sterile conditions must be maintained at all times and all culture work should be performed under a laminar flow hood. Although it is customary to add antimicrobial agents to culture media to prevent contamination, it is not completely necessary. Indeed, prolonged usage may cause cell lines to develop antibiotic resistance or lead to cytotoxicity. Moreover, certain combinations of antimicrobial substances may be incompatible. For most aging cell types, an antibiotic/antimycotic solution of streptomycin, amphotericin B, and penicillin (Mediatech, Inc., http://www.cellgro.com) added to the media at a final concentration of 1% adequately hinders the growth of bacteria, fungi, and yeast. All incubations and long-term culturing must be done in a humidified 37°C, 5% CO2 incubator. Media, trypsin/EDTA solutions, and phosphate-buffered saline (PBS) should be warmed to 37°C in a water bath before use. Cells are grown and subcultured in appropriate media (see Table 1 for culture media components for common cell types).
Table 1.
Culture Media Components of Common Cell Types
| Cell line | Base media | Supplements | Serum |
|---|---|---|---|
| MRC-5, WI-38, IMR-90 fetal lung fibroblasts, BJ foreskin fibroblasts | Minimum Essential Medium (MEM) Eagle + Earle's Basic Salt Solution (BSS) | 2X concentration of essential & nonessential amino acids and vitamins 2 mM l-glutamine, 1.5 g/L sodium bicarbonate, 1.0 mM sodium pyruvate | 15% FBS, uninactivated |
| CCL-39 Chinese hamster lung fibroblasts | McCoy's 5a | 10% FBS | |
| A9 L mouse fibroblasts | Dulbecco's Minimum Essential Medium (DMEM) | 4 mM l-glutamine, 1.5 g/L sodium bicarbonate, 4.5 g/L glucose | 10% FBS |
| C2C12 mouse myoblasts | Dulbecco's Minimum Essential Medium (DMEM) | 4 mM l-glutamine, 1.5 g/L sodium bicarbonate, 4.5 g/L glucose | 10% FBS (or 10% horse serum to enhance myotube formation) |
| RBL-2H3 rat basophilic leukemia | Minimum Essential Medium (MEM) Eagle + Earle's Basic Salt Solution (BSS) | 2 mM l-glutamine, 1.5 g/L sodium bicarbonate, 0.1 mM nonessential amino acids, 1.0 mM sodium pyruvate | 15% FBS, heat-inactivated |
2.1.2. Methods
The methods outlined as follows describe the thaw and recovery of frozen cells, the trypsinization and subcultivation of monolayer cells, and the freezing of monolayer cells.
2.1.2.1. Thaw and Recovery
Remove cryovial from liquid nitrogen and place in a 37°C water bath. Agitate gently until thawed.
Wipe vial with 70% ethanol before putting under hood and opening.
Transfer cells to a sterile 15 mL conical tube containing medium prewarmed to 37°C. Centrifuge 5–10 min at 150–200g (approx 1000 rpm in a swinging bucket or 45° fixed-angle rotor). Discard supernatant containing residual dimethylsulfoxide (DMSO).
Resuspend pellet in 1 mL complete medium and transfer to culture plate/flask containing the appropriate amount of medium. Place in incubator. Check cells for adherence after 24 h.
2.1.2.2. Trypsinization and Subcultivation
Cells should be subcultured upon approx 90% confluence to avoid contact inhibition or transformation.
Remove media from primary culture with a sterile Pasteur pipet. Wash adherent cells with a small volume of sterile PBS to remove residual fetal bovine serum (FBS), which may inhibit trypsin.
Add sufficient 37°C trypsin/EDTA solution (see Note 1) to cover the cell monolayer. Place in incubator for 1–2 min (see Note 2). Check cells with an inverted microscope to make sure cells are detached.
Add appropriate amount (2–6 mL) of warmed culture medium and disperse cells by gently pipetting up and down.
Add appropriate aliquots of the cell suspension to new culture vessels.
Incubate cultures, checking for adherence after 24 h.
Renew medium every 2–4 d.
2.1.2.3. Freezing
Cells to be frozen should be in late log phase growth.
Dissociate monolayers with trypsin/EDTA (see Note 1) and resuspend cells in complete medium.
Count resuspended cells to determine viability and number (see Note 3).
Gently pellet cells for 5 min at approx 300–350g (1500 rpm in a swinging bucket or 45° fixed-angle rotor) and remove and discard supernatant above the pellet.
Resuspend cells in freezing medium (see Note 4) at a concentration of approx 5 × 106 cells/mL. Aliquot cell suspension into labeled cryovials and freeze immediately.
Cells can be frozen at −80°C for short-term storage. (Optional: place cryovials into a styrofoam container or slow freezer such as the Nalgene® Cryo Freezing Container filled with isopropanol, which has a repeatable −1°C/min cooling rate to prevent the formation of damaging ice crystals.)
Transfer cells in cryovials to liquid nitrogen (−196°C) after 24 h for long-term storage.
2.2. Aging Cell Culture: Major Problems and Experimental Considerations
2.2.1. Relationship of Replicative Senescence to In Vivo Senescence
One major question in the study of aging is whether the changes we observe in replicative senescence correlate with the pathways and mechanisms of cell senescence in situ. Support for the direct relationship between replicative senescence and cell senescence in situ has come from the decline in the propagative lifespan of skin fibroblasts in culture as a function of donor age (9-11). However, consistent findings confirming the inverse relationship of donor age to proliferative capacity have not been shown, which has been problematic for defining the relevance of the cell culture model to aging of the entire organism (9).
A major source of controversy regarding replicative senescence is the fact that many authors have modulated certain pathways and cell culture conditions leading to the senescent phenotype (reviewed in ref. 12), showing that the senescent phenotype can be achieved despite proliferation. This evidence suggests that the senescent phenotype is a final, common pathway for actively dividing cells in which signaling and/or metabolic imbalances occur. This has led to the conclusion that cells may not be able to achieve their true differentiated fate in vivo because of signals missing from the culture medium or failure to process these signals (12). However, establishing this direct relationship is not a requirement for utilizing cell culture as a tool for studying aging mechanisms. In fact, fibroblasts in culture express human genetic, metabolic, and regulatory behavior allowing the cells to undergo changes in a predictable and reproducibly constant environment (9).
2.2.2. Extra-Culture Conditions
Cells maintained in culture are continuously exposed to full-spectrum fluorescent light, which can be detrimental to stable cell conditions. This light can affect culture conditions as well as the cells themselves. It has been illustrated that exposure to fluorescent light can damage photoactive components of cell culture media (13). Also, repeated exposure to standard fluorescent light greatly enhances the frequency of single-stranded DNA breaks (14,15). There are several other conditions that can induce a premature senescent phenotype, such as hydrogen peroxide (16), tert-butylhydroperoxide (17), and ultraviolet (18) and gamma radiation (19). Another source of cellular stress is the trypsinization procedure utilized to remove adherent cells from the substratum. It was shown by comparing different frequencies of trypsinizations to a nonenzymatic technique that weekly trypsinizations do not affect the proliferative capacity of the mass culture (20).
2.2.3. Timeline
There are some inherent drawbacks to studying aging in vitro that should be considered when outlining a study. Aging studies are often time-consuming. For accuracy, it is better to investigate one serially cultured sample at different ages than to examine a few different cultured samples at different time periods. This often takes months of assaying young cells, growing the same culture to near-senescence, and assaying the aged cells. One solution is to start multiple cultures offset by a few days so that cells can be cultured and assayed according to their experimental design simultaneously. While serially culturing aging cells, it is important to store cells at various ages. Freezing cells in liquid nitrogen is a good way to keep cells at a certain age (see Subheading 2.1.2.3. and Note 4). Should a problem arise with a culture of a certain age, re-freezing enables the researcher to begin the study again. This also obviates frequent re-ordering of cells at specific ages, which is contingent on their availability. The Coriell Institute (http://ccr.coriell.org/nia/) provides many different cell lines used in aging research; however, not all ages are available consistently.
3. In Vitro Signs of Aging
3.1. Morphological Signs of Aging
Evidence for the correlation of aging cells in culture with cellular aging in vivo exists, especially in light of the events that occur both in vitro and in vivo. Macieira-Coelho (4) reviews the overlapping events, which include the loss of division potential; chromatin alterations; loss of the capacity to migrate; increase in cell size, volume, and protein content; and decreased mitogenic response to growth factors. Characteristic morphological changes accompany replicative senescence observed in culture, namely increased cell size, nuclear size, nucleolar size, number of multinucleated cells, prominent Golgi apparati, increased number of vacuoles in the endoplasmic reticulum and cytoplasm, increased numbers of cytoplasmic microfilaments, and large lysosomal bodies (reviewed in ref. 21). These cells also seem to exhibit an increased sensitivity to cell contact (21), perhaps as a result of changes in interactions with the extra-cellular matrix (ECM) or expression of secreted proteins (reviewed in ref. 12), resulting in reduced harvest and saturation densities (22). Although the synthesis of macromolecules decreases with increasing age, the intracellular content of RNA and proteins increases. These elevations could contribute to the increased cell and nuclear size and numbers of inclusion bodies observed in late-passage cells, and might be caused by reduced protein degradation by proteosome-mediated pathways, declined RNA turnover, and the uncoupling of cell growth from cell division (and putative block of senescent cells in late G1).
3.2. Bio-Markers of Aging
Some important biological markers of aging exist that could help identify senescent cells in culture and in vivo. Some of these markers are outlined in Table 2, and include IGF-1, EGF, c-fos, and senescence-associated β-galactosidase (see Table 2 for references). IGF-1 is produced by many cell types and plays an important role in regulation of cell proliferation. Ferber et al. (23) investigated the production of IGF-1 and its ligand, the IGF-1 receptor, in normal diploid fibroblasts that were both presenescent and senescent. The results illustrated that IGF-1 mRNA production was lowered to undetectable levels in senescent cells whereas IGF-1 receptor mRNA production remained at detectable levels. Another biological marker that is differentially regulated in senescent cells is epidermal growth factor (EGF). EGF signaling has been postulated to be impaired in nonproliferating senescent human diploid fibroblasts downstream of receptor binding. Carlin et al. (24) compared lysates from young and senescent WI-38 cells for proteolytic activity directed toward the EGF receptor. Their results indicate a protease that cleaves the 170 kDa EGF receptor and is produced only in senescent fibroblasts. The production of the proto-oncogene c-fos is important in growth regulation, as it is part of the AP-1 transcriptional activator. In 1990, Seshadri and Campisi (25) demonstrated a loss of c-fos inducibility in senescent WI-38 cells, suggesting that lack of proliferation was in some part due to selective repression of c-fos. Staining for β-galactosidase is a technique to label senescent cells in situ and has been used in many studies to determine the amount of senescence achieved in culture (26-28). Principles and techniques for senescence-associated β-gal staining (SA-β-gal) are discussed in Chapter 4.
Table 2.
Molecular Markers of Aging
| Cell line tested | Aging bio-marker |
Description | Reference |
|---|---|---|---|
| WI-38, HS74, IMR-90 | IGF-1 | Senescent cells do not produce mRNA for IGF-1 | Ferber et al., 1993 (23) |
| WI-38 | EGF | Altered form of EGF produced in senescent cells | Carlin et al., 1994 (24) |
| WI-38 | c-fos | Repressed in late passage cells | Seshadri and Campisi, 1990 (25) |
| WI-38, IDH4, NHEK, CMV-MJ, HCA2 | SA-β-Gal | Expressed in senescent cells but not in quiescent or terminally differentiated cells | Dimri et al., 1995 (26) Itahana et al. (Chapter 4, this volume) |
3.3. Quantification of Cellular Aging
The aging process in vitro is accompanied by a loss in proliferative potential, which results in a decline in cellular replication. The factors involved in this reduction are as yet unknown. During aging studies, it is necessary to detail growth and keep track of cellular age in terms of population doublings, or the number of times a cell population doubles after a predesignated period of time. Generally, population doublings can be recorded by plating a specific number of cells and then counting those cells after a defined period of growth. If the population has doubled, for example a plating of 1.0 × 106 cells is followed by a growth phase and a count of 2.0 × 106, one population doubling is added to the current age of the cells. However, because even counts as specified above are almost never achieved, a more precise method is needed for accuracy. Figure 1 outlines an equation developed in our lab that takes into account all the cells counted and results in a more exact population doubling.
Fig. 1.
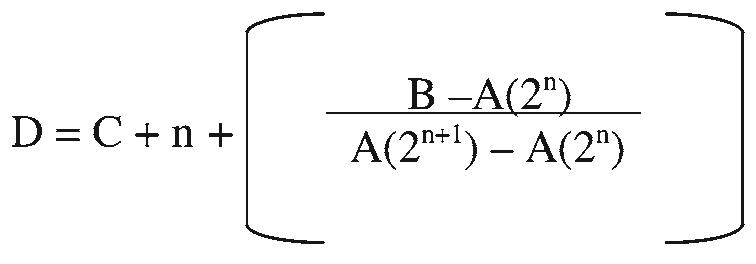
Equation to determine population doubling of aging cells in culture. Where A equals no. of cells plated, B equals no. of cells counted after growth period, C equals old population doubling, D equals new population doubling and n equals the largest number that satisfies the equation A(2n) ≤ B.
Acknowledgments
This work was supported in part by the NSF GK-12 Fellowship (to S. M. O. Phipps), the Cancer Prevention and Control Training Program (to J. B. Berletch), the UAB Ovarian SPORE, and the UAB Evelyn McKnight Brain Institute grant.
Footnotes
Most cell types require a 0.25% (w/v) trypsin/0.2% (w/v) EDTA solution prepared in sterile Hank's Balanced Salt Solution (HBSS) or 0.9% (w/v) NaCl to detach cells and chelate Ca2+ and Mg2+ ions that could hinder the action of trypsin.
With the exception of BJ fibroblasts and other primary cell cultures, 1–2 min submerged in trypsin at room temperature or 37°C should be sufficient to disperse cells. BJs and other difficult cell types may require 5–15 min of incubation at 37°C, with occasional checks under an inverted microscope to ensure that cells are detached. To avoid clumping of cells with fibroblast-like morphology, do not agitate the cells by hitting or shaking the flask/culture vessel. Continue to place cells at 37°C until they are no longer adherent or until 15 mins have passed (extended trypsin exposure damages cells).
Freezing medium is composed of 90–95% complete culture medium supplemented with 5–10% DMSO or glycerol.
References
- 1.Rubin H. Cell aging in vivo and in vitro. Mech. Ageing Dev. 1997;98:1–35. doi: 10.1016/s0047-6374(97)00067-5. [DOI] [PubMed] [Google Scholar]
- 2.Lai SR, Phipps SMO, Andrews LG, Tollefsbol TO. Epigenetic control of telomerase and modes of telomere maintenance in aging and abnormal systems. Front. Biosci. 2005;10:1779–1796. doi: 10.2741/1661. [DOI] [PubMed] [Google Scholar]
- 3.Hadley EC, Lakatta EG, Borrison-Bogorad M, Warner HR, Hodes RJ. The future of aging therapies. Cell. 2005;120:557–567. doi: 10.1016/j.cell.2005.01.030. [DOI] [PubMed] [Google Scholar]
- 4.Macieira-Coelho A. Behavior of cells in culture and physiopathology of organism. In: von Hahn HP, editor. Biology of Normal Proliferating Cells in vitro. Relevance for in vivo Aging. Karger; Basel, Switzerland: 1988. pp. 6–20. [Google Scholar]
- 5.Rubin H. A substance in conditioned medium which enhances the growth of chick embryo cells in tissue culture. Exp. Cell Res. 1966;41:138–148. doi: 10.1016/0014-4827(66)90554-4. [DOI] [PubMed] [Google Scholar]
- 6.Macieira-Coelho A. Ups and downs of aging studies in vitro: The crooked path of science. Gerontol. 2000;46:55–63. doi: 10.1159/000022135. [DOI] [PubMed] [Google Scholar]
- 7.Biessmann H, Mason JM. Telomere maintenance without telomerase. Chromosoma. 1997;106:63–69. doi: 10.1007/s004120050225. [DOI] [PubMed] [Google Scholar]
- 8.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 9.Cristofalo VJ, Beck J, Allen RG. Cell Senescence: An evaluation of replicative senescence in culture as a model for cell aging in situ, commentary and author's response to commentary. J. Gerontol. 2003;58A:776–781. doi: 10.1093/gerona/58.9.b776. [DOI] [PubMed] [Google Scholar]
- 10.Martin GM, Sprague CA, Epstein CJ. Replicative life-span of cultivated human cells. Lab Invest. 1970;23:86–92. [PubMed] [Google Scholar]
- 11.Schneider EC, Mitsui Y. The relationship between in vitro cellular aging and in vivo human age. Proc. Natl. Acad. Sci. USA. 1976;73:3584–3588. doi: 10.1073/pnas.73.10.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cristofalo VJ, Lorenzini A, Allen RG, Torres C, Tresini M. Replicative senescence: a critical review. Mech. Ageing Dev. 2004;125:827–848. doi: 10.1016/j.mad.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 13.Wang RJ. Effect of room fluorescent light on the deterioration of tissue culture medium. In Vitro. 1976;12:19–22. doi: 10.1007/BF02832788. [DOI] [PubMed] [Google Scholar]
- 14.Bradley MO, Sharkley NA. Mutagenicity and toxicity of visible fluorescent light to cultured mammalian cells. Nature. 1976;266:724–727. doi: 10.1038/266724a0. [DOI] [PubMed] [Google Scholar]
- 15.Bradley MO, Hsu IC, Harris CC. Relationship between sister chromatid exchange and mutagenicity, toxicity and DNA damage. Nature. 1979;282:318–320. doi: 10.1038/282318a0. [DOI] [PubMed] [Google Scholar]
- 16.Frippiat C, Chen QM, Remacle J, Toussaint O. Cell cycle regulation in H2O2 induced premature senescence of human diploid fibroblasts and regulatory control exerted by the papilloma virus E6 and E7 proteins. Exp. Gerontol. 2000;35:733–745. doi: 10.1016/s0531-5565(00)00167-4. [DOI] [PubMed] [Google Scholar]
- 17.Dumont P, Burton M, Chen QM, et al. Induction of replicative senescence biomarkers by sublethal oxidative stresses in normal human fibroblasts. Free Radic. Med. 2000;28:361–373. doi: 10.1016/s0891-5849(99)00249-x. [DOI] [PubMed] [Google Scholar]
- 18.Ma W, Wlaschek M, Hommel C, Schneider LA, Scharffetter-Koehanek K. Psoralen plus UVA (PUVA) induced premature senescence as a model for stress-induced premature senescence. Exp. Gerontol. 2002;37:1197–1201. doi: 10.1016/s0531-5565(02)00143-2. [DOI] [PubMed] [Google Scholar]
- 19.Seidita G, Polizzi D, Costanzo G, Costa S, Di Leonardo A. Differential gene expression in p53-mediated G(1) arrest of human fibroblasts after gamma-irradiation or N-phosphoacetyl-L-aspartate treatment. Carcinogenesis. 2000;21:2203–2210. doi: 10.1093/carcin/21.12.2203. [DOI] [PubMed] [Google Scholar]
- 20.Hadley EC, Kruss ED, Cristofalo VJ. Trypsinization frequency and loss of proliferative capacity in WI-38 cells. J. Gerontol. 1979;34:170–176. doi: 10.1093/geronj/34.2.170. [DOI] [PubMed] [Google Scholar]
- 21.Cristofalo VJ, Pignolo RJ. Replicative senescence of human fibro-blast-like cells in culture. Physiological Reviews. 1993;73:617–638. doi: 10.1152/physrev.1993.73.3.617. [DOI] [PubMed] [Google Scholar]
- 22.Cristofalo VJ. Cellular biomarkers of aging. Exp. Gerontol. 1988;23:297–307. doi: 10.1016/0531-5565(88)90032-0. [DOI] [PubMed] [Google Scholar]
- 23.Ferber A, Chang C, Sell C, et al. Failure of senescent human fibroblasts to express the insulin-like growth factor-1 gene. J. Biol. Chem. 1993;265:17,883–17,888. [PubMed] [Google Scholar]
- 24.Carlin C, Phillips PD, Brooks-Frederich K, Knowles BB, Cristofalo VJ. Cleavage of the epidermal growth factor receptor by a membrane-bound leupeptin-sensitive protease active in nonionic detergent lysates of senescent but not young human diploid fibroblasts. J. Cell Physiol. 1994;160:427–434. doi: 10.1002/jcp.1041600305. [DOI] [PubMed] [Google Scholar]
- 25.Seshadri T, Campisi J. Repression of c-fos transcription and an altered genetic program in senescent human fibroblasts. Science Wash. DC. 1990;247:205–209. doi: 10.1126/science.2104680. [DOI] [PubMed] [Google Scholar]
- 26.Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alexander K, Yang HS, Hinds PW. Cellular senescence requires CDK5 repression of Rac1 activity. Mol. Cell Biol. 2004;24:2808–2819. doi: 10.1128/MCB.24.7.2808-2819.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stenderup K, Justesen J, Clausen C, Kassem M. Aging is associated with decreased maximal life span and accelerated senescence of bone marrow stromal cells. B. Bone. 2003;33:919–926. doi: 10.1016/j.bone.2003.07.005. [DOI] [PubMed] [Google Scholar]