

Abstract
AKR1D1 (steroid 5β-reductase) reduces all Δ4-3-ketosteroids to form 5β-dihydrosteroids, a first step in the clearance of steroid hormones and an essential step in the synthesis of all bile acids. The reduction of the carbon-carbon double bond in an α,β-unsaturated ketone by 5β-reductase is a unique reaction in steroid enzymology because hydride transfer from NADPH to the β-face of a Δ4-3-ketosteroid yields a cis-A/B-ring configuration with an ∼90° bend in steroid structure. Here, we report the first x-ray crystal structure of a mammalian steroid hormone carbon-carbon double bond reductase, human Δ4-3-ketosteroid 5β-reductase (AKR1D1), and its complexes with intact substrates. We have determined the structures of AKR1D1 complexes with NADP+ at 1.79- and 1.35-Å resolution (HEPES bound in the active site), NADP+ and cortisone at 1.90-Å resolution, NADP+ and progesterone at 2.03-Å resolution, and NADP+ and testosterone at 1.62-Å resolution. Complexes with cortisone and progesterone reveal productive substrate binding orientations based on the proximity of each steroid carbon-carbon double bond to the re-face of the nicotinamide ring of NADP+. This orientation would permit 4-pro-(R)-hydride transfer from NADPH. Each steroid carbonyl accepts hydrogen bonds from catalytic residues Tyr58 and Glu120. The Y58F and E120A mutants are devoid of activity, supporting a role for this dyad in the catalytic mechanism. Intriguingly, testosterone binds nonproductively, thereby rationalizing the substrate inhibition observed with this particular steroid. The locations of disease-linked mutations thought to be responsible for bile acid deficiency are also revealed.
The Δ4-3-ketosteroid functionality is present in all steroid hormones except estrogens. The first step in their metabolism involves the reduction of the Δ4-ene to produce either 5α- or 5β-dihydrosteroids in reactions catalyzed by steroid 5α- or 5β-reductase, respectively (1). These two enzymes belong to distinct gene superfamilies (2, 3), and structural information on these enzymes is currently lacking.
Δ4-3-Ketosteroid 5β-reductase is a soluble monomeric NADPH-dependent enzyme and a member of the aldo-keto reductase (AKR)3 superfamily and is designated AKR1D1 in humans (3, 4). In utilizing NADPH as hydride donor, the enzymatic reaction introduces a 90° bend at the steroid A/B-ring junction, which adopts a cis-A/B- or β-configuration (Fig. 1) (5-7). In the absence of enzyme catalysis, this reaction is extremely difficult to perform with chemical reductants. Treatment with borohydride favors formation of the 3β-allylic alcohol, and under harsher conditions, the 3β,5α-tetrahydrosteroid is formed (8, 9). It is possible to generate a 5β-cholestane from the corresponding Δ4-3-ketosteroid only if a large directing group is placed at C-4 or C-7 to direct the face of hydride addition or catalytic hydrogenation (10, 11). Notably, AKR1D1 exclusively generates the reduced steroid bearing a thermodynamically less favorable 5β-configuration. Moreover, the enzyme reduces only the Δ4-ene, leaving the 3-oxo group intact. Thus, AKR1D1 catalyzes a reaction that is unique in steroid enzymology, and it accomplishes this with ease in comparison with chemical methods.
FIGURE 1.

Reduction of the carbon-carbon double bond at C-5 of a Δ4-3-ketosteroid to form a 5β-dihydrosteroid as catalyzed by AKR1D1. Steroid rings and selected carbon atoms are labeled according to standard steroid nomenclature. The structures of testosterone and 5β-dihydrotestosterone (R = OH) illustrate the remarkable configurational differences between the substrate and product of this reaction (atomic coordinates retrieved from Cambridge Structural Database entries TESTOM and JEPJET, respectively) (41).
Rat Δ4-3-ketosteroid 5β-reductase (AKR1D2) was first purified to homogeneity from liver as a 37-kDa protein (6). The cDNA for the corresponding human enzyme was cloned but characterized only in mammalian cell expression assays (3, 12). Recently, this enzyme has been cloned and expressed in Escherichia coli and completely characterized (13). These studies showed that a single enzyme could catalyze the reduction of C18-27Δ4-3-ketosteroids, yielding the corresponding 5β-dihydrosteroids. This enzyme thus plays an important role in the biosynthesis of bile acids (14, 15). For example, AKR1D1 reduces Δ4-cholesten-7α-ol-3-one and Δ4-cholesten-7α,12α-diol-3-one to 5β-cholestan-7α-ol-3-one and 5β-cholestan-7α,12α-diol-3-one, respectively; in turn, these two 5β-dihydrosteroids yield the bile acid precursors 5β-cholestan-3α,7α-diol and 5β-cholestan-3α,7α,12α-triol in reactions catalyzed by 3α-hydroxysteroid dehydrogenases that belong to the same gene family, AKR1C1-AKR1C4 (4, 15-17). Subsequent oxidation of the side chain of 5β-cholestan-3α,7α-diol and 5β-cholestan-3α,7α,12α-triol yields the primary bile acids chenodeoxycholate and cholic acid, respectively.
AKR1D1 deficiency results in accumulation of C27 bile acid precursors bearing intact Δ4-3-oxo groups, which, upon reduction by steroid 5α-reductase, can be converted to allo-bile acids (15, 18). These sterols are hepatotoxic, and their accumulation causes liver failure (19). Steroid 5β-reductase deficiency, which is characterized by single point mutations in the enzyme (L106F, P198L, P133R, and R261C) (20-22), is effectively treated with oral bile acid therapy (19). However, the effects of these mutations on enzyme structure and function remain to be delineated.
To date, the crystal structures of many of the human AKRs have been reported, including aldehyde reductase (AKR1A1) (23) and aldose reductase (AKR1B1) (24). The structures of relevant hydroxysteroid dehydrogenases (AKR1C1-AKR1C4) have also been reported: AKR1C1, human 20α-hydroxysteroid dehydrogenase (25); AKR1C2, rat and human type III 3α-hydroxysteroid dehydrogenase/bile acid-binding protein (26, 27); AKR1C3, human type II 3α-hydroxysteroid dehydrogenase and type V 17β-hydroxysteroid dehydrogenase/prostaglandin F synthase (28); AKR1C4, human 3α-hydroxysteroid dehydrogenase type I (Protein Data Bank code 2FVL)4; AKR1C5, rabbit 20α-hydroxysteroid dehydrogenase type 3 (30); and AKR1C9, rat liver 3α-dihydroxysteroid dehydrogenase (31-33). Even though these hydroxysteroid dehydrogenases share significant amino acid sequence identity, these crystal structures illuminate the roles of specific residues that contribute to the differing substrate specificities among these enzymes. All AKR structures to date contain a conserved catalytic tetrad of Tyr55, Lys84, His117, and Asp50 (numbering scheme based on AKR1C9, rat 3α-hydroxysteroid dehydrogenase). In contrast, AKR1D1 is the only AKR to have a modified catalytic tetrad: His117 is replaced by Glu120 and is proposed to play an important role in AKR1D1 catalysis (34). In addition to the above-mentioned hydroxysteroid dehydrogenase structures, the crystal structure of a novel short chain dehydrogenase/reductase, 5β-reductase from Digitalis lanata, has been reported recently (35). However, this reductase is unrelated in that it adopts a Rossmann fold.
Here, we report the first x-ray crystal structure of a mammalian steroid hormone carbon-carbon double bond reductase, human Δ4-3-ketosteroid 5β-reductase (AKR1D1). We have determined the structures of AKR1D1 complexed with NADP+ at 1.79-Å resolution, NADP+ and cortisone at 1.90-Å resolution, NADP+ and progesterone at 2.03-Å resolution, and NADP+ and testosterone at 1.62-Å resolution. Additionally, we report the crystal structures of the binary complex with NADP+ at 1.35-Å resolution (containing a HEPES buffer molecule bound in the active site). These structures provide valuable inferences regarding the catalytic mechanism and also illuminate the possible effects of single point mutations responsible for bile acid deficiency.
EXPERIMENTAL PROCEDURES
Materials—The vectors pET16b and pET28a were purchased from Novagen. E. coli strain C41(DE3) was provided by Dr. J. E. Walker (Medical Research Council Laboratory of Molecular Biology, Cambridge, UK). The QuikChange II site-directed mutagenesis kit was purchased from Stratagene. Restriction endonucleases were purchased from New England Biolabs. Synthetic oligonucleotides were obtained from Invitrogen. NADPH was obtained from Roche Applied Science. Steroids were purchased from Steraloids, Inc. [4-14C]Testosterone (50 mCi/mmol) was obtained from PerkinElmer Life Sciences. Nickel-Sepharose 6 Fast Flow was purchased from Amersham Biosciences. All other reagents were of ACS quality or higher.
Construction of Expression Vectors—Previously, we reported the purification and characterization of recombinant AKR1D1 expressed from a pET16b vector (13). This enzyme was refractory to crystallization, and therefore, a different expression vector was generated. The pET16b-AKR1D1 expression vector was digested with NdeI and BamHI to remove the cDNA for AKR1D1. The resultant fragment was subcloned into the expression vector pET28a. The AKR1D1 insert was verified by dideoxy sequencing.
The expression vectors for AKR1D1(Y58F) and AKR1D1 (E120A) were made using the pET16b-AKR1D1 construct as a template by conducting site-directed mutagenesis using the QuikChange method following the manufacturer's protocol. The following forward and reverse primers were used (where the boldface nucleotides indicate the mutation introduced): for the Y58F mutant, 5′-dGGG GCC TAC ATC TTC CAA AAT GAA CAC GAA GTT GG-3′ and 5′-dCC AAC TTC GTG TTC ATT TTG GAA GAT GTA GGC CCC-3′; and for the E120A mutant, 5′-dG GAT CTT TAC ATC ATT GCA GTA CCA ATG GCC TTT AAG C-3′ and 5′-dG CTT AAA GGC CAT TGG TAC TGC AAT GAT GTA AAG ATC C-3′. The introduction of these mutations into the pET16b-AKR1D1 construct was verified by dideoxy sequencing.
Expression and Purification of AKR1D1—The pET28a-AKR1D1 expression vector was used to transform competent E. coli C41(DE3) cells. The cells were grown in 4-liter cultures of Luria-Bertani medium at 37 °C (containing 100 μg/ml ampicillin). Upon reaching A600 = 0.6, 1 mm isopropyl 1-thio-β-d-galactopyranoside was added to induce enzyme expression overnight. The following day, the culture was centrifuged for 15 min at 10,000 × g, and the pellets were resuspended in 20 mm Tris-HCl (pH 7.9) and 5 mm imidazole. Resuspensions were lysed by sonication and centrifuged for 15 min at 10,000 × g, and the supernatant was dialyzed overnight in 20 mm Tris-HCl (pH 7.9), 20 mm imidazole, and 0.5 m NaCl. The dialyzed fraction was loaded onto a nickel-Sepharose column equilibrated with the dialysis buffer, and the column was washed with the same buffer. Bound protein was eluted with a linear gradient of 20-400 mm imidazole. Active fractions containing AKR1D1 were identified by monitoring the conversion of [4-14C]testosterone to 5β-dihydrotestosterone by discontinuous assay using standard assay conditions and by visualization of the protein content of each fraction by SDS-PAGE. Peak fractions were pooled and dialyzed overnight in 20 mm Tris-HCl (pH 7.0) containing 1 mm EDTA. The final specific activity of the recombinant enzyme was 80 nmol of testosterone reduced per min/mg. The mutant proteins AKR1D1(Y58F) and AKR1D1(E120A) were purified in a similar manner.
Standard Radiometric Assay—The reduction of [4-14C]testosterone was used to monitor 5β-reductase activity collected during purification and for activity measurements. Reactions contained 2 μm [4-14C]testosterone (40,000 dpm), 8 μm unlabeled testosterone, 5% acetonitrile, and 100 mm potassium phosphate buffer (pH 6.0). Reactions were initiated by the addition of 200 μm NADPH and performed at 37 °C. The substrate and product of the quenched reaction were separated by TLC and quantitated by scintillation counting.
Crystallography—The ternary complexes of AKR1D1·NADP+·testosterone, AKR1D1·NADP+·cortisone, AKR1D1·NADP+·progesterone, and AKR1D1·NADP+·HEPES were crystallized by the hanging drop vapor diffusion method at 4 °C. In a typical experiment, drops containing 4.0 μl of enzyme solution (5.0 mg/ml AKR1D1, 2.0 mm NADP+, 2.0 mm steroid, and 10.0 mm Tris (pH 7.4)) and 4.0 μl of precipitant buffer (0.1 m HEPES/Tris-HCl (pH 7.0), 10-20% (w/v) polyethylene glycol 4000, and 10% isopropyl alcohol) were equilibrated against a 1-ml reservoir of precipitant buffer. Diamond-like crystals appeared in ∼2-3 days with typical dimensions of 0.2 × 0.3 × 0.5 mm. Crystals of each AKR1D1·NADP+·steroid complex were further soaked for 1 week in the same mother liquor solution augmented with 5.0 mm NADP+ and 5.0 mm steroid to ensure complete ligand occupancy. Following transfer to a 28% glycerol cryoprotectant solution and flash-cooling, crystals of the AKR1D1·NADP+·testosterone complex yielded diffraction data to 1.62-Å resolution at the Advanced Light Source (beamline 8.2.2, λ = 1.000 Å, 100 K). Diffraction intensities measured from these crystals indicated a space group P212121 with unit cell parameters a = 49.8, b = 110.1, and c = 129.4 Å. There were two molecules in the asymmetric unit and the Matthews coefficient VM = 2.4 Å3/Da, corresponding to a solvent content of 48.5%. Crystals of the AKR1D1·NADP+·cortisone, AKR1D1·NADP+·progesterone, and AKR1D1·NADP+·HEPES complexes yielded diffraction data to 1.90-, 2.03-, and 1.35-Å resolution, respectively, at Brookhaven National Laboratory (beamlines X6A and X29A, λ = 1.000 Å, 100 K) and were found to be isomorphous with crystals of the AKR1D1·NADP+·testosterone complex (Table 1). Data reduction was achieved with HKL2000 (36) and Scalepack (36) (Table 1).
TABLE 1.
Data collection and refinement statistics
PDB, Protein Data Bank; r.m.s., root mean square.
| Structure | AKR1D1·NADP+·testosterone complex | AKR1D1·NADP+·cortisone complex | AKR1D1·NADP+·progesterone complex | AKR1D1·NADP+·HEPES complex | AKR1D1·NADP+ complex |
|---|---|---|---|---|---|
| Data collection | |||||
| PDB code | 3BUR | 3CMF | 3COT | 3BUV | 3BV7 |
| Resolution range (Å) | 50.0-1.62 | 50.0-1.90 | 30.0-2.03 | 50.0-1.35 | 50.0-1.79 |
| Unique reflections measured | 86,037 (7,952) | 51,994 (4,640) | 44,101 (4,301) | 147,700 (13,665) | 66,774 (6,684) |
| Rmergea | 0.103 (0.36)b | 0.112 (0.50)b | 0.131 (0.58)b | 0.094 (0.41)b | 0.109 (0.39)b |
| I/σ(I) | 20.3 (2.7)b | 10.9 (2.5)b | 12.2 (2.0)b | 22.6 (1.9)b | 20.5 (3.4)b |
|
Completeness (%)
|
96.0
(90.0)b |
93.7
(84.9)b |
97.6
(96.9)b |
95.7
(89.6)b |
98.6
(100.0)b |
| Refinement statistics | |||||
| Reflections used in refinement (test set) | 81,663/4,306 | 49,006/2,451 | 41,289/1,866 | 140,579/7,075 | 63,520/2,800 |
| R/Rfreec | 0.228/0.248 | 0.195/0.223 | 0.194/0.234 | 0.218/0.232 | 0.236/0.257 |
| Protein atomsd | 5,254 | 5,254 | 5,254 | 5,254 | 5,254 |
| Water moleculesd | 498 | 616 | 497 | 556 | 304 |
| NADP+ moleculesd | 2 | 2 | 2 | 2 | 2 |
| Glycerol moleculesd | 10 | 2 | 3 | ||
| Testosterone moleculesd | 2 | ||||
| Cortisone moleculesd | 2 | ||||
| Progesterone moleculesd | 2 | ||||
| HEPES moleculesd | 1 | ||||
| r.m.s. deviations | |||||
| Bond lengths (Å) | 0.007 | 0.006 | 0.007 | 0.005 | 0.012 |
| Bond angles | 1.1° | 1.2° | 1.1° | 1.2° | 1.1° |
| Average B-factors (Å2) | |||||
| Main chain atoms | 17 | 15 | 26 | 16 | 20 |
| Side chain atoms | 22 | 17 | 28 | 19 | 25 |
| Water molecules | 31 | 27 | 35 | 26 | 25 |
| NADP+ | 14 | 10 | 20 | 12 | 16 |
| Testosterone | 34 | ||||
| Cortisone | 41 | ||||
| Progesterone | 54 | ||||
| HEPES | 34 | ||||
| Glycerol | 42 | 43 | 41 | ||
| Ramachandran statisticse | |||||
| Allowed (%) | 90.4 | 89.2 | 89.0 | 89.9 | 91.1 |
| Additionally allowed (%) | 9.3 | 10.5 | 10.7 | 9.8 | 8.6 |
| Generously allowed (%) | 0.0 | 0.3 | 0.3 | 0.2 | 0.2 |
| Disallowed (%) | 0.2 | 0.0 | 0.0 | 0.2 | 0.2 |
Rmerge = ∑|I - <I>|/∑I, where I is the observed intensity and <I> is the average intensity calculated for replicate data.
The number in parentheses refer to the outer 0.1-Å shell of data.
Crystallographic R-factor, R = ∑(|Fo| - |Fc|)/∑|Fo| for reflections contained in the working set. Free R-factor, Rfree = ∑(|Fo| - |Fc|)/∑|Fo| for reflections contained in the test set excluded from refinement. |Fo| and |Fc| are the observed and calculated structure factor amplitudes, respectively.
Per asymmetric unit.
Ramachandran statistics were calculated with PROCHECK (29).
For crystallization of the AKR1D1·NADP+ complex, hanging drops containing 4.0 μl of enzyme solution (5.0 mg/ml AKR1D1, 2 mm NADPH, 2.0 mm 5β-cholestan-3-one, and 10.0 mm Tris (pH 7.4)) and 4.0 μl of precipitant buffer (0.1 m Tris/HEPES (pH 7.5), 10-20% (w/v) polyethylene glycol 4000, and 10% isopropyl alcohol) were equilibrated against a 1-ml reservoir of precipitant buffer. Crystals of the AKR1D1·NADP+ complex were soaked for 1 week in the same mother liquor solution augmented with 5.0 mm 5β-cholestan-3-one, a steroid product containing a cis-A/B-ring fusion. Following transfer to a cryoprotectant solution containing 28% glycerol, crystals of the AKR1D1·NADP+ complex yielded diffraction data to 1.80-Å resolution at the Advanced Photon Source (beamline 22 BM, λ = 1.000 Å, 100 K) and were isomorphous with crystals of the ternary complexes discussed above (Table 1).
The structure of the AKR1D1·NADP+·testosterone complex was solved by molecular replacement using one monomer of the AKR1C2·NADP+·testosterone complex (Protein Data Bank code 1J96) (27) less the atoms of NADP+·testosterone and solvent as an initial search probe. The program PHASER (37) was used to perform the molecular replacement calculations; the optimal solution for the positioning of the two monomers in the asymmetric unit yielded a total log likelihood gain of 1832, a rotation function Z score of 12.3, and a translational function Z score of 19.5 for the first monomer and a rotation function Z score of 13.2 and a translational function Z score of 42.5 for the second monomer using data in the 50-2.5-Å resolution range. The analysis of this solution showed reasonable packing interactions in the unit cell. The initial electron density map clearly revealed the presence of NADP+ bound in the active site. Using the programs O (38) and CNS (39) for model fitting and refinement, respectively, the model was manually rebuilt and refined. In the later stages of refinement with CNS, the non-crystallographic symmetry restraints were released, after which the program SHELX was used to complete the refinement (40). Additional solvent molecules were introduced during refinement with SHELX, and the electron density map of testosterone became very clearly defined. In the final stage of refinement, the atomic coordinates of testosterone were retrieved from entry TESTOM in the Cambridge Structural Database (41), built into the electron density map, and refined with full occupancy. The quality of the final model was checked with Verify3d (42); the analysis of the structure was performed with PISA (43).
The crystal structures of the AKR1D1·NADP+·cortisone, AKR1D1·NADP+·progesterone, AKR1D1·NADP+·HEPES, and AKR1D1·NADP+ complexes were solved by the difference Fourier method. Refinement was performed as described above using CNS (39).
Twenty-one residues at the N terminus (the uncleaved expression tag from the pET28a vector) were disordered and therefore excluded from the final model of each structure. The exclusion of 21 of 346 residues in each monomer (6.1% of the scattering matter) likely contributed to the slightly elevated R and Rfree values recorded in Table 1.
RESULTS
Crystal Structure of the AKR1D1·NADP+ Complex—The asymmetric unit of the unit cell contains two monomers of AKR1D1. Each monomer consists of a 325-residue polypeptide chain that adopts an (α/β)8-barrel fold typical of AKRs. The top of the β-barrel is capped by loops A (Ile119-Leu147), B (Tyr219-Leu238), and C (Leu302-Tyr326), which enclose the active site (Fig. 2a). Notably, the Pro133-Lys139 segment in loop A of monomer A exhibits higher thermal B-factors compared with the Pro133-Lys139 segment in monomer B, where this segment appears to be stabilized in part by interlattice contacts. However, the loop A conformations in monomers A and B are essentially identical (data not shown).
FIGURE 2.

AKR1D1·NADP+ complex. a, AKR1D1 adopts the (α/β)8-barrel fold conserved among members of the AKR superfamily. Loops A, B, and C (blue) flank the steroid-binding pocket. Atoms of the catalytic tetrad and NADP+ are labeled and color-coded as follows: carbon, black; oxygen, red; and nitrogen, blue. The water molecule hydrogen-bonded between Tyr58 and Glu120 is indicated by a red sphere. Hydrogen bonds are indicated by red dashed lines. b, superposition of AKR1D1 (yellow), AKR1C9 (red), and AKR1C2 (blue) reveals significant conformational differences in loops A, B, and C.
Given the amino acid sequence identities of 56% between AKR1D1 and AKR1C2 (27) and 58% between AKR1D1 and AKR1C9 (33), the overall tertiary structures of these enzymes are quite similar. The root mean square deviation of 307 C-α atoms between AKR1D1 in complex with NADP+ and between AKR1C2 in complex with NADP+ and testosterone (Protein Data Bank Code 1J96) (27) is 1.0 Å, and that of 317 C-α atoms between AKR1D1 and AKR1C9 complexed with NADP+ and testosterone (Protein Data Bank code 1AFS) is 0.78 Å (33). However, significant conformational differences in loops A, B, and C are observed when the structures of AKR1D1, AKR1C2, and AKR1C9 are compared (Fig. 2b). The majority of amino acid substitutions between AKR1D1 and these enzymes are found in these loops. The Val309-Phe322 segment of loop C and its corresponding segment in AKR1C2 exhibit significant C-α deviations of ∼4Å. Because these loops play an important role in substrate binding, it is likely that their sequence and structural differences reflect differences in steroid substrate specificity and catalysis among these enzymes.
The NADP+ cofactor in the AKR1D1·NADP+ complex adopts an extended anti-conformation as observed in other AKR structures and is located in a long tunnel with the adenine group exposed to solvent and the nicotinamide ring located deep inside the tunnel oriented toward the central active site cavity (Fig. 2a). Approximately 84% of the accessible surface area of NADP+ is buried inside the tunnel. The NADP+ cofactor participates in a significant number of hydrogen bond interactions (Fig. 3). Notably, the cofactor binding mode is essentially conserved with respect to that observed in AKR1C9 (32) or AKR1C2 (27). In complex with AKR1D1, the nicotinamide head-group of NADP+ is stacked against Tyr219, and the carboxamide group makes contacts with Asn170, Ser169, and Gln193. Additionally, the phosphate group of 2′-AMP makes an electrostatic link with Arg279. However, these structures do not reveal a hydrogen bond interaction between Asn227 and Lys30 and Lys273 that would correspond to the salt link interaction between Asp217 and Lys22 and Lys263 observed in the AKR1A1 structure (23) or the salt link interactions between Asp216 and Lys21 and Lys262 observed in the AKR1B1 structure (24). These salt links create a “safety belt” that contributes to the high affinity (∼10 nm) of cofactor binding to AKR1A1 and AKR1B1. Because NADP+ binding to AKR1D1 lacks this safety belt, binding affinity should be weaker and more comparable with cofactor binding to AKRs similarly lacking a safety belt (100-200 nm), and this is what has been observed (13).
FIGURE 3.

AKR1D1·NADP+·HEPES complex. a, shown is the difference electron density map of HEPES bound in the active site of AKR1D1 prior to the inclusion of HEPES in the refinement (contoured at 2.2σ; red). The electron-rich sulfur atom of HEPES is indicated by its stronger electron density contoured at 6.7σ (green). b, hydrogen bond interactions in the AKR1D1·NADP+·HEPES complex are indicated by red dashed lines. Atoms are color-coded as described for Fig. 2a except that protein carbon atoms are yellow, HEPES carbon atoms are black, and its sulfur atom is green.
Interestingly, when crystals of the AKR1D1·NADP+ complex were grown in the presence of 5β-cholestan-3-one, the binding of this product-like steroid containing a cis-A/B-ring fusion was not observed. Instead, either a molecule of glycerol from the cryoprotectant solution bound in the active site of monomer A (1.79-Å resolution structure) (data not shown), or a HEPES molecule from the crystallization buffer bound in the active site of monomer B (1.35-Å resolution structure) (Fig. 3). Thus, these two structures represent those of the binary AKR1D1·NADP+ complex.
Crystal Structures of the AKR1D1·NADP+·Cortisone and AKR1D1·NADP+·Progesterone Complexes—In these enzyme·substrate complexes, the electron density of each steroid substrate is clear and unambiguous (Fig. 4, a and b), showing that each steroid lies perpendicular to the NADP+ cofactor. The binding of cortisone is very similar to the binding of progesterone even though these steroids bear different pendant groups on their D-rings.
FIGURE 4.

AKR1D1·NADP+-steroid complexes. Two map orientations are shown for each steroid, and hydrogen bond interactions are indicated by red dashed lines. Atoms are color-coded as described for Fig. 2a except that protein carbon atoms are yellow. For clarity, the orientation of the protein is rotated ∼180° horizontally relative to the orientation shown in Fig. 2a. a, difference electron density map of cortisone contoured at 2.7σ. b, difference electron density map of progesterone contoured at 2.6σ. c, difference electron density map of testosterone contoured at 2.3σ. Note the dramatically different, nonproductive binding mode of testosterone; in contrast with the binding of cortisone and progesterone, the A-ring of testosterone is oriented away from the catalytic tetrad and the nicotinamide ring of the cofactor.
Overall, the crystal structure of each ternary complex is very similar to that of the AKR1D1·NADP+ complex, with root mean square deviations of 0.26 and 0.13 Å, respectively, for 325 C-α atoms. However, important conformational differences are evident in loops A and B. Significantly, cortisone and progesterone binding triggers the movement of Ser225-Val231 in loop B away from the active site, with C-α deviations ranging from 0.3 to 2 Å. Furthermore, an ∼7-Å movement of the side chain of Trp230 accommodates substrate binding (illustrated for cortisone binding in Fig. 5). Thus, Trp230 appears to play a key role in packing against the β-face of the steroid ring system in the active site of human AKR1D1, as also observed for the corresponding tryptophan residue in the structure of the AKR1C9·NADP+·testosterone complex (33).
FIGURE 5.

Least-squares superposition of the AKR1D1·NADP+ (yellow), AKR1D1·NADP+·cortisone (red), and AKR1D1·NADP+·testosterone (green) complexes. The distance of ∼3.7 Å between the anomeric carbon of NADP+ and the olefinic C-4 of cortisone is indicated by a red dashed line, which would represent the trajectory of hydride transfer from NADPH. The position of the water molecule in the unliganded structure is indicated by a yellow sphere. Loops A, B, and C are labeled; note that loop B and Trp230 in particular must undergo a significant conformational change to accommodate productive substrate binding as represented by cortisone (red). The indole ring of Trp230 remains in its substrate-free conformation (yellow) to accommodate the nonproductive binding mode of testosterone (green).
In the AKR1D1 active site, ∼80% of the accessible surface area of each steroid substrate is buried in the hydrophobic binding pocket. Each steroid substrate is positioned with its β-face oriented toward the re-face of the nicotinamide ring of NADP+ such that the steroid carbon-carbon double bond would be adjacent to the 4-pro-(R)-hydrogen of NADPH (Fig. 4, a and b). The C-3 carbonyl oxygen of each steroid substrate accepts hydrogen bonds from the phenolic hydroxyl group of Tyr58 and the anti-oriented conformer of the carboxylic acid side chain of Glu120. Notably, the C-3 carbonyl oxygen of each substrate occupies the same position as the water molecule that hydrogen bonds with Tyr58 and Glu120 in the AKR1D1·NADP+ complex (Figs. 2 and 3). Site-directed mutagenesis of these residues to Y58F and E120A supports their role in catalysis because neither mutant has any detectable activity in the reduction of testosterone using the standard radiometric assay, where the limit of detection is 0.002 nmol/min.
Crystal Structure of the AKR1D1·NADP+·Testosterone Complex—In this enzyme·substrate complex, the electron density of testosterone is extremely well defined. However, the arrangement of NADP+ and testosterone differs from that seen in the other ternary complexes because they are parallel and not perpendicular. Testosterone also binds with a “backward” orientation in which its D-ring binds in the same region occupied by the A-rings of cortisone and progesterone in their respective complexes with AKR1D1 (Fig. 4c). Consequently, the carbon-carbon double bond of testosterone is distant from the nicotinamide ring of the cofactor, and this binding orientation is judged to be nonproductive. The side chain of Trp230 does not undergo a conformational change to accommodate testosterone binding and instead remains in the conformation observed in the structure of the substrate-free AKR1D1·NADP+ complex (Fig. 5). Approximately 85% of the accessible surface area of testosterone is buried in the hydrophobic binding pocket. The testosterone C-3 carbonyl group accepts hydrogen bonds from Asn227 and Ser225, whereas the testosterone hydroxyl group makes a hydrogen bond interaction with Tyr132.
The nonproductive testosterone binding modes observed with AKR1C2 and AKR1D1 suggest that this particular steroid can occupy more than one position and that at the high steroid concentrations used in the crystallization trials, the nonproductive binding mode is favored. This is reflected in substrate inhibition observed with AKR1C2 (44, 45). We also observed substrate inhibition of AKR1D1 by high concentrations of testosterone. Although the Km value for this steroid is close to 2.0 μm, concentrations of testosterone that exceed 10 μm produced marked inhibition (Fig. 6).
FIGURE 6.

Substrate inhibition of AKR1D1 by testosterone. A velocity versus substrate plot for AKR1D1 shows substrate inhibition using testosterone. Assays contained 0.078 μm enzyme, 1-40 μm testosterone, 15 μm NADPH, and 4% acetonitrile in 100 mm potassium phosphate buffer (pH 6.0) in a final volume of 1 ml. Reactions were monitored fluorometrically using an excitation wavelength of 340 nm (slit width of 5 nm) and an emission wavelength of 450 nm (slit width of 10 nm) at 37 °C. Kinetic analysis of initial velocities obtained was performed using the Henri-Michaelis-Menten equation for uncompetitive substrate inhibition, v = (Vmax× [S])/(Km + [S] + [S]2/Ki), and fit using the program GraFit. The iterative fits provided estimates of Vmax, Km, and Ki and associated S.E. values.
Disease-linked Mutations—Four point mutations in AKR1D1 are associated with bile acid deficiency: L106F, P198L, P133R, and R261C (20-22). As predicted from other AKR structures, these residues are not located in the cofactor- or steroid-binding sites and are not involved in catalysis. The locations of these residues in the AKR1D1 structure are shown in Fig. 7. The P133R substitution in loop A is closest to the substrate-binding site, and it is conceivable that this substitution perturbs the conformational changes in loop A that accompany steroid binding. The remaining mutations may exert a deleterious effect on catalysis by causing structural changes that propagate through the protein scaffold to perturb substrate and/or cofactor binding or by otherwise destabilizing the folded conformation of the protein.
FIGURE 7.

AKR1D1·NADP+·cortisone complex: positions of the natural mutations L106F, P198L, P133R, and R261C. Cortisone, Leu106, Pro198, Pro133, and Arg261 atoms are black, and NADP+ atoms are magenta. Among these four residues, Pro133 in loop A is closest to the substrate-binding site.
DISCUSSION
The x-ray crystal structure of human Δ4-3-ketosteroid 5β-reductase (AKR1D1) is the first structure of a mammalian steroid carbon-carbon double bond reductase. As a member of the AKR superfamily, it is not surprising that the (α/β)8-barrel fold and the NADP+ cofactor-binding site of AKR1D1 are highly conserved with those other members of the superfamily. However, the structures of AKR1D1·substrate complexes reported herein illuminate new features of steroid substrate recognition, the catalytic mechanism, and the positions of natural mutations associated with bile acid deficiency.
Substrate Recognition—AKR1D1 binds Δ4-3-ketosteroids productively and nonproductively. In the productive binding mode observed with cortisone and progesterone, the two substrates are bound similarly in a pocket defined by residues from the β2-α2 loop, loop A, loop B, and loop C. Alignments of pocket and loop residues of AKR1D1 with those of other AKR1C structures containing bound steroids are shown in supplemental Tables S1 and S2. Of the 10 pocket residues assigned, Tyr58 is catalytic, and Tyr26 and Trp230 are highly conserved. The indole ring of Trp230 in the AKR1D1 structures is versatile in that rotation around both side chain torsion angles and movement of the associated polypeptide backbone of loop B allow the indole ring to pack differently, i.e. against the β-face of a productively bound steroid substrate or against the α-face of a nonproductively bound steroid substrate (Fig. 5).
Interestingly, superposition of the AKR1D1·NADP+·cortisone and AKR1C9·NADP+·testosterone complexes, in which both substrates adopt productive binding orientations, reveals that the substrate is immobilized more deeply in the active site of AKR1D1 compared with AKR1C9 (Fig. 8). In part, this appears to result from the removal of the steric bulk of the His120 imidazole side chain by the substitution of Glu120 in AKR1D1. As a result, the 4-pro-(R)-hydride of the NADPH cofactor would be adjacent to the substrate C-3 carbonyl group in the AKR1C9 active site, and the 4-pro-(R)-hydride of the NADPH cofactor would be adjacent to C-5 of the substrate carbon-carbon double bond in the AKR1D1 active site (Fig. 8).
FIGURE 8.

Comparisons of steroid binding modes in catalysis. a, superposition of the AKR1D1·NADP+·cortisone complex (red) on the AKR1C9·NADP+·testosterone complex (green) showing that the A-ring of the substrate binds ∼1 Å deeper in the active site of the former enzyme relative to the position of the nicotinamide ring of NADP+. Presuming that NADPH binds similarly to NADP+, the 4-pro-(R)-hydride of NADPH would be adjacent to the substrate C-3 carbonyl in the AKR1C9 active site (green dotted lines), whereas the 4-pro-(R)-hydride of the NADPH cofactor would be adjacent to C-5 of the substrate carbon-carbon double bond in the AKR1D1 active site (red dotted lines). b, same orientation as a, but a close-up view showing all atoms.
Catalytic Mechanism—The four catalytic residues Asp53, Tyr58, Lys87, and Glu120 are located in the center of the β-barrel (Fig. 2a). The side chain of Lys87 donates hydrogen bonds to Asp53 and Tyr58. In the substrate-free enzyme, the phenolic hydroxyl group of Tyr58 donates a hydrogen bond to a water molecule, which in turn hydrogen bonds with Glu120. Because the C-3 carbonyl oxygen atoms of the substrates cortisone and progesterone displace this water molecule in their complexes with AKR1D1, the side chain of Glu120 must be protonated as the anti-oriented carboxylic acid to donate a hydrogen bond to the substrate carbonyl oxygen (and possibly also the water molecule in the substrate-free enzyme). Furthermore, given that carboxylate groups that serve as general bases generally do so with their more basic, syn-oriented lone electron pairs (46), it is reasonable to conclude that protonated carboxylic acids that serve as general acids or hydrogen bond donors may do so with their more acidic (pKa ∼0.5) anti-oriented conformers. Thus, in AKR1D1, the anti-oriented conformer of Glu120 may serve as a superacidic hydrogen bond donor to help activate the α,β-unsaturated ketone moiety of the substrate for carbon-carbon bond reduction by NADPH.
That Glu120 appears in AKR1D1 represents a unique exception for AKRs having an (α/β)8-barrel fold. All other AKRs, including AKR1C9 (33) and AKR1C2 (26), have a histidine residue at the corresponding position. The side chain conformations of Asp53, Tyr58, and Lys87 are mostly conserved with respect to the conformations of corresponding residues in the AKR1C2·NADP+·testosterone complex; the side chain of Glu120 is coplanar with respect to the orientation of the corresponding histidine residue in this complex (27). Interestingly, the hydrogen-bonded water molecule between Tyr58 and Glu120 occupies a position close to that of an acetate oxygen atom observed in the AKR1C2·NADP+·testosterone complex (27), and a corresponding water molecule is also observed in the crystal structure of AKR1C9 (32). It is proposed that the binding of this water molecule in AKR1C9 mimics the binding of the carbonyl oxygen of a 3-ketosteroid substrate (33).
In AKR1D1, the interactions of Tyr58 and Glu120 with
the carbonyl groups of steroid substrates suggest that both residues
participate in the catalytic mechanism of carbon-carbon double bond reduction.
In AKR1B1 and AKR1C9, site-directed mutagenesis studies provide compelling
evidence that the catalytic tyrosine acts as the general acid-base
(47,
48). Moreover, site-directed
mutagenesis studies of AKR1C9 also show that the corresponding H117E
substitution introduces 5β-reductase activity into this enzyme
(34). Based on
kcat and kcat/Km
pH-rate profiles for wild-type AKR1C9 and AKR1C9(H117E) enzymes, a
facilitatory role for the glutamic acid residue is proposed in which it alters
the pK of Tyr55, which functions as the general acid. This
facilitatory role is based on an acid shift in the pKb of
Tyr55 in the H117E mutant. These data are interpreted to indicate
that Tyr55 has more 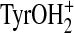 character (i.e. it may behave as a “superacid”), which in
turn polarizes the α,β-unsaturated ketone so that hydride transfer
can occur at C-5.
character (i.e. it may behave as a “superacid”), which in
turn polarizes the α,β-unsaturated ketone so that hydride transfer
can occur at C-5.
The carboxylic acid side chain of Glu120 may also serve as a superacid to help polarize the C-3 ketone of a productively bound substrate such as cortisone or progesterone (Fig. 4, a and b). The non-enzymatic mechanism of carbon-carbon double bond reduction in an α,β-unsaturated ketone requires a superacid such as HF-SbF5 (49), and it is possible that AKR1D1 adopts a similar strategy for catalysis. Generation of the AKR1D1(Y58F) and AKR1D1(E120A) mutants abolishes all enzyme activity, consistent with the crucial role of these residues in catalysis. Once 4-pro-(R)-hydride transfer from the re-face of NADPH to the carbon-carbon double bond of the substrate is achieved, the substrate will adopt a bent, cis-A/B-ring conformation. Either an enzyme-bound residue or a solvent molecule will protonate the C-4 atom of the substrate in the final step of the steroid reduction reaction. A proposed mechanism that incorporates these structural data in light of available enzymological measurements is presented in Fig. 9.
FIGURE 9.

Proposed catalytic mechanism for steroid double bond reduction. The 4-pro-(R)-hydride transfer from the re-face of NADPH to C-5 of the Δ4-ene is proposed while the C-3 carbonyl is polarized by both Tyr58 and Glu120, which create a superacidic oxyanion hole as predicted by Jez and Penning (34).
Disease-linked Mutations—The four point mutations associated with inherited bile acid deficiency are located in regions independent of substrate recognition and catalysis. If these mutations are responsible for bile acid deficiency, then it is likely that they affect conformational changes in loop A that accompany substrate binding (P133R) and/or otherwise compromise enzyme stability. Future studies of these mutant recombinant proteins and their expression in mammalian cells will allow us to probe the structural and functional consequences of these mutations.
Supplementary Material
Acknowledgments
We thank the Brookhaven National Laboratory (beamlines X6A, X12B, and X29A; Upton, NY), the Advanced Light Source (beamline 8.2.2; Berkeley, CA), and the Advanced Photon Source (beamline 22 BM, Chicago, IL) for beamtime access.
This paper is dedicated to Dr. Paul Talalay, The John Jacob-Abel Distinguished Service Professor, Department of Pharmacology and Molecular Sciences, Johns Hopkins School of Medicine, in honor of his 85th birthday. Paul has been a pioneer in steroid hormone enzymology throughout his career.
The atomic coordinates and structure factors (code 3BUR, 3CMF, 3COT, 3BUV, and 3BV7) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported, in whole or in part, by National Institutes of Health Research Grant R01-GM56838 (to D. W. C.) and Grants R01-DK47015 and P30-ES013508 (to T. M. P.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1 and S2.
This article was selected as a Paper of the Week.
Footnotes
The abbreviation used is: AKR, aldo-keto reductase.
E. Ugochukwu, C. Smee, K. Guo, P. Lukacik, K. Kavanagh, J. E. Debreczeni, F. von Delft, J. Weigelt, M. Sundstrom, C. Arrowsmith, A. Edwards, and U. Oppermann, unpublished data.
References
- 1.Tomkins, G. (1956) Recent Prog. Horm. Res. 12 125-133 [PubMed] [Google Scholar]
- 2.Russell, D. W., and Wilson, J. D. (1994) Annu. Rev. Biochem. 63 25-61 [DOI] [PubMed] [Google Scholar]
- 3.Kondo, K.-H., Kai, M. H., Setoguchi, Y., Eggersten, G., Sjöblom, P., Setoguchi, Y., Okuda, K. I., and Björkhem, J. (1994) Eur. J. Biochem. 219 357-363 [DOI] [PubMed] [Google Scholar]
- 4.Jez, J. M., Bennett, M. J., Schelgel, B. P., Lewis, M., and Penning, T. M. (1997) Biochem. J. 325 625-636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berséus, O., Danielsson, H., and Einarsson, K. (1967) J. Biol. Chem. 242 1211-1219 [PubMed] [Google Scholar]
- 6.Okuda, K., and Okuda, K. (1984) J. Biol. Chem. 259 7519-7524 [PubMed] [Google Scholar]
- 7.Onishi, Y., Noshiro, M., Shimosato, T., and Okuda, K. (1991) FEBS Lett. 283 215-218 [DOI] [PubMed] [Google Scholar]
- 8.Norymberski, J. K., and Woods, G. F. (1955) J. Chem. Soc. (Lond.) 3426-3430
- 9.March, J. (1984) in Advanced Organic Chemistry: Reactions, Mechanisms and Structure (Smith, M. B., and March, J., eds) pp. 694-695, John Wiley & Sons, Inc., New York
- 10.Michalak, K., Stepanenko, W., and Wicha, J. (2000) J. Chem. Soc. Perkin Trans. I 1587-1594
- 11.Takeda, K., Osaka, H., Akio, H., Shionogi, et al. (1961) Yakugaku Zasshi 81 325-330 [Google Scholar]
- 12.Charbonneau, A., and Luu-The, V. (2001) Biochim. Biophys. Acta 1517 228-235 [DOI] [PubMed] [Google Scholar]
- 13.Drury, J. E., and Penning, T. M. (2007) in Enzymology and Molecular Biology of Carbonyl Metabolism (Weiner, H., Maser, E., Lindhal, R., and Plapp, B., eds) pp. 332-340, Purdue University Press, West Lafayette, IN
- 14.Russell, D. W., and Setchell, K. D. (1992) Biochemistry 31 4737-4749 [DOI] [PubMed] [Google Scholar]
- 15.Russell, D. W. (2003) Annu. Rev. Biochem. 72 137-174 [DOI] [PubMed] [Google Scholar]
- 16.Penning, T. M., Burczynski, M. E., Jez, J. M., Hung, C.-F., Lin, H.-K., Ma, H., Moore, M., Palackal, N., and Ratnam, K. (2000) Biochem. J. 351 67-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deyashiki, Y., Taniguchi, H., Amano, T., Nakayama, T., Hara, A., and Sawada, H. (1992) Biochem. J. 282 741-746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Setchell, K. D., Suchy, F. J., Welsh, M. B., Zimmer-Nechemias, L., Heubi, J., and Balistreri, W. F. (1988) J. Clin. Investig. 82 2148-2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clayton, P. T., Mills, K. A., Johnson, A. W., Barabino, A., and Marazzi, M. G. (1996) Gut 38 623-628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kimura, A., Kondo, K.-H., Okuda, K. I., Higashi, S., Suzuki, M., Kurosawa, T., Tohma, M., Inoue, T., Nishiyori, A., Yoshino, M., Kato, H., and Setoguchi, T. (1998) Eur. J. Pediatr. 157 386-390 [DOI] [PubMed] [Google Scholar]
- 21.Gonzales, E., Cresteil, D., Baussan, C., Dabadie, A., Gerhardt, M.-F., and Jacquemin, E. (2004) J. Hepatol. 40 716-718 [DOI] [PubMed] [Google Scholar]
- 22.Lemonde, H. A., Custard, E. J., Bouquet, J., Duran, M., Overmars, H., Scambler, P. J., and Clayton, P. T. (2003) Gut 52 1494-1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.El-Kabbani, O., Green, N. C., Lin, G., Carson, M., Narayana, S. V., Moore, K. M., Flynn, T. G., and DeLucas, L. J. (1994) Acta Crystallogr. Sect. D Biol. Crystallogr. 50 859-868 [DOI] [PubMed] [Google Scholar]
- 24.Wilson, D. K., Bohren, K. M., Gabbay, K. H., and Quiocho, F. A. (1992) Science 257 81-84 [DOI] [PubMed] [Google Scholar]
- 25.Couture, J.-F., Legrand, P., Cantin, L., Luu-The, V., Labrie, F., and Breton, R. (2003) J. Mol. Biol. 331 593-604 [DOI] [PubMed] [Google Scholar]
- 26.Jin, Y., Stayrook, S. E., Albert, R. H., Palackal, N. T., Penning, T. M., and Lewis, M. (2001) Biochemistry 40 10161-10168 [DOI] [PubMed] [Google Scholar]
- 27.Nahoum, V., Gangloff, A., Legrand, P., Zhu, D. W., Cantin, L., Zhorov, B. S., Luu-The, V., Labrie, F., Breton, R., and Lin, S. X. (2001) J. Biol. Chem. 276 42091-42098 [DOI] [PubMed] [Google Scholar]
- 28.Komoto, J., Yamada, T., Watanabe, K., and Takusagawa, F. (2004) Biochemistry 43 2188-2198 [DOI] [PubMed] [Google Scholar]
- 29.Laskowski, R. A., MacArthur, M. W., Moss, D. S., and Thornton, J. M. (1993) J. Appl. Crystallogr. 26 283-291 [Google Scholar]
- 30.Couture, J.-F., Legrand, P., Cantin, L., Labrie, F., Luu-The, V., and Breton, R. (2004) J. Mol. Biol. 339 89-102 [DOI] [PubMed] [Google Scholar]
- 31.Hoog, S. S., Pawlowski, J. E., Alzari, P. M., Penning, T. M., and Lewis, M. (1994) Proc. Natl. Acad. Sci. U. S. A. 91 2517-2521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bennett, M. J., Schlegel, B. P., Jez, J. M., Penning, T. M., and Lewis, M. (1996) Biochemistry 35 10702-10711 [DOI] [PubMed] [Google Scholar]
- 33.Bennett, M. J., Albert, R. H., Jez, J. M., Ma, H., Penning, T. M., and Lewis, M. (1997) Structure (Camb.) 5 799-812 [DOI] [PubMed] [Google Scholar]
- 34.Jez, J. M., and Penning, T. M. (1998) Biochemistry 37 9695-9703 [DOI] [PubMed] [Google Scholar]
- 35.Thorn, A., Egerer-Sieber, C., Jäeger, C. M., Herl, V., Müeller-Uri, F., Kreis, W., and Muller, Y. A. (November 21, 2007) J. Biol. Chem. 10.1074/jbc.M706185200 [DOI] [PubMed]
- 36.Otwinowski, Z., and Minor, M. (1997) Methods Enzymol. 276 307-326 [DOI] [PubMed] [Google Scholar]
- 37.McCoy, A. J., Grosse-kunstleve, R. W., Storni, L. C., and Read, R. J. (2005) Acta Crystallogr. Sect. D Biol. Crystallogr. 61 458-464 [DOI] [PubMed] [Google Scholar]
- 38.Jones, T. A., Zou, J.-Y., Cowan, S. W., and Kjeldgaard, M. (1991) Acta Crystallogr. Sect. A Found. Crystallogr. 47 110-119 [DOI] [PubMed] [Google Scholar]
- 39.Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T., and Warren, G. L. (1998) Acta Crystallogr. Sect. D Biol. Crystallogr. 54 905-921 [DOI] [PubMed] [Google Scholar]
- 40.Sheldrick, G., and Schneider, T. (1997) Methods Enzymol. 277 319-343 [PubMed] [Google Scholar]
- 41.Allen, F. H. (2002) Acta Crystallogr. Sect. D Biol. Crystallogr. 58 380-388 [DOI] [PubMed] [Google Scholar]
- 42.Eisenberg, D., Lüthy, R., and Bowie, J. U. (1997) Methods Enzymol. 277 396-404 [DOI] [PubMed] [Google Scholar]
- 43.Krissinel, E., and Henrick, K. (2007) J. Mol. Biol. 372 774-797 [DOI] [PubMed] [Google Scholar]
- 44.Steckelbroeck, S., Jin, Y., Oyesanmi, B., Kloosterboer, H. J., and Penning, T. M. (2004) Mol. Pharmacol. 66 1702-1711 [DOI] [PubMed] [Google Scholar]
- 45.Jin, Y., and Penning, T. M. (2006) Steroids 71 380-391 [DOI] [PubMed] [Google Scholar]
- 46.Gandour, R. (1981) Bioorg. Chem. 10 169-176 [Google Scholar]
- 47.Grimshaw, C. E., Bohren, K. M., Lai, C. J., and Gabbay, K. H. (1995) Biochemistry 34 14374-14384 [DOI] [PubMed] [Google Scholar]
- 48.Schlegel, B. P., Jez, J. M., and Penning, T. M. (1998) Biochemistry 37 3538-3548 [DOI] [PubMed] [Google Scholar]
- 49.Coustard, J.-M., Douteau, M.-H., Jacquesy, J.-C., and Jacquesy, R. (1975) Tetrahedron Lett. 25 2029-2030 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.