

Abstract
Ligand binding to enzymes and antibodies is often accompanied by protein conformational changes. Although such structural adjustments may be conducive to enzyme catalysis, much less is known about their effect on reactions promoted by engineered catalytic antibodies. Crystallographic and pre-steady state kinetic analyses of antibody 34E4, which efficiently promotes the conversion of benzisoxazoles to salicylonitriles, show that the resting catalyst adopts two interconverting active-site conformations, only one of which is competent to bind substrate. In the predominant isomer, the indole side chain of TrpL91 occupies the binding site and blocks ligand access. Slow conformational isomerization of this residue, on the same time scale as catalytic turnover, creates a deep and narrow binding site that can accommodate substrate and promote proton transfer using GluH50 as a carboxylate base. Although 34E4 is among the best catalysts for the deprotonation of benzisoxazoles, its efficiency appears to be significantly limited by this conformational plasticity of its active site. Future efforts to improve this antibody might profitably focus on stabilizing the active conformation of the catalyst. Analogous strategies may also be relevant to other engineered proteins that are limited by an unfavorable conformational pre-equilibrium.
Tailored antibody catalysts have been generated for a wide variety of reactions using transition state analogs or other appropriately designed template molecules as antigens (1). Although these proteins exhibit many of the properties of authentic enzymes, including rate accelerations, substrate specificity, regioselectivity, and stereoselectivity, their efficiency generally lags behind that of their natural counterparts. Among the many factors that contribute to antibody inefficiency (2), suboptimal conformational properties of the immunoglobulin scaffold have been cited as a potentially significant limitation (3, 4).
Proteins are innately flexible, undergoing conformational changes over a wide range of time scales and amplitudes. Such flexibility is believed to be important for enzyme function (5–8). Dynamic structural fluctuations can influence substrate and product binding. They also enable the catalyst to adjust to changes in the substrate as the reaction coordinate is traversed, and they provide a means to position functional groups for efficient electrostatic, nucleophilic, and acid-base catalysis. Conformational changes may even shape the effective barrier of the catalyzed reaction in some cases (9).
Antibodies undergo a similar range of conformational changes as enzymes. Switches between different rotamers of individual side chains, segmental movements of hypervariable loops, and alterations in the relative disposition of the VH and VL domains have all been observed (3, 10, 11). These structural adjustments increase the effective diversity of the primary immunological repertoire and are important for achieving high affinity and selective molecular recognition (12). However, such conformational changes are difficult to exploit intentionally for antibody catalysis, given the indirect nature of the immunological selection process, which optimizes binding to an imperfect transition state mimic rather than catalytic activity. In fact, affinity maturation reduces conformational flexibility in some antibodies and also increases specificity (13–16). Comparisons of germ line and mature antibodies catalyzing an oxy-Cope rearrangement indicate that such rigidification can be deleterious to catalytic efficiency (17). Nevertheless, investigations of a family of esterolytic antibodies (18) provide evidence that conformational changes can be beneficial in some instances and contribute to higher rate accelerations. In other cases, structural dynamics may influence substrate binding or product release. For example, crystallographic snapshots of the complete reaction cycle of antibody cocaine degradation visualized significant conformational changes in the active site along the reaction coordinate (19). Although substrate and products were bound in partially open conformations, the antibody active site engulfed the transition state analog more tightly, thus enabling transition state stabilization through hydrogen bonding (19).
In this study, crystallographic and kinetic approaches were employed to characterize structural changes in a catalytic antibody promoting the conversion of benzisoxazoles to salicylonitriles (Fig. 1, 1 → 3). This reaction, known as the Kemp elimination, is a valuable model system for studying proton transfer from carbon (20–22). Antibody 34E4 was generated against the cationic 2-aminobenzimidazolium hapten 4 and catalyzes this transformation with high rates (kcat = 0.7 s-1, kcat/Km = 6 × 103 m-1 s-1) and multiple turnovers (>103) (23). Combined modeling (24), mutagenesis (25), and structural (26) work implicate the carboxylate group of the heavy-chain glutamate 50 (GluH50, Kabat numbering) as the catalytic base. These studies further suggest that high catalytic efficiency in this system derives, at least in part, from successful exploitation of extensive hydrogen bonding, π-stacking, and van der Waals interactions to align base and substrate.
FIGURE 1.

Base-catalyzed Kemp elimination of 5-nitrobenzisoxazole (1). The base abstracts the C-3 proton from the substrate (1) and induces its decomposition. Antibody 34E4 was raised against hapten 4 and efficiently catalyzes the Kemp elimination. Compound 5 is a hapten derivative, which was used for measuring the hapten binding kinetics, and compound 6 is a noncleavable substrate analog, which served as a model compound to determine the substrate binding kinetics.
The possibility that conformational changes might be important in 34E4 is suggested by sequence comparisons with the recently characterized, noncatalytic antibody SPE7 (27, 28). This antibody was raised against a 2,4-dinitrophenyl hapten, but is a highly dynamic receptor, which binds multiple ligands via different active-site conformations (27). The variable λ light chains of 34E4 and SPE7 are 93% identical, and the backbone atoms in their respective framework regions have a root mean square deviation of only 0.4 Å. Although their variable heavy chain domains do not appear to be closely related (52% identity, 1.1 Å root mean square deviation), key residues lining the binding pocket are also conserved. Indeed, consistent with these similarities, we find that 34E4 is subject to conformational changes that adversely affect catalytic efficiency. Specifically, the apo antibody exists predominantly in an unreactive form that must undergo slow and significant structural rearrangement to accommodate its substrate. Comparisons with other structurally characterized catalytic antibodies suggest that this behavior may be quite general. By stabilizing the active form of the catalyst, it may be possible to enhance efficacy in these systems significantly.
EXPERIMENTAL PROCEDURES
34E4 Fab Preparation, Crystallization, and Data Collection—The apo 34E4 Fab4 was produced and purified as a murine-human chimera, as described previously (25). The Fab, concentrated to 15 mg/ml, was crystallized by the sitting drop vapor diffusion method at 22.5 °C. The Fab crystallized from 30% MPEG 2000, 0.1 m sodium acetate (pH 4.3), 0.2 m (NH4)2SO4 in orthorhombic space group P212121 (crystal form A) and from 32% PEG 2000, 0.2 m CdCl2, 0.1 m Bistris propane (pH 7.0), in triclinic space group P1 (crystal form B). For data collection, the crystals were flash-cooled to 100 K using 25% glycerol as cryoprotectant. Data were collected at ALS Berkeley and SSRL Stanford. Data processing and scaling were performed in HKL2000 (29). The data processing statistics are summarized in Table 1.
TABLE 1.
Crystallographic data collection and refinement statistics
| 34E4 crystal form A | 34E4 crystal form B | |
|---|---|---|
| Space group | P212121 | P1 |
| Unit cell dimensions (Å,°) | a = 81.1, b = 114.5, c = 212.1, α = β = γ = 90.0 | a = 81.1, b = 106.3, c = 116.1, α = 89.9, β = 90.0, γ = 89.5 |
| No. of Fabs/water molecules in asua | 4/277 | 8/518 |
| Resolution range (Å) | 50.0–2.6 (2.69–2.60)b | 50.0–2.6 (2.66–2.60) |
| Unique reflections | 58,339 (6,285) | 112,380 (7,869) |
| Completeness (%) | 99.7 (99.9) | 95.3 (94.7) |
| Rsymc | 0.108 (0.628) | 0.064 (0.465) |
| 〈I/σ〉 | 11.9 (2.0) | 11.3 (1.5) |
| Redundancy | 4.1 (4.1) | 1.6 (1.5) |
| Rcrystd/Rfreee | 0.20/0.25 | 0.22/0.24 |
| r.m.s.d.f bond length (Å)/angle (°) | 0.013/1.4 | 0.012/1.3 |
| Ramachandran plot, most favored/additionally allowed/generously allowed/disallowedg (%) | 89.9/9.4/0.2/0.5 | 88.7/10.7/0.2/0.4 |
asu means asymmetric unit
Data indicate the highest resolution shell
Rsym = ΣhklΣi|Ii(hkl) – 〈Ii(hkl)〉|/ΣhklΣi|Ii(hkl)|
Rcryst = Σhkl||Fc(hkl)| – |Fo(hkl)||/Σhkl|Fo(hkl)|
Rfree is calculated as for Rcryst, but from 5% of the data that was not used for refinement
r.m.s.d. means root mean square deviation
ThrL51 and SerL93 are the only residues in a disallowed region, as observed in the hapten complex structures. ThrL51 is in a well defined γ-turn, as in almost all other antibodies (46)
Structure Determination and Refinement—The structures of unliganded 34E4 Fab were determined by molecular replacement using the coordinates of the previously determined hapten complex (26) with the program Phaser (30). The structures were refined by alternating cycles of model building with the program O (31) and refinement with Refmac5 (32). During refinement, tight noncrystallographic symmetry restraints were applied to all main-chain atoms of the Fab molecules in the asymmetric unit, except for some loop regions. The final refinement statistics are shown in Table 1. The quality of the structures was constantly monitored using the programs Mol-Probity (33), WHAT IF (34), and PROCHECK (35).
Fluorescence Titration—Thermodynamic dissociation constants (Kd) for the ligand·Fab complex were determined as described previously (36). A titration curve was generated by stepwise addition of ligand (0–3 μm) to a dilute solution of the chimeric Fab fragment (50 nm) and subsequent measurement of the fluorescence. The excitation and emission wavelengths were 290 and 340 nm, respectively, and the corresponding band pass was either 8 or 16 nm. The high voltage of the detector was set to 800–900 V. These measurements were carried out in 20 mm Tris-HCl (pH 7.0) and 100 mm NaCl at 20.0 ± 0.1 °C on an Aminco-Bowman Series 2 luminescence spectrometer (SLM Aminco, Urbana, IL). From the measurements of the observed antibody fluorescence (F) at various concentrations of the ligand (LT), the dissociation constant (Kd) was calculated by nonlinear least squares fitting of the data to Equation 1,
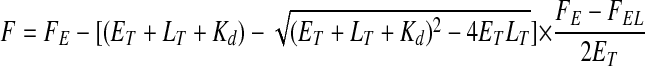 |
(Eq. 1) |
where ET is the total Fab concentration; FE is the observed fluorescence intensity without ligand, and FEL is the fluorescence intensity of the Fab·ligand complex at infinite ligand concentration.
Stopped-flow Methods—Kinetic measurements were performed on an SX.18MV stopped-flow fluorescence instrument (Applied Photophysics, Leatherhead, UK). The Fab fragment (25–50 nm) was allowed to react with ligand that was in at least 10-fold excess at equilibrium. Reactions were performed in 20 mm Tris-HCl (pH 7.0) and 100 mm NaCl at 20.0 ± 0.1 °C. Samples were excited at 290 nm, and fluorescence was monitored through a 320 nm cutoff filter. Data points from 19 to 35 reactions were averaged to improve the signal-to-noise ratio. Averaged data were fit to a single exponential or, where systematic deviations in the residuals from the fitted curve were observed, to a double exponential, using Origin 6.1 (Origin Lab Corp., Northampton, MA). In particular, the slow phase of hapten binding (Fig. 5B) is in excellent agreement with a double exponential function, as confirmed by decreased χ2 and increased R2 values with respect to a single exponential fit. The extracted rate constants, designated as kfast, kslow, and kslowest (kobs = 1/τ, where τ is the reciprocal relaxation time), are functions of a combination of terms, including ligand concentration and microscopic rate constants for individual kinetic steps. Values of kfast were fitted to a standard bimolecular association model as follows: 1/τ1 = kfast = k-2 + k2[L], where [L] is the ligand concentration. Values of kslow were fitted to a pre-equilibrium model as follows: 1/τ2 = kslow = k1 + k-1 (Ks/([L] + Ks)), where Ks = k-2/k2 (37).
FIGURE 5.

Triphasic kinetics of hapten binding. A, initial phase; B, subsequent phase, fitted to single and double exponentials, respectively. The lack of correspondence between the terminal fluorescence of the curve in A and the initial fluorescence of the curve in B is because of different settings of the instrument in two separate reactions to achieve the highest sensitivity. The three phases (fast, slow, and very slow) roughly separate into time scales of 0.04, 5, and 20 s, respectively (kfast = 54 ± 4 s-1, kslow = 0.79 ± 0.03 s-1, and kslowest = 0.130 ± 0.005 s-1).
RESULTS
Conformational Isomerism in 34E4—The crystal structure of the unliganded Fab fragment of antibody 34E4 was determined from an orthorhombic crystal form at 2.6 Å resolution and from a related triclinic form at 2.6 Å resolution by molecular replacement (Table 1). Both crystal forms contain a large number of noncrystallographic symmetry-related Fab molecules (four and eight, respectively) in the crystal asymmetric unit, which allows structural differences due to variable crystal packing environments to be distinguished from conformational changes associated with hapten binding.
A detailed quantitative comparison of the free and hapten-bound (26) antibody shows that ligand binding does not shift the relative disposition of the VL and VH domains or cause any major movement of individual CDR loops. The most striking structural difference between liganded and unliganded 34E4 is the orientation of the TrpL91 indole ring (Figs. 2 and 3). In 11 copies of the 12 unliganded Fabs, the side chain of TrpL91 predominantly assumes the m95° rotamer, with χ1 of -60° and χ2 of +95° (38). This rotamer represents the most common side-chain conformation of tryptophan in a data base of 240 high quality protein structures at 1.7 Å resolution or better (38). In only one of the 12 unliganded Fabs does TrpL91 adopt the less common t-105° rotamer, with χ1 of 180° and χ2 of -105° (38). This t-105° rotamer is the only one observed for TrpL91 in the previously determined hapten complex (26). The switch between these two m95° and t-105° rotamers substantially remodels the binding pocket and converts it from a shallow indentation on the surface of the protein to a deep cavity that can accommodate ligand (Fig. 4, A and B). Because the electron density of the TrpL91 side chain of some apo Fabs was somewhat weaker than that of the surrounding residues, indicating greater mobility and/or multiple conformations, final placement of the indole side chain into the electron density was guided by a difference Fourier omit map, where the TrpL91 side chain was truncated to the C-β atom. The resulting model then reflects the most prevalent conformation for this residue. Nearby HisH100B is the only other active-site residue to undergo significant movement. However, the rearrangement of its side chain is less pronounced than that of TrpL91.
FIGURE 2.

Antibody combining site of 34E4 in its unliganded state. The heavy and light chains are colored in blue and green, respectively. The 2Fo - 1Fc σA-weighted electron density maps are contoured at 1.1σ. Antibody 34E4 predominantly exists in the displayed closed conformation, where TrpL91 swings into the binding pocket, occluding the active site. The electron density of TrpL91 is less well defined than surrounding residues, indicating increased flexibility and/or multiple conformations.
FIGURE 3.

Superimposition of the free (blue, four independent copies of the orthorhombic crystal form) and hapten-bound (yellow, four independent copies) 34E4. The complementarity determining regions (CDRs) of the light and heavy chains, which are labeled L1–L3 and H1–H3, respectively, undergo only minor changes upon ligand binding, whereas TrpL91 swings into the binding pocket and serves as surrogate ligand in the apo form.
FIGURE 4.

Molecular surface representation of free (A) and hapten-bound (B) 34E4. The hapten is displayed as pink stick model. The architecture of the antibody combining site is dramatically different in the two states. The shallow indentation of the predominant closed conformation of the antibody in the unliganded form (A) can convert to an open conformation that can then harbor the hapten in a deep cavity (B).
Triphasic Kinetics of Hapten Binding—Fab 34E4 binds hapten derivative 5 (Fig. 1) with high affinity (Kd = 1.5 nm) (25). Ligand association is accompanied by changes in intrinsic Fab fluorescence that are resolvable by stopped-flow techniques. Typical kinetic traces are shown in Fig. 5. The fluorescence transients can be roughly divided into three phases, each of which is described by a distinct exponential function. The fast phase of the reaction appears to be complete within ∼0.1 s with an observed rate constant, kfast, of 54 ± 4 s-1 (Fig. 5A). Attempts to fit the subsequent slower process to a single exponential resulted in systematic deviations in the residuals, whereas a double exponential function gave an excellent fit to the data (Fig. 5B), with rate constants kslow = 0.79 ± 0.03 s-1 and kslowest = 0.130 ± 0.005 s-1 for the two additional phases.
The observation of triphasic transients is consistent with two possible mechanisms. In one (Fig. 6A), two conformations of the antibody, Fab and Fab*, with different reactivity toward the ligand, are in equilibrium with each other and with the respective ligand-bound complexes (12). Induced fit (Fig. 6A, clockwise from top left) and pre-equilibrium (counterclockwise from top left) mechanisms constitute the two limiting cases of this cyclic scheme. In an alternative three-step linear mechanism (Fig. 6B), only one antibody conformer (Fab*) is capable of ligand binding. Once the pocket is occupied, the initially formed Fab*·L complex subsequently undergoes an additional isomerization to form Fab**·L. In this scheme, the central binding step in conjunction with either the initial or final equilibria alone would correspond to the limiting pre-equilibrium and induced fit mechanisms, respectively.
FIGURE 6.

Various mechanisms of ligand association. The structural and kinetic data of antibody 34E4 are exclusively consistent with mechanism B. Fab, Fab*, and Fab** represent different conformations of the antibody fragment Fab of 34E4. A, cyclic combination of induced fit (upper arm) and pre-existing equilibrium (lower arm). B, linear combination of pre-existing equilibrium with subsequent induced fit isomerization.
In principle, an induced fit mechanism can be distinguished from a pre-equilibrium mechanism from the dependence of the slow phase on ligand concentration (37). However, because the change in kslow is most pronounced at ligand concentrations below the dissociation constant of the Fab·ligand complex, and negligible at saturating ligand concentrations (12), it was not possible to resolve this issue with the tight binding hapten 5.
Biphasic Kinetics of Substrate Analog Binding—Benztriazole 6 (Fig. 1) is a substrate analog that binds to the 34E4 Fab fragment with lower affinity (Kd = 643 ± 26 nm) than the hapten, as judged by fluorescence titration. The weaker affinity of this ligand was exploited to examine the concentration dependence of the rate constants associated with binding. For complex formation between 34E4 and 6, only the fast (∼0.1 s) and slow (∼5 s) phases were observed (Fig. 7A). At 5 μm, kfast and kslow were 69.0 ± 0.5 and 0.68 ± 0.03 s-1, respectively, in good agreement with kfast and kslow values obtained with 5. Although a very slow phase with low signal amplitude cannot be definitively ruled out, the available data support a simpler mechanism, such as an induced fit or pre-equilibrium pathway, for association of the substrate analog.
FIGURE 7.

Biphasic kinetics of substrate analog binding. A, pre-steady state fluorescence quenching of 34E4 Fab on addition of substrate analog 6 (5 μm). The two phases roughly separate into time scales of 0.1 and 5 s, respectively. Similar measurements were performed at different ligand concentrations ranging from 0.75 to 10 μm. The two phases were fit independently to single exponentials. At 5 μm, kfast and kslow were 69.0 ± 0.5 and 0.68 ± 0.03 s-1, respectively. B, dependence of kfast on ligand concentration. These data were fitted to a standard bimolecular association model. C, dependence of kslow on ligand concentration. These data were fitted to a pre-equilibrium model.
To distinguish between the induced fit and pre-equilibrium mechanisms, the influence of ligand concentration on the fast and slow phases was determined. The fast phase of the reaction was linearly dependent on the concentration of the substrate analog (Fig. 7B), consistent with a simple bimolecular association step for ligand binding to the antibody with a second-order association rate constant (k2) of (1.23 ± 0.01) × 107 m-1 s-1 and a dissociation rate constant (k-2) of 6.96 ± 0.12 s-1 (39). The resulting affinity constant (K2 = k2/k-2) for the Fab·6 complex is (1.77 ± 0.01) × 106 m-1. By contrast, the rate constant for the slow phase, kslow, decreased with increasing concentration of the substrate analog (Fig. 7C), which is consistent with a pre-equilibrium mechanism (37). The slow phase when fitted to a pre-equilibrium model gave values of 0.50 ± 0.02 s-1 for the forward rate constant (k1) and 1.55 ± 0.08 s-1 for the reverse rate constant (k-1).
Thus, the Fab apparently exists as two conformational isomers in solution in an equilibrium ratio of ∼3:1, which favors the nonbinding form. The overall association constant, defined as Ka = K2/(1 + K1-1) (39), is (4.3 ± 0.5) × 105 m-1, which corresponds to a dissociation constant (Kd = 1/Ka) of 2.3 ± 0.3 μm. An increase in kslow at very high ligand concentrations relative to Kd (data not shown) suggests that the “inactive” Fab conformation may have weak affinity for the ligand or that ligand may induce another conformational change (12). This finding may account for the small difference between the calculated dissociation constant and the equilibrium value determined by fluorescence titration.
DISCUSSION
Ligand binding to 34E4 does not conform precisely to standard “lock and key” or “induced fit” mechanisms. Instead, the antibody exists as a mixture of “closed” and “open” forms in its resting state, only the latter of which is competent to bind ligand. In the closed state, which predominates in the absence of ligand, the bulky aromatic side chain of TrpL91 occupies the binding pocket, thereby blocking substrate access. When the indole moves out of the binding site, a deep pocket is created that can accommodate substrate and, hence, position it appropriately for rapid proton abstraction by GluH50.
The initial burst phase detected by stopped flow, which is completed in about 50 ms, can be attributed to ligand binding to the small fraction of antibody present in its open conformation at equilibrium. The change in fluorescence upon complex formation during this phase (an increase in the case of cationic hapten derivative 5 and a large decrease with the neutral substrate analog 6) is consistent with stacking of the ligand against TrpL91 in the resulting complex. In the subsequent slow phase, which proceeds over several seconds, association of the ligand depends on re-equilibration of the isomeric antibody conformers. The structural data argue against the latter process being induced by ligand, because binding to the closed form of the antibody would hinder the TrpL91 rotameric switch. In contrast, the very slow phase, which is apparent only with the original hapten, probably reflects an induced fit adjustment of the protein around the bound ligand to form the high-affinity complex.
The binding properties of 34E4 resemble those of the noncatalytic antibody SPE7 (27, 28), which exploits multiple conformations to permit binding to several ligands with micromolar to nanomolar affinity. Consistent with their nearly identical light chains and similarly configured active sites, both antibodies exhibit triphasic kinetics for binding their cognate haptens and simpler biphasic kinetics with lower affinity ligands. The relative magnitudes of the derived microscopic rate constants are similar, although the rates of the pre-equilibrium and the very slow induced fit steps are roughly 10-fold slower in 34E4 than SPE7. In structural terms, the conformational changes in 34E4 are less dramatic than in SPE7. However, in both antibodies, the switch between the t-105° and m95° rotamers of structurally analogous tryptophans controls access of the aromatic haptens to the active site. Moreover, once bound, the planar ligands are sandwiched between this indole ring (TrpL91 or TrpL93) and a tyrosine (TyrH100D in 34E4 or TyrH105 in SPE7).
What are the consequences of a pre-equilibrium mechanism of 34E4 for catalytic efficiency? In the absence of ligand, the inactive form of the antibody predominates and diminishes the concentration of functional active sites. As a consequence, catalytic efficiency (kcat/Km) will be reduced by a factor of at least 3 or 4. Furthermore, conversion of the inactive into the active conformer is relatively slow. Given the similarity of the rate constant for this process (0.5 s-1) and the kcat value for proton transfer for the Fab (0.45 s-1) (25), conformational isomerism may (partially) limit turnover of the catalyst.5 Whether conformational dynamics have a direct influence on the actual proton transfer step is uncertain, but the very slow induced fit adjustment of the binding site to hapten derivative 5 is suggestive. The complementarity between the protein and the substrate would be similarly expected to increase as the proton is transferred to GluH50 in the transition state, which could lead to a tightening of the structure.
Unliganded and liganded forms of 18 other catalytic antibodies have been characterized structurally and deposited in the Protein Data Bank (40). Conformational changes are evident in nine. These recapitulate the structural adjustments seen in many noncatalytic antibodies (3, 10, 11) and include rotations of single side chains to rigid-body movements of entire CDR loops. In three catalysts, an aromatic side chain flips into the binding pocket as in 34E4, occupying or partly overlapping with the substrate-binding site. In antibody 4C6, which catalyzes a cationic cyclization reaction, the TrpL89 indole regulates ligand binding in this way (41). In antibodies CNJ206 (42, 43) and 5C8 (44), which promote an esterolytic reaction and a disfavored cyclization of trans-epoxy alcohols, respectively, the mobile residue is a tyrosine. Although these catalysts have not been characterized by pre-steady state kinetics, they would also be expected to display a pre-equilibrium mechanism like 34E4.
Achieving enzyme-like efficiency in an immunoglobulin scaffold still remains a formidable challenge. General problems associated with imperfect hapten design, indirect selection, and limited screening procedures have been recognized (2). Here we show that efficacy can also be compromised by an unfavorable conformational equilibrium that transforms the active form of the catalyst into an inactive form that cannot bind substrate. As aromatic moieties are prevalent in haptens used to elicit high-affinity antibodies, such structural rearrangements may be inevitable (45). With respect to catalysis by antibody 34E4, stabilization of the ligand-competent form by active-site redesign to fix the TrpL91 in place may augment its catalytic power. More generally, protein engineering to attenuate the problem of adverse structural rearrangements promises to yield better antibody, as well as other protein catalysts.
Supplementary Material
Acknowledgments
We thank the ALS staff at beamline 8.2.1 and the SSRL staff at beamline 9-2 for their assistance.
The atomic coordinates and structure factors (codes 3CFJ and 3CFK) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported, in whole or in part, by National Institutes of Health Grant GM38273 (to I. A. W.). This work was also supported by a Skaggs predoctoral fellowship and a Jairo H. Arévalo fellowship from the The Scripps Research Institute graduate program (to E. W. D.) and the ETH Zürich (to D. H.). This is publication 18451-MB from the Scripps Research Institute. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains a supplemental table.
Footnotes
The abbreviations used are: Fab, fragment antigen binding; CDR, complementarity determining region; Bistris propane, 1,3-bis[tris(hydroxymethyl)methylamino]propane.
Consistent with the presence of a minor population of active catalyst in solution, preliminary stopped-flow measurements of the antibody-catalyzed decomposition of 5-nitrobenzisoxazole indicate a small initial burst in activity prior to attainment of the steady state. The effect is modest, however, suggesting that the rate constant for proton transfer is not much larger (∼2 times) than the rate constant k1 for conversion of the inactive into the active form of the antibody.
References
- 1.Schultz, P. G., and Lerner, R. A. (1995) Science 269 1835-1842 [DOI] [PubMed] [Google Scholar]
- 2.Hilvert, D. (2000) Annu. Rev. Biochem. 69 751-793 [DOI] [PubMed] [Google Scholar]
- 3.Padlan, E. A. (1994) Mol. Immunol. 31 169-217 [DOI] [PubMed] [Google Scholar]
- 4.Stewart, J. D., and Benkovic, S. J. (1995) Nature 375 388-391 [DOI] [PubMed] [Google Scholar]
- 5.Karplus, M. (1984) Adv. Biophys. 18 165-190 [DOI] [PubMed] [Google Scholar]
- 6.Hammes, G. G. (2002) Biochemistry 41 8221-8228 [DOI] [PubMed] [Google Scholar]
- 7.Boehr, D. D., Dyson, H. J., and Wright, P. E. (2006) Chem. Rev. 106 3055-3079 [DOI] [PubMed] [Google Scholar]
- 8.Hammes-Schiffer, S., and Benkovic, S. J. (2006) Annu. Rev. Biochem. 75 519-541 [DOI] [PubMed] [Google Scholar]
- 9.Olsson, M. H. M., Parson, W. W., and Warshel, A. (2006) Chem. Rev. 106 1737-1756 [DOI] [PubMed] [Google Scholar]
- 10.Wilson, I. A., and Stanfield, R. L. (1994) Curr. Opin. Struct. Biol. 4 857-867 [DOI] [PubMed] [Google Scholar]
- 11.Sundberg, E. J., and Mariuzza, R. A. (2002) Adv. Protein Chem. 61 119-160 [DOI] [PubMed] [Google Scholar]
- 12.Foote, J., and Milstein, C. (1994) Proc. Natl. Acad. Sci. U. S. A. 91 10370-10374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wedemayer, G. J., Patten, P. A., Wang, L. H., Schultz, P. G., and Stevens, R. C. (1997) Science 276 1665-1669 [DOI] [PubMed] [Google Scholar]
- 14.Wedemayer, G. J., Wang, L. H., Patten, P. A., Schultz, P. G., and Stevens, R. C. (1997) J. Mol. Biol. 268 390-400 [DOI] [PubMed] [Google Scholar]
- 15.Yin, J., Beuscher, A. E., Andryski, S. E., Stevens, R. C., and Schultz, P. G. (2003) J. Mol. Biol. 330 651-656 [DOI] [PubMed] [Google Scholar]
- 16.Zimmermann, J., Oakman, E. L., Thorpe, I. F., Shi, X., Abbyad, P., Brooks, C. L., III, Boxer, S. G., and Romesberg, F. E. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 13722-13727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ulrich, H. D., Mundorff, E., Santarsiero, B. D., Driggers, E. M., Stevens, R. C., and Schultz, P. G. (1997) Nature 389 271-275 [DOI] [PubMed] [Google Scholar]
- 18.Lindner, A. B., Eshhar, Z., and Tawfik, D. S. (1999) J. Mol. Biol. 285 421-430 [DOI] [PubMed] [Google Scholar]
- 19.Zhu, X., Dickerson, T. J., Rogers, C. J., Kaufmann, G. F., Mee, J. M., McKenzie, K. M., Janda, K. D., and Wilson, I. A. (2006) Structure (Camb.) 14 205-216 [DOI] [PubMed] [Google Scholar]
- 20.Casey, M. L., Kemp, D. S., Paul, K. G., and Cox, D. D. (1973) J. Org. Chem. 38 2294-2301 [Google Scholar]
- 21.Kemp, D. S., and Casey, M. L. (1973) J. Am. Chem. Soc. 95 6670-6680 [Google Scholar]
- 22.Kemp, D. S., Cox, D. D., and Paul, K. G. (1975) J. Am. Chem. Soc. 97 7312-7318 [Google Scholar]
- 23.Thorn, S. N., Daniels, R. G., Auditor, M. T., and Hilvert, D. (1995) Nature 373 228-230 [DOI] [PubMed] [Google Scholar]
- 24.Hu, Y., Houk, K. N., Kikuchi, K., Hotta, K., and Hilvert, D. (2004) J. Am. Chem. Soc. 126 8197-8205 [DOI] [PubMed] [Google Scholar]
- 25.Seebeck, F. P., and Hilvert, D. (2005) J. Am. Chem. Soc. 127 1307-1312 [DOI] [PubMed] [Google Scholar]
- 26.Debler, E. W., Ito, S., Seebeck, F. P., Heine, A., Hilvert, D., and Wilson, I. A. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 4984-4989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.James, L. C., Roversi, P., and Tawfik, D. S. (2003) Science 299 1362-1367 [DOI] [PubMed] [Google Scholar]
- 28.James, L. C., and Tawfik, D. S. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 12730-12735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Otwinowski, Z., and Minor, W. (1997) Methods Enzymol. 276 307-326 [DOI] [PubMed] [Google Scholar]
- 30.Storoni, L. C., McCoy, A. J., and Read, R. J. (2004) Acta Crystallogr. Sect. D Biol. Crystallogr. 60 432-438 [DOI] [PubMed] [Google Scholar]
- 31.Jones, T. A., Zou, J. Y., Cowan, S. W., and Kjeldgaard, M. (1991) Acta Crystallogr. Sect. A. 47 110-119 [DOI] [PubMed] [Google Scholar]
- 32.Murshudov, G. N., Vagin, A. A., and Dodson, E. J. (1997) Acta Crystallogr. Sect. D Biol. Crystallogr. 53 240-255 [DOI] [PubMed] [Google Scholar]
- 33.Lovell, S. C., Davis, I. W., Arendall, W. B., III, de Bakker, P. I., Word, J. M., Prisant, M. G., Richardson, J. S., and Richardson, D. C. (2003) Proteins 50 437-450 [DOI] [PubMed] [Google Scholar]
- 34.Vriend, G. (1990) J. Mol. Graphics 8 52-56 [DOI] [PubMed] [Google Scholar]
- 35.Laskowski, R. A., MacArthur, M. W., Moss, D. S., and Thornton, J. M. (1993) J. Appl. Crystallogr. 26 283-291 [Google Scholar]
- 36.Taira, K., and Benkovic, S. J. (1988) J. Med. Chem. 31 129-137 [DOI] [PubMed] [Google Scholar]
- 37.Fersht, A. (1998) Structure and Mechanism in Protein Science, pp. 147-149, W. H. Freeman & Co., New York
- 38.Lovell, S. C., Word, J. M., Richardson, J. S., and Richardson, D. C. (2000) Proteins 40 389-408 [PubMed] [Google Scholar]
- 39.Lancet, D., and Pecht, I. (1976) Proc. Natl. Acad. Sci. U. S. A. 73 3549-3553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N., and Bourne, P. E. (2000) Nucleic Acids Res. 28 235-242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu, X., Heine, A., Monnat, F., Houk, K. N., Janda, K. D., and Wilson, I. A. (2003) J. Mol. Biol. 329 69-83 [DOI] [PubMed] [Google Scholar]
- 42.Golinelli-Pimpaneau, B., Gigant, B., Bizebard, T., Navaza, J., Saludjian, P., Zemel, R., Tawfik, D. S., Eshhar, Z., Green, B. S., and Knossow, M. (1994) Structure (Cambridge) 2 175-183 [DOI] [PubMed] [Google Scholar]
- 43.Charbonnier, J. B., Carpenter, E., Gigant, B., Golinelli-Pimpaneau, B., Eshhar, Z., Green, B. S., and Knossow, M. (1995) Proc. Natl. Acad. Sci. U. S. A. 92 11721-11725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gruber, K., Zhou, B., Houk, K. N., Lerner, R. A., Shevlin, C. G., and Wilson, I. A. (1999) Biochemistry 38 7062-7074 [DOI] [PubMed] [Google Scholar]
- 45.Padlan, E. A. (1996) Adv. Protein Chem. 49 57-133 [DOI] [PubMed] [Google Scholar]
- 46.Al-Lazikani, B., Lesk, A. M., and Chothia, C. (1997) J. Mol. Biol. 273 927-948 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.