

Abstract
Inflammatory bowel disease arises from the interplay between luminal bacteria and the colonic mucosa. Targeted inhibition of pro-inflammatory pathways without global immunosuppression is highly desirable. Apolipoprotein (apo) E has immunomodulatory effects and synthetically derived apoE-mimetic peptides are beneficial in models of sepsis and neuroinflammation. Citrobacter rodentium is the rodent equivalent of enteropathogenic Escherichia coli, and it causes colitis in mice by colonizing the surface of colonic epithelial cells and inducing signaling events. We have reported that mice deficient in inducible nitric-oxide (NO) synthase (iNOS) have attenuated C. rodentium-induced colitis. We used young adult mouse colon (YAMC) cells that mimic primary colonic epithelial cells to study effects of an antennapedia-linked apoE-mimetic peptide, COG112, on C. rodentium-activated cells. COG112 significantly attenuated induction of NO production, and iNOS mRNA and protein expression, in a concentration-dependent manner. COG112 inhibited the C. rodentium-stimulated induction of iNOS and the CXC chemokines KC and MIP-2 to the same degree as the NF-κB inhibitors MG132 or BAY 11-7082, and there was no additive effect when COG112 and these inhibitors were combined. COG112 significantly reduced nuclear translocation of NF-κB, when assessed by electromobility shift assay, immunoblotting, and immunofluorescence for p65. This correlated with inhibition of both C. rodentium-stimulated IκB-α phosphorylation and degradation, and IκB kinase activity, which occurred by inhibition of IκB kinase complex formation rather than by a direct effect on the enzyme itself. These studies indicate that apoE-mimetic peptides may have novel therapeutic potential by inhibiting NF-κB-driven proinflammatory epithelial responses to pathogenic colonic bacteria.
Human inflammatory bowel disease (IBD)3 includes ulcerative colitis and Crohn disease. The prevalence of IBD is high in industrialized nations; it is estimated that between 1.1 and 1.4 million people are affected in the United States (1). There are an assortment of treatment options, the most prevalent of which are immunosuppressive agents and biologic therapies, such as antibodies to tumor necrosis factor (TNF)-α. These treatments are often complicated by multiple side effects and can be very expensive. Additionally, even the most aggressive use of biologic therapies still fails to produce remission in half of the patients that are treated (2). Therefore, new approaches to treatment of IBD are warranted.
Apolipoprotein (apo) E is known for its function in cholesterol and lipid metabolism, and independent of this role, has been shown to alter both innate and adaptive immune responses (3). ApoE-deficient mice exhibited increased systemic inflammatory response and higher mortality following lipopolysaccharide (LPS) injection, and apoE treatment reduced mortality and suppressed inflammatory cytokine release (4). Based on the anti-inflammatory properties of apoE, COG133 was created from amino acid residues 133–149 located in the receptor binding region of the 299-amino acid apoE holoprotein (5). COG133 inhibited LPS-stimulated TNF-α and NO production in microglial cells in vitro (5) and suppressed brain and systemic inflammatory responses in LPS-injected mice (6). To enhance transmembrane permeability, COG133 has been fused to a protein transduction domain derived from the Drosophila antennapedia protein to create COG112 (7). This molecular fusion has been shown to enhance the bioactivity of COG133 such that there was substantial clinical improvement and protection from inflammation and demyelination injury in the spinal cord in the murine experimental autoimmune encephalomyelitis (EAE) model of multiple sclerosis (7). In peritoneal macrophages isolated from EAE mice, pretreatment with COG112 attenuated production of NO and secretion of TNF-α and IL-6 in response to stimulation with LPS plus IFN-γ ex vivo (7).
We therefore hypothesized that COG112 could have benefit in models of colonic inflammation. We were specifically interested in studying the role of COG112 in inhibiting bacterial activation of colonic epithelial cells that is involved in the pathophysiology of colitis and diarrhea. In the case of the mouse-adapted pathogen, C. rodentium (8), this bacterium forms attaching and effacing lesions on colonic epithelial cells leading to the development of colitis that is associated with a vigorous T helper cell 1-driven immune response (9–11). We have reported that C. rodentium-induced colitis results in a marked up-regulation of iNOS in the colonic epithelium in vivo, and that iNOS-/- mice are significantly protected from disease (11). We have, therefore, now tested the effect of C. rodentium on mouse colonic epithelial cells in vitro and found that this resulted in a marked induction of iNOS mRNA and protein and NO production. Using this model, we show that COG112 effectively inhibited iNOS expression and NO production.
Activation of colonic epithelial cells by enteropathogenic Escherichia coli in humans is very similar to that caused by C. rodentium and has been strongly linked to activation of the transcription factor NF-κB (12, 13), leading us to speculate that the inhibitory effect of COG112 was occurring via an effect on NF-κB. We now report that COG112 had a similar effect as the NF-κB inhibitors MG132 or BAY 11-7082 on NO production and on iNOS and pro-inflammatory CXC chemokine expression levels induced by C. rodentium, and that there was no additive effect of COG112 with either of these inhibitors.
NF-κB is composed of homodimers and heterodimers of five family members, namely RelA (p65), c-Rel, RelB, p50, and p52 (14). Activation of NF-κB requires phosphorylation of the inhibitory protein IκB-α by an IκB kinase, IKK (15), which leads to IκB-α degradation and subsequent release of NF-κB proteins into the cytosol (16). IKK activity requires formation of a complex composed of IKK-α and IKK-β (15, 17, 18), and the regulatory subunit IKK-γ (19). Activation of the complex depends on phosphorylation of IKK-α and IKK-β (20). In response to bacterial stimulation, p65 can be translocated to the nucleus where it can bind to promoter response elements and facilitate transcription of pro-inflammatory genes (21). We now report that exposure of colonic epithelial cells to C. rodentium results in activation of NF-κB that is potently inhibited by COG112. We show that COG112 inhibits the accumulation of p65 in the nucleus, the phosphorylation and degradation of IκB-α, and the activity of IKK, through an effect on the formation of the IKK complex. This is the first report to demonstrate the inhibitory effect of an apoE-mimetic peptide on the canonical NF-κB pathway, and this has direct significance for understanding the potential benefits of these agents in colitis and other inflammatory diseases.
EXPERIMENTAL PROCEDURES
Reagents—Reagents for cell culture and RNA extraction were from Invitrogen (Carlsbad, CA). Real-time PCR reagents were from Bio-Rad. BAY 11-7082 was from Calbiochem/EMD Biosciences (San Diego, CA). All other chemicals were purchased from Sigma.
Bacteria, Cells, and Culture Conditions—A wild-type strain of C. rodentium (DBS100) was used as described (11). For each experiment, C. rodentium were cultured on Luria agar plates and then transferred to Luria broth in a standing culture overnight. For co-culture experiments, C. rodentium was washed with phosphate-buffered saline (PBS) and resuspended in Roswell Park Memorial Institute (RPMI) 1640 medium. The bacterial concentration was determined by optical density at 650 nm and confirmed by serial dilution and culture (11).
Young adult mouse colon (YAMC) cells are derived from colonic crypts from the immortomouse, such that they are conditionally immortalized with an SV40 large T-antigen with a temperature-sensitive interferon (IFN)-γ inducible promoter (22). The YAMC cells were maintained under the permissive growth conditions in RPMI 1640 medium supplemented with 10% fetal bovine serum, 2 mm glutamine, 50 μg/ml gentamicin, 100 units/ml penicillin, 100 μg/ml streptomycin, and 5 units/ml IFN-γ in a humidified incubator with 5% CO2 at 33 °C. For experiments, cells were incubated at 33 °C in IFN-γ-containing medium for 24 h and then transferred to 37 °C in IFN-γ-free RPMI 1640 medium for 24 h. Cells were then washed and placed in RPMI 1640 medium containing 10% serum and 2 mm glutamine, without antibiotics or IFN-γ. For all stimulation studies, C. rodentium was added at a multiplicity of infection (MOI) of 200, and RPMI 1640 vehicle was added to control wells.
ApoE-mimetic Peptide—COG112 is a chimeric peptide containing the antennapedia protein transduction domain (RQIKIWFQNRRMKWKKC) followed by COG133 (LRVRLASHLRKLRKRLL) (5). The resulting sequence of COG112 (acetyl-RQIKIWFQNRRMKWKKCLRVRLASHLRKLRKRLL-amide) (7) was synthesized using standard Fmoc chemistries and purified by NeoMPS, Inc. (San Diego, CA). In all experiments, YAMC cells were pretreated with COG112 for 1 h prior to activation. In some experiments, the antennapedia peptide alone was used as a control.
NO Production—Concentration of the stable metabolite of NO, NO-2, was measured in culture supernatants by the Griess reaction, using a Griess reagent kit from Promega (Madison, WI). For experiments, 1 × 106 cells/ml were plated in 24-well plates and NO-2 was measured after 24 h of stimulation by C. rodentium with or without COG112, MG132, a proteosomal inhibitor that blocks NF-κB activation, or BAY11-7082, a specific NF-κB inhibitor.
Determination of Cell Viability—Cell viability in YAMC cells was measured by a colorimetric assay as described (23), in which the conversion of the tetrazolium salt 2,3-bis[2-methoxy-4-nitro-5-sulfophenyl]-2H-tetrazolium-5-carboxyaniline (XTT) to formazan by intact cellular mitochondrial dehydrogenases is detected spectrophotometrically.
mRNA Analysis—RNA was extracted from control and activated YAMC cells using TRIzol reagent (23). 1 μg of RNA was reverse-transcribed using an iScript™ cDNA synthesis kit (Bio-Rad). Each PCR was performed with 2 μl of cDNA using iQ™ SYBR Green Supermix (Bio-Rad). For iNOS the primers were: sense, 5′-CACCTTGGAGTTCACCCAGT-3′ and antisense, 5′-ACCACTCGTACTTGGGATGC-3′. For keratinocyte-derived chemokine (KC; CXCL1) the primer sequences were: sense, 5′-GCTGGGATTCACCTCAAGAA-3′ and antisense, 5′-CTTGGGGACACCTTTTAGCA-3′, and for macrophage inflammatory protein (MIP)-2 (CXCL2), the primer sequences were: sense, 5′-GCCAAGGGTTGACTTCA-3′ and antisense, 5′-TGTCTGGGCGCAGTG-3′. The thermal cycling conditions and the method used to calculate relative expression have been described previously (23–25).
Western Blot Analysis—For iNOS, total YAMC cell lysates were prepared and immunoblotted with polyclonal antibody to iNOS at 1:2500 as described (25, 26). For p65, nuclear and cytosolic extracts were prepared using an extraction kit from Pierce/Thermo Scientific. Rabbit polyclonal antibody to a synthetic peptide corresponding to amino acids near the C-terminal domain of human p65 (RelA) from EMD Biosciences was used at a dilution of 1:1000. For IκB-α studies, the cytosolic extract was used. For phospho-IκB-α, a phosphospecific rabbit polyclonal antibody (Invitrogen) to the region of the human IκB-α that contains Ser32 and Ser36 was used at a dilution of 1:1000. Total IκB-α was assessed with rabbit polyclonal antibody (EMD Biosciences) to a synthetic peptide corresponding to amino acids surrounding Ser32 of human IκB-α at a dilution of 1:1000. β-Actin immunoblotting in cytosolic and total cell lysates was performed as described (23–26). All proteins were detected by chemiluminescence as described (23–26).
Electrophoretic Mobility Shift Assay (EMSA)—Nuclear extracts were prepared as above. 22-bp oligonucleotide NF-κB consensus binding sequences were used as probes for EMSA: sense, 5′-AGTTGAGGGGACTTTCCCAGGC-3′ and antisense, 3′-TCAACTCCCCTGAAAGGGTCCG-5′. 50 ng of double-stranded oligonucleotides were labeled with [γ-32P]ATP using T4 polynucleotide kinase (Promega). Binding reactions were carried out with 20,000 cpm labeled oligonucleotides and 5 μg of nuclear protein. For competition experiments, 100-fold excess of cold oligonucleotides with either the NF-κB binding sequence or the SP-1 binding sequence was used. The entire reaction was loaded on a 5% polyacrylamide gel and run at room temperature at 60 mA for 1 h. Gels were dried and visualized using a phosphorimager (26).
Immunofluorescence Staining for p65—2 × 104 YAMC cells were plated in 4-well chamber slides. After treatments, cells were washed with PBS and fixed in 3.7% formaldehyde. Cells were then permeabilized in PBS containing 50 mm NH4Cl and 0.2% Triton X-100, washed, and blocked in 5% goat serum for 1 h at room temperature. Cells were incubated at 4 °C for 16 h with primary antibody to p65, described above, at 1:1000 dilution, followed by incubation with fluorescein isothiocyanate (FITC)-labeled goat anti-rabbit secondary antibody (KPL, Gaithersburg, MD) for 2 h at room temperature. Cells were counterstained with DAPI for nuclear staining. Slides were dried and mounted in Permount (Fisher Scientific). Slides were visualized using a Nikon E800 fluorescence microscope, and images were acquired using a Spot RT Slider Digital Camera and Spot Advanced software (Diagnostic Instruments, Inc., Sterling Heights, MI) as performed previously (23).
IκB Kinase Activity Assay—An ELISA assay (K-LISA™ detection kit, Calbiochem/EMD Biosciences) was performed for the IκB kinase (IKK) that phosphorylates IκB-α at Ser32 and Ser36. Cytosolic cell extracts were incubated in glutathionecoated wells of a 96-well plate with a glutathione S-transferase (GST)-tagged IκB-α fusion polypeptide substrate that includes the Ser32 and Ser36 IκB-α kinase phosphorylation sites. Phosphorylated GST-IκB-α substrate was detected using horseradish peroxidase-conjugated antiphospho-IκB-α antibody. The optical density proportional to kinase activity was measured at 450 nm in a Molecular Devices plate reader (SpectraMax Plus, Sunnyvale, CA). In some experiments, recombinant His-Tag IKK-β enzyme was incubated with GST-IκB-α substrate in the absence or presence of COG112 or a small molecule inhibitor of IKK-β, supplied by the manufacturer in the K-LISA™ kit with the product name IKK-2 inhibitor IV. This agent is also known as 2-[(aminocarbonyl)amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide or TPCA-1 (27).
Immunoprecipitation of IKK Complex—Total YAMC cell lysates were prepared as above, and 100 μg of protein were immunoprecipitated as previously described (26) with 1 μg of rabbit polyclonal antibody to IKK-α (Cell Signaling, Danvers, MA). Immunoblotting was performed on the immunoprecipitate using rabbit polyclonal antibodies to IKK-β, IKK-γ, and phospho-IKK-α(Ser180)/β(Ser181), all from Cell Signaling and each used at a dilution of 1:1000 and detected by chemiluminescence.
Statistical Analysis—Quantitative data are shown as the mean ± S.E. Comparisons between multiple groups were made by using analysis of variance with the Student-Newman-Keuls post-hoc multiple comparisons test. When comparisons between only two groups were made, a Student's paired t test was performed.
RESULTS
C. rodentium Stimulates Induction of NO Production and iNOS mRNA and
Protein Expression That Are Inhibited by COG112—We have reported
that C. rodentium infection of mice results in expression of colonic
iNOS, and that iNOS-/- knock-out mice exhibited attenuation of
C. rodentium-associated colitis. Because we found that iNOS
expression localized to colonic epithelial cells in the colitis tissues
(11) and C. rodentium
exerts its pathogenicity by adhering to colonic epithelial cells
(9,
10,
28), we tested the effect of
C. rodentium on iNOS expression and activity in vitro. We
used conditionally immortalized YAMC cells that exhibit characteristics of
non-transformed cells at 37 °C. When these cells were exposed to C.
rodentium for 4 h and NO production was assessed after a total of 24 h,
there was an 18.2 ± 0.6-fold increase in NO levels, measured as
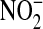 , compared with control levels
(p < 0.001, n = 10 experiments performed in triplicate).
Addition of the ApoE-mimetic peptide, COG112, inhibited NO production in a
concentration-dependent manner, as shown in
Fig. 1. To exclude the
possibility of a toxic effect of COG112 as the cause for this inhibition, we
also assessed cell viability by XTT assay. When compared with cells exposed to
C. rodentium without COG112, there was no difference in cell
viability at 1, 5, or 10 μm concentrations of COG112. Only at 20
μm COG112, at which point the inhibitory effect on NO production
had plateaued, was there evidence of cellular cytotoxicity. At the 10
μm concentration of COG112, there was a 53.3 ± 1.3%
inhibition of C. rodentium-stimulated NO production. It should also
be noted that when we have performed experiments using the antennapedia
peptide alone without the fused COG 133 peptide, there was no effect on C.
rodentium-stimulated NO production (data not shown).
, compared with control levels
(p < 0.001, n = 10 experiments performed in triplicate).
Addition of the ApoE-mimetic peptide, COG112, inhibited NO production in a
concentration-dependent manner, as shown in
Fig. 1. To exclude the
possibility of a toxic effect of COG112 as the cause for this inhibition, we
also assessed cell viability by XTT assay. When compared with cells exposed to
C. rodentium without COG112, there was no difference in cell
viability at 1, 5, or 10 μm concentrations of COG112. Only at 20
μm COG112, at which point the inhibitory effect on NO production
had plateaued, was there evidence of cellular cytotoxicity. At the 10
μm concentration of COG112, there was a 53.3 ± 1.3%
inhibition of C. rodentium-stimulated NO production. It should also
be noted that when we have performed experiments using the antennapedia
peptide alone without the fused COG 133 peptide, there was no effect on C.
rodentium-stimulated NO production (data not shown).
FIGURE 1.

Inhibitory effect of COG112 on NO production in YAMC cells stimulated with C. rodentium. Conditionally immortalized YAMC cells were cultured at the non-permissive condition at 37 °C for 24 h. Cells were then activated with C. rodentium at an MOI of 200 for 4 h, washed three times with PBS, and cultured an additional 20 h. When COG112 was used, it was added 1 h before addition of C. rodentium and was re-added after washing away the bacteria. NO-2 levels and cell viability were assessed by the Griess and XTT assays, respectively, in C. rodentium-stimulated cells in the presence of increasing concentrations of COG112. The level of NO-2 in unstimulated YAMC cells was 0.68 ± 0.06 μm. §§, p < 0.01 for COG112 treatment versus C. rodentium alone for the NO-2 levels, and ##, p < 0.01 for COG112 versus C. rodentium alone for the cell viability levels. In the NO-2 studies, n = 31 for control, C. rodentium alone, and C. rodentium + 10 μm COG112; n = 25 for C. rodentium + 1 μm COG112, and n = 6 for C. rodentium + COG112 concentrations of 3, 5, 7, and 20 μm. In the XTT studies, n = 6 for each of the values shown.
When iNOS mRNA was assessed, C. rodentium stimulated a 22.7 ± 2.7-fold increase in YAMC cells by real-time PCR (Fig. 2A). This was inhibited by 35.2-± 7.2% and 74.0 ± 4.8% with 1 and 10 μm COG112, respectively. When iNOS protein expression was assessed by Western blotting, there was a marked increase with C. rodentium that was partially inhibited with 1 μm COG112 and attenuated with 10 μm COG112 (Fig. 2B).
FIGURE 2.

Effect of COG112 on iNOS mRNA and protein expression in YAMC cells activated with C. rodentium. A, YAMC cells were co-cultured with C. rodentium at an MOI of 200 in the absence or presence of COG112 added 1 h before bacterial stimulation. Bacterial co-culture with YAMC cells was continued for 4 h, and iNOS mRNA levels were assessed by real-time PCR using SYBR green with standardization to β-actin levels. **, p < 0.01 versus control; §§, p < 0.01 versus C. rodentium alone. B, Western blot analysis. Cells were co-cultured with C. rodentium at an MOI of 200 for 4 h, washed, and cultured an additional 20 h. When COG112 was used, it was added 1 h before addition of C. rodentium, and was re-added after washing away the bacteria as described in the legend to Fig. 1. iNOS protein (130 kDa) and β-actin (42 kDa) were assessed from the same membrane. 30 μg of total cellular lysate protein were run per lane. Similar results were observed in three experiments.
Similar Inhibition of iNOS by COG112 and the Proteosomal Inhibitor MG132—Because activation of the transcription factor NF-κB has been implicated in iNOS expression in general and attaching and effacing bacteria are known to activate NF-κB (12, 13), we hypothesized that the inhibitory effect of COG112 on iNOS may be due to an effect on NF-κB. We compared the ability of COG112 and MG132 to inhibit iNOS; the latter is a proteosomal inhibitor that blocks the degradation of IκB-α needed to allow release of active subunits of NF-κB into the cytoplasm that is required prior to nuclear translocation. As shown in Fig. 3A, both COG112 and MG132 inhibited C. rodentium-stimulated NO production to an equal degree. There was a slight degree of further inhibition when both agents were combined, but when iNOS mRNA was assessed by real-time PCR, there was no additive inhibitory effect of COG112 and MG132 (Fig. 3B). Taken together, these data suggest that COG112 and MG132 may be acting upon the same pathway.
FIGURE 3.

Effect of COG112 and MG132 on NO production, and iNOS, KC, and MIP-2 mRNA levels in YAMC cells activated with C. rodentium. A, NO-2 levels were assessed at 24 h after stimulation, as described in the legend to Fig. 1. B, iNOS mRNA levels were assessed by real-time PCR 4 h after stimulation, as described in the legend to Fig. 2. C, KC mRNA levels and D, MIP-2 mRNA levels, both were assessed by real-time PCR at 4 h after stimulation. In A–D, the concentrations of COG112 and MG132 were 10 μm and 20 μm, respectively. **, p < 0.01 versus unstimulated control cells; §§, p < 0.01 versus C. rodentium alone; n = 9 for each condition in A and n = 3 experiments for each condition in B–D.
Similar Inhibition of KC and MIP-2 by COG112 and MG132—To further assess the possibility that COG112 could be affecting NF-κB activation, we determined the effect of COG112 and MG132 on C. rodentium-stimulated chemokine expression. We focused on the murine equivalents to human interleukin-8, the CXC chemokines KC (CXCL1) and MIP-2 (CXCL2), the expression of which has been been linked to NF-κB activation (29, 30). When assessed by real-time PCR, C. rodentium induced a 12.0 ± 0.7-fold increase in KC mRNA expression that was inhibited by 64.8 ± 5.6 and 64.1 ± 0.7% with COG112 and MG132, respectively, and by 59.5 ± 2.7% with both agents combined (Fig. 3C). In the case of MIP-2, C. rodentium stimulated an 11.7 ± 1.0-fold increase in MIP-2 mRNA levels that were inhibited by 61.4 ± 1.7 and 52.0 ± 3.7% for COG112 and MG132, respectively, and 59.8 ± 3.2% for both agents combined (Fig. 3D). These data provide further evidence that COG112 may act to inhibit NF-κB activation.
Similar Inhibition of iNOS, KC, and MIP-2 by COG112 and the NF-κB Inhibitor BAY 11-7082—To confirm the findings with MG132, we also utilized a more specific NF-κB inhibitor, BAY 11-7082. This agent was originally described as an irreversible inhibitor of TNF-α-induced IκB-α phosphorylation (31) and has been used in numerous studies as an inhibitor of NF-κB (32, 33). We determined that in our system of C. rodentium-stimulated YAMC cells, BAY 11-7082 inhibited NO production by 57.0 ± 2.4%, which was similar to the inhibition by COG112 by 57.9 ± 5.0% and that of COG112 + BAY 11-7082 combined of 61.6 ± 3.2% (Fig. 4A). When iNOS mRNA was assessed the same effect was observed, such that the inhibitory effects of COG112 (67.5 ± 4.0%), BAY 11-7082 (68.5 ± 2.5%), and COG112 + BAY 11-7082 (66.2 ± 7.2%) were nearly identical (Fig. 4B). In the same manner, when KC (Fig. 4C) and MIP-2 (Fig. 4D) were assessed, COG112 and BAY 11-7082 both inhibited the expression of these genes, and there was no additive inhibition with COG112 and BAY 11-7082. Therefore, these data corroborate the findings with MG132 and again indicate that COG112 is likely to be acting upon NF-κB.
FIGURE 4.

Effect of COG112 and BAY 11-7082 on NO production, and iNOS, KC, and MIP-2 mRNA levels in YAMC cells activated with C. rodentium. A, NO-2 levels were assessed at 24 h after stimulation. B, iNOS mRNA levels were assessed by real-time PCR 4 h after stimulation. C, KC mRNA levels and D, MIP-2 mRNA levels, both were assessed by real-time PCR at 4 h after stimulation. In A–D, the concentrations of COG112 and BAY 11-7082 were 10 μm and 20 μm, respectively. **, p < 0.01 versus unstimulated control cells; §§, p < 0.01 versus C. rodentium alone; n = 4 for each condition in A, and n = 3 experiments for each condition in B–D.
C. rodentium Induces Nuclear Translocation of Active NF-κB That Is Inhibited by COG112—Because the MG132 and BAY 11-7082 data implicated NF-κB activation in the induction of iNOS, KC, and MIP-2 by C. rodentium, we directly assessed the presence of activated NF-κB in the nucleus of C. rodentium-stimulated YAMC cells by EMSA. As shown in Fig. 5A, there was a time-dependent increase in binding of a radiolabeled NF-κB-specific oligonucleotide to nuclear proteins from cells activated with C. rodentium, which peaked at 40 min after inoculation of cells with bacteria. The specificity of this binding was demonstrated by the finding that excess cold probe competed out the binding of the radiolabeled NF-κB-specific oligonucleotide to the nuclear extracts. Furthermore, addition of excess cold SP-1 oligonucleotide did not affect the stimulated NF-κB binding. When cells were treated with COG112, the induction of binding of activated proteins in the nucleus to the NF-κB consensus binding sequence was abolished (Fig. 5B).
FIGURE 5.

Effect of C. rodentium on NF-κB activity in YAMC cells and inhibition by COG112 as assessed by EMSA. Nuclear extracts were prepared, probed with a [γ-32P]ATP-labeled oligonucleotide probe for the consensus binding sequence for NF-κB, electrophoresed on polyacrylamide gels, and phosphorimaged. A, time course of NF-κB activation. Cells were co-cultured with C. rodentium at an MOI of 200 for the times indicated. In lane 7, excess unlabeled (cold) probe for NF-κB competed away binding of the labeled probe, while in lane 8, excess unlabeled nonspecific competitor, SP-1, did not. B, cells were co-cultured with C. rodentium for 40 min in the absence or presence of COG112 (10 μm) that was added 1 h before stimulation with bacteria. Nucl, nuclear. Similar results were observed in four separate experiments.
COG112 Inhibits Nuclear Translocation of p65—Because we had observed that C. rodentium induced NF-κB binding to nuclear proteins, we determined whether p65, the active subunit of NF-κB, translocates to the nucleus in response to this bacterial stimulus. In the absence of COG112, there was a modest increase in cytosolic p65 from 5 to 60 min, but a marked, time-dependent increase in p65 levels in the nuclear extracts that peaked at 40 min after C. rodentium stimulation (Fig. 6). In the presence of COG112, this nuclear accumulation was prevented.
FIGURE 6.

Induction of p65 activation and nuclear accumulation by C. rodentium in YAMC cells and the inhibitory effect of COG112. YAMC cells were cultured in the absence or presence of C. rodentium at an MOI of 200 for the times indicated. In addition, cells were also pretreated with COG112 (10 μm) for 1 h where indicated. Cells were lysed, and cytosolic and nuclear fractions prepared by centrifugation. Western blotting was performed using 20 μg of protein/lane. For both the cytosolic and nuclear fractions, blots generated from protein samples from the cells without (left panel) or with (right panel) COG112 were incubated in the same container with primary antibody and then secondary antibody. After addition of chemiluminescence reagents, membranes were exposed together on the same film. Cytos, cytosolic; Nucl, nuclear. Similar results were observed in two separate experiments.
To confirm these findings, we performed immunofluorescence staining for p65 (Fig. 7). Compared with uninfected cells, C. rodentium stimulation resulted in an increase in both cytoplasmic and nuclear staining at 20 min after stimulation and more intense nuclear staining and reduction of cytoplasmic staining at 40 min. In the presence of COG112, there was complete inhibition of both cytoplasmic and nuclear p65 staining at 20 min, and a significant reduction in the nuclear staining at 40 min. Taken together, these data indicate that p65 is rapidly translocated to the nucleus with C. rodentium activation, and that this is efficiently blocked by COG112.
FIGURE 7.

Immunofluorescence staining demonstrates that C. rodentium stimulates nuclear translocation of p65 that is inhibited by COG112. YAMC cells were cultured in chamber slides in the absence or presence of C. rodentium at an MOI of 200. Where indicated, cells were pretreated with COG112 (10 μm) for 1 h. At the times shown, YAMC cells were fixed and permeabilized, followed by staining with polyclonal antibody to p65 detected with FITC-conjugated secondary antibody. DAPI was added for labeling of nuclei prior to mounting slides. All images shown were photographed at a magnification of ×600. Similar results were observed in three separate experiments.
COG112 Inhibits C. rodentium-stimulated Phosphorylation of IκB-α and Prevents the Degradation of IκB-α—p65 translocation requires phosphorylation of the inhibitory protein IκB-α and its dissociation from a cytoplasmic complex with p65. We therefore assessed the effect of C. rodentium and COG112 on IκB-α phosphorylation and total IκB-α levels. As shown in Fig. 8, C. rodentium induces a rapid phosphorylation of IκB-α that results in a marked reduction of total IκB-α, indicative of IκB-α degradation. In the presence of COG112, the phosphorylation of IκB-α is inhibited, and IκB-α is significantly less degraded.
FIGURE 8.

Stimulation of IκB-α phosphorylation and degradation by C. rodentium and the inhibitory effect of COG112. YAMC cells were cultured in the absence or presence of C. rodentium for the times indicated. Cells were also pretreated with COG112 (10 μm) for 1 h where indicated. Immunoblotting was performed with antibodies specific to phospho-IκB-α (40 kDa), total IκB-α (40 kDa), and β-actin (42 kDa). Blots from cells activated without (left panel) or with COG112 (right panel) were incubated in the same container with primary and then secondary antibody. After addition of chemiluminescence reagents, they were exposed to the same film. Similar results were observed in three separate experiments.
COG112 Inhibits IκB Kinase Activity in C. rodentium-stimulated YAMC Cells—Because we had identified that COG112 significantly attenuated C. rodentium-stimulated IκB-α phosphorylation, we determined whether this was due to an effect of COG112 on the IKK complex that acts to phosphorylate IκB-α. In Fig. 9A, IKK activity was assessed by ELISA in cytoplasmic cellular lysates from YAMC cells stimulated with C. rodentium in the presence or absence of COG112. Beginning 5 min after activation with C. rodentium, the IKK activity was significantly increased and peaked at 10 min with a 2.8 ± 0.5-fold increase. When cell lysates from COG112-treated cells were used, the kinase activity remained similar to the basal level of untreated YAMC cells for the first 10 min after C. rodentium stimulation and was significantly reduced at each time point after activation, out to 80 min.
FIGURE 9.

C. rodentium stimulates IκB kinase activity that is inhibited by COG112 by an effect on the formation of the IKK complex. In A–D, an ELISA assay for IKK activity was performed. In A, the time course of IKK activity in YAMC cells stimulated with C. rodentium in the absence (open squares) or presence of COG112 pretreatment (closed squares) was determined. Time 0 indicates cells that were not stimulated with C. rodentium.§, p < 0.05; §§, p < 0.01 versus C. rodentium alone; n = 3 separate experiments performed in duplicate. B, IKK activity was assessed at 20 min after stimulation of YAMC cells with C. rodentium in the absence or presence of the inhibitors COG112 (10 μm), BAY 11-7082 (20 μm), SB203580 (1 μm), PD98059 (5 μm), and SP600125 (5 μm). **, p < 0.01 versus control; §§, p < 0.01 versus C. rodentium alone; n = 2–3 experiments performed in duplicate. C, lysates from unstimulated and C. rodentium-stimulated YAMC cells were preincubated with COG112 (10 μm) for 1 h at 37 °C prior to addition to the IKK activity assay. **, p < 0.01 versus control; n = 2 experiments performed in duplicate. D, recombinant IKK-β enzyme provided by the manufacturer of the IKK enzyme assay kit was used at the concentrations indicated in the absence or presence of COG112 (10 μm) or the IKK-β inhibitor (IKK-2 inhibitor IV; 20 μm) provided by the kit's manufacturer. §, p < 0.05; §§, p < 0.01 versus no inhibitor; n = 2–3 experiments performed in duplicate. E, IKK complex formation was assessed in lysates of YAMC cells in the absence or presence of C. rodentium stimulation in the absence or presence of COG112 pretreatment (10 μm) by immunoprecipitation with antibody to IKK-α followed by immunoblotting with antibody to IKK-β (top panel), IKK-γ (middle panel), and phospho-IKK-α/β (bottom panel). Data shown are representative of two separate experiments.
It is notable that COG112 inhibited IKK activity to a similar degree as BAY 11-7082 (Fig. 9B), which is known to inhibit IKK (33). Because MAP kinases have been implicated in NF-κB activation in colonic epithelial cells (34, 35), we considered the possibility that this could be a mechanism for upstream inhibition of NF-κB by COG112. However, when we used the inhibitors of p38 MAP kinase (SB203580), pERK1/2 (PD98059), and JNK (SP600125), none had any effect on the C. rodentium-stimulated IKK activity (Fig. 9B), suggesting that these pathways are not involved in the activation of NF-κBby C. rodentium that we have identified.
COG112 Does Not Directly Inhibit the IκB Kinase—Because it is possible that COG112 could have a direct effect on the IKK enzyme itself, we conducted studies in which cellular lysates from unstimulated and C. rodentium-stimulated YAMC cells were incubated with COG112 for 1 h prior to addition to the kinase assay reaction mixture (Fig. 9C). This preincubation was performed because it has been reported that preincubation with other inhibitors may be necessary to observe an effect on IKK activity (15). However, our data demonstrate that the addition of COG112 had no effect on IKK activity in the cell-free system after lysates from stimulated cells had been prepared (Fig. 9C).
IKK-β is the activated form of IKK that is required for the rapid degradation of IκB-α associated with innate immunity in general (20) and as we observed in Fig. 8. Therefore, we analyzed IKK-β activity specifically, with the addition of increasing concentrations of recombinant IKK-β in our kinase assay that measures the phosphorylation of IκB-α (Fig. 9D). We found that there was a concentration-dependent increase in IKK-β activity that was not affected by addition of COG112 but was inhibited by the IKK-β inhibitor (IKK-2 inhibitor IV) supplied by the manufacturer of the K-LISA IKK activity assay kit.
COG112 Inhibits the Formation of the IκB Kinase (IKK) Complex in C. rodentium-stimulated YAMC Cells—Because COG112 inhibited IκB-α phosphorylation and IKK activity without a direct effect on the enzyme itself, we determined whether COG112 could affect the formation of the IKK enzyme complex, which is known to consist of three subunits (IKK-α, IKK-β, and IKK-γ), with assembly and phosphorylation of IKK-α/β required for activity (15, 17, 18). Therefore, we used antibody to IKK-α to immunoprecipitate the IKK complex from lysates of YAMC cells stimulated with C. rodentium in the absence or presence of COG112 (Fig. 9E). Immunoblotting with antibody to IKK-β (top panel) demonstrated that C. rodentium stimulated an increase in IKK-α/β complex formation that was reduced by COG112 to basal levels. Immunoblotting of the IKK-α-precipitated protein with antibody to IKK-γ demonstrated that while the IKK-α/γ complex was not increased by C. rodentium, formation of this complex was inhibited in the C. rodentium-stimulated cells by COG112. When the IKK-α-precipitated protein was immunoblotted with antibody to phospho-IKK-α/β, there was an increase in the phosphorylation of the IKK-α/β complex with C. rodentium stimulation that was inhibited by COG112. Taken together, these data indicate that C. rodentium stimulates formation of a functional IKK complex in colonic epithelial cells, and that COG112 inhibits the canonical pathway of NF-κB activation by blocking the formation of this active complex that is required for phosphorylation of IκB-α.
DISCUSSION
In the current report, we have demonstrated that the apoE-mimetic peptide COG112 inhibits bacterially induced expression of iNOS mRNA and protein and NO production in colonic epithelial cells in a concentration-dependent manner. This is important because the pathogen we have studied, C. rodentium, causes colitis in mice that closely mimics human IBD, and deletion of iNOS protects against disease while inhibition of the competing arginase pathway exacerbates colitis (11). Furthermore, uncontrolled up-regulation of iNOS has been associated with a causal role in colonic inflammation in numerous rodent model studies (36). The relevance of our co-culture model is also supported by the fact that we and others (11, 37) have demonstrated that iNOS immunolocalizes to the colonic epithelium during C. rodentium colitis.
We also demonstrated that COG112 inhibits C. rodentium-induced expression of the CXC chemokines, KC and MIP-2, which are similar to human IL-8. This is also important because epithelial chemokine expression has long been recognized as a key early step in the pathophysiology of inflammatory responses to numerous colonic pathogens (38) and has been directly linked to the pathophysiology of human IBD (39). Thus the ability of COG112 to down-regulate these neutrophil chemoattractants is predictive of a potential benefit in various forms of colonic inflammation.
A common thread that links induction of chemokines and iNOS in the epithelium is the activation of NF-κB (40). In fact, we found that the proteosomal inhibitor MG132 that blocks NF-κB activation inhibited expression of iNOS and the chemokines KC and MIP-2. It was striking that the degree of inhibition of each transcript with MG132 was identical to that of COG112, suggesting a convergent, common mechanism of inhibition of gene expression that was corroborated by the finding that the combination of COG112 and MG132 exhibited no additivity in the inhibition of these genes. We also observed nearly identical results when a specific NF-κB inhibitor, BAY 11-7082, was used. Because MG132 and BAY 11-7082 act at different sites in the NF-κB pathway, the similar findings in these additivity experiments provide strong evidence that COG112 inhibits the degradation of IκB-α. It should be noted that neither COG112, MG132, nor BAY 11-7082 completely blocked expression of iNOS, KC, or MIP-2. Although this finding may reflect to some degree the limitations of these inhibitors in terms of cytotoxicity versus efficacy, these data suggest that there may also be NF-κB-independent components involved in the induction of these pro-inflammatory genes.
Our studies confirmed an important role for COG112 in the inhibition of NF-κB activation in response to C. rodentium activation. We have shown that there is C. rodentium-induced accumulation of nuclear proteins that bind to NF-κB consensus recognition sites by EMSA that is potently inhibited by COG112. We verified that there was nuclear translocation of p65 by both immunoblotting and immunofluorescence, and that COG112 impressively blocked the nuclear accumulation of p65. We also further defined the COG112 effect by demonstrating that it effectively blocked IκB-α phosphorylation and degradation, and the activity of the upstream IκB kinase. COG112 did not have any direct effect on IKK activity, because incubation of COG112 with either cell lysates from C. rodentium-stimulated cells or recombinant IKK-β did not impair the enzyme activity in a specific assay. However, COG112 markedly attenuated the formation of the IKK-α/β complex and its phosphorylation induced by C. rodentium and reduced the formation of the complex of the regulatory subunit IKK-γ with IKK-α that has also been implicated in IKK-β kinase activity (19). Rapid activation of NF-κB as we have observed in our studies is consistent with canonical NF-κB activation and formation of the IKK complex as a key regulatory step (20). Therefore our data strongly suggest that COG112 acts on canonical signaling, and it is unlikely that COG112 blocks NF-κB activation by an effect outside of the NF-κB pathway. Investigation into the mechanism of inhibition of the IKK complex formation by COG112 should prove to be an important area of future studies.
It is unlikely that the salutary effects of COG112 that we have observed are nonspecific, because previous work has demonstrated that scrambled apoE-mimetic peptides have no effect in vitro (5) or in vivo (41), and we tested the antennapedia moiety of COG112 alone and it had no inhibitory effects. The relevance of our studies is supported by a recent report that C. rodentium infection in vivo is associated with IκB-α degradation and p65 nuclear translocation in colonic crypts (42). Furthermore, the activation of NF-κB that we observed is similar to what has been described in studies using EPEC in vitro (12, 13).
These reports and our data suggest that the effect of COG112 may be generalizable to the in vivo condition and to other models. In fact, we have recently found that COG112 attenuates IL-8 mRNA expression in Helicobacter pylori-stimulated gastric epithelial cells.4 It has also been reported that macrophages exposed to C. rodentium in vitro demonstrate nuclear translocation of p65 (43). Consistent with this, in preliminary studies we have found that COG112 effectively inhibits iNOS mRNA expression and NO production in RAW 264.7 murine macrophages stimulated with C. rodentium lysates.5 Given that mice with toll-like receptor (TLR) 4 deletion exhibit attenuation of C. rodentium colitis through an effect attributed to reduced NF-κB activation in macrophages (43), it is likely that an inhibitory effect of COG112 in macrophages may also be important in colitis models. The studies in the current report complement previous work demonstrating that mice with apoE deficiency have increased susceptibility to inflammation (4) and that treatment with apoE (4) and the apoE-mimetic peptides COG133 (5) and COG112 (7) reduces activation of immune cells. Our studies provide new insight into the potential role of apoE in the response of epithelial cells to enteric pathogens. Because C. rodentium likely activates epithelial cells in an LPS- and TLR-4-independent manner (43), our studies highlight the potential importance of both endogenous apoE and therapeutic administration of exogenous apoE-mimetic peptides in the regulation of inflammatory responses in epithelial cells.
Our data may have direct relevance to IBD. It has long been recognized that there is activation of NF-κB in human IBD tissues (44). Anti-inflammatory drugs used to treat the disease, such as corticosteroids (45), salicylates (46), and sulfasalazine (47) are inhibitors of NF-κB. Intriguingly, it has also been recently reported that decoy oligonucleotides targeting the NF-κB consensus binding sequence attenuate both T helper cell 1- and T helper cell 2-mediated forms of experimental colitis (48). Based on our results presented herein and the striking effectiveness of COG112 in vivo in the EAE model of neuroinflammation (7), we have initiated experiments administering COG112 to mice in colitis models. In the dextran sulfate sodium model of colitis, impairment of innate immunity in TLR4-/- and myeloid differentiation marker (MyD)88-/- mice results in exacerbation of colitis (49), raising the possibility that an agent like COG112 that attenuates innate immune response genes, such as iNOS and chemokines, could actually be deleterious. However, we have found that in mice treated with DSS for 7 days, there is significant amelioration of body weight loss and reduction in histologic injury with daily injection of COG112 (1 mg/kg).5 Further, in preliminary studies in the C. rodentium model of colitis we have also observed reduction in colitis severity.6 Finally, NF-κB activation has been directly implicated in the causation of colitis-associated carcinoma in the azoxymethane-dextran sulfate sodium mouse model (50, 51). This suggests that apoE-mimetic peptides may have benefit in the chemoprevention of inflammation-associated carcinogenesis in a variety of tissues, and studies along these lines should prove to be a fruitful area of investigation.
Acknowledgments
This study utilized the Novel Cell Line Development Core of the Vanderbilt University Medical Center Digestive Disease Research Center supported by National Institutes of Health Grant P30DK058404.
This work was supported, in whole or in part, by National Institutes of Health Grants R41 DK075161 (to K. T. W. and M. P. V.) and R01 DK053620 (to K. T. W.). This work was also supported by the Office of Medical Research, Dept. of Veterans Affairs (to K. T. W.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: IBD, inflammatory bowel disease; apoE, apolipoprotein E; EAE, experimental autoimmune encephalomyelitis; EMSA, electromobility shift assay; FITC, fluorescein isothiocyanate; IFN, interferon; IL, interleukin; iNOS, inducible nitric-oxide synthase; KC, keratinocytederived chemokine; LPS, lipopolysaccharide; MIP, macrophage inflammatory protein; NO, nitric oxide; PBS, phosphate-buffered saline; TNF, tumor necrosis factor; YAMC, young adult mouse colon; MOI, multiplicity of infection; ELISA, enzyme-linked immunosorbent assay; GST, glutathione S-transferase; DAPI, 4′,6-diamidino-2-phenylindole.
S. M. Cauble, M. Asim, R. Chaturvedi, D. P. Barry, N. D. Lewis, K. Singh, M. P. Vitek, and K. T. Wilson. Abstract presented at the 2008 Digestive Week Meeting of the American Gastroenterological Association, May 2008.
K. Singh, R. Chaturvedi, D. P. Barry, M. Asim, R. J. Wadia, N. D. Lewis, M. K. Washington, M. P. Vitek, and K. T. Wilson. Abstract presented at the 2008 Digestive Week Meeting of the American Gastroenterological Association, May 2008.
K. Singh, R. Chaturvedi, D. P. Barry, M. Asim, N. D. Lewis, M. P. Vitek, and K. T. Wilson, unpublished data.
References
- 1.Loftus, C. G., Loftus, E. V., Jr., Harmsen, W. S., Zinsmeister, A. R., Tremaine, W. J., Melton, L. J., 3rd, and Sandborn, W. J. (2007) Inflamm. Bowel Dis. 13 254-261 [DOI] [PubMed] [Google Scholar]
- 2.Clark, M., Colombel, J. F., Feagan, B. C., Fedorak, R. N., Hanauer, S. B., Kamm, M. A., Mayer, L., Regueiro, C., Rutgeerts, P., Sandborn, W. J., Sands, B. E., Schreiber, S., Targan, S., Travis, S., and Vermeire, S. (2007) Gastroenterology 133 312-339 [DOI] [PubMed] [Google Scholar]
- 3.Laskowitz, D. T., Lee, D. M., Schmechel, D., and Staats, H. F. (2000) J. Lipid Res. 41 613-620 [PubMed] [Google Scholar]
- 4.Van Oosten, M., Rensen, P. C., Van Amersfoort, E. S., Van Eck, M., Van Dam, A. M., Breve, J. J., Vogel, T., Panet, A., Van Berkel, T. J., and Kuiper, J. (2001) J. Biol. Chem. 276 8820-8824 [DOI] [PubMed] [Google Scholar]
- 5.Laskowitz, D. T., Thekdi, A. D., Thekdi, S. D., Han, S. K., Myers, J. K., Pizzo, S. V., and Bennett, E. R. (2001) Exp. Neurol. 167 74-85 [DOI] [PubMed] [Google Scholar]
- 6.Lynch, J. R., Tang, W., Wang, H., Vitek, M. P., Bennett, E. R., Sullivan, P. M., Warner, D. S., and Laskowitz, D. T. (2003) J. Biol. Chem. 278 48529-48533 [DOI] [PubMed] [Google Scholar]
- 7.Li, F. Q., Sempowski, G. D., McKenna, S. E., Laskowitz, D. T., Colton, C. A., and Vitek, M. P. (2006) J. Pharmacol. Exp. Ther. 318 956-965 [DOI] [PubMed] [Google Scholar]
- 8.Schauer, D. B., Zabel, B. A., Pedraza, I. F., O'Hara, C. M., Steigerwalt, A. G., and Brenner, D. J. (1995) J. Clin. Microbiol. 33 2064-2068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Higgins, L. M., Frankel, G., Connerton, I., Goncalves, N. S., Dougan, G., and MacDonald, T. T. (1999) Science 285 588-591 [DOI] [PubMed] [Google Scholar]
- 10.Higgins, L. M., Frankel, G., Douce, G., Dougan, G., and MacDonald, T. T. (1999) Infect. Immun. 67 3031-3039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gobert, A. P., Cheng, Y., Akhtar, M., Mersey, B. D., Blumberg, D. R., Cross, R. K., Chaturvedi, R., Drachenberg, C. B., Boucher, J. L., Hacker, A., Casero, R. A., Jr., and Wilson, K. T. (2004) J. Immunol. 173 2109-2117 [DOI] [PubMed] [Google Scholar]
- 12.Savkovic, S. D., Koutsouris, A., and Hecht, G. (1997) Am. J. Physiol. 273 C1160-C1167 [DOI] [PubMed] [Google Scholar]
- 13.Savkovic, S. D., Koutsouris, A., and Hecht, G. (2003) Am. J. Physiol. Cell Physiol. 285 C512-C521 [DOI] [PubMed] [Google Scholar]
- 14.Baeuerle, P. A., and Henkel, T. (1994) Annu. Rev. Immunol. 12 141-179 [DOI] [PubMed] [Google Scholar]
- 15.DiDonato, J. A., Hayakawa, M., Rothwarf, D. M., Zandi, E., and Karin, M. (1997) Nature 388 548-554 [DOI] [PubMed] [Google Scholar]
- 16.Chiao, P. J., Miyamoto, S., and Verma, I. M. (1994) Proc. Natl. Acad. Sci. U. S. A. 91 28-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mercurio, F., Zhu, H., Murray, B. W., Shevchenko, A., Bennett, B. L., Li, J., Young, D. B., Barbosa, M., Mann, M., Manning, A., and Rao, A. (1997) Science 278 860-866 [DOI] [PubMed] [Google Scholar]
- 18.Zandi, E., Rothwarf, D. M., Delhase, M., Hayakawa, M., and Karin, M. (1997) Cell 91 243-252 [DOI] [PubMed] [Google Scholar]
- 19.Rothwarf, D. M., Zandi, E., Natoli, G., and Karin, M. (1998) Nature 395 297-300 [DOI] [PubMed] [Google Scholar]
- 20.Hacker, H., and Karin, M. (2006) Sci. STKE 2006, re13. [DOI] [PubMed]
- 21.Ghosh, S., and Karin, M. (2002) Cell 109 (suppl.) S81-S96 [DOI] [PubMed] [Google Scholar]
- 22.Whitehead, R. H., VanEeden, P. E., Noble, M. D., Ataliotis, P., and Jat, P. S. (1993) Proc. Natl. Acad. Sci. U. S. A. 90 587-591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chaturvedi, R., Cheng, Y., Asim, M., Bussiere, F. I., Xu, H., Gobert, A. P., Hacker, A., Casero, R. A., Jr., and Wilson, K. T. (2004) J. Biol. Chem. 279 40161-40173 [DOI] [PubMed] [Google Scholar]
- 24.Cheng, Y., Chaturvedi, R., Asim, M., Bussiere, F. I., Xu, H., Casero, R. A., Jr., and Wilson, K. T. (2005) J. Biol. Chem. 280 22492-22496 [DOI] [PubMed] [Google Scholar]
- 25.Chaturvedi, R., Asim, M., Lewis, N. D., Algood, H. M., Cover, T. L., Kim, P. Y., and Wilson, K. T. (2007) Infect. Immun. 75 4305-4315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bussiere, F. I., Chaturvedi, R., Cheng, Y., Gobert, A. P., Asim, M., Blumberg, D. R., Xu, H., Kim, P. Y., Hacker, A., Casero, R. A., Jr., and Wilson, K. T. (2005) J. Biol. Chem. 280 2409-2412 [DOI] [PubMed] [Google Scholar]
- 27.Podolin, P. L., Callahan, J. F., Bolognese, B. J., Li, Y. H., Carlson, K., Davis, T. G., Mellor, G. W., Evans, C., and Roshak, A. K. (2005) J. Pharmacol. Exp. Ther. 312 373-381 [DOI] [PubMed] [Google Scholar]
- 28.Luperchio, S. A., and Schauer, D. B. (2001) Microbes Infect. 3 333-340 [DOI] [PubMed] [Google Scholar]
- 29.Tebo, J. M., Datta, S., Kishore, R., Kolosov, M., Major, J. A., Ohmori, Y., and Hamilton, T. A. (2000) J. Biol. Chem. 275 12987-12993 [DOI] [PubMed] [Google Scholar]
- 30.Widmer, U., Manogue, K. R., Cerami, A., and Sherry, B. (1993) J. Immunol. 150 4996-5012 [PubMed] [Google Scholar]
- 31.Pierce, J. W., Schoenleber, R., Jesmok, G., Best, J., Moore, S. A., Collins, T., and Gerritsen, M. E. (1997) J. Biol. Chem. 272 21096-21103 [DOI] [PubMed] [Google Scholar]
- 32.Kamthong, P. J., and Wu, M. (2001) Biochem. J. 356 525-530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kundu, J. K., Shin, Y. K., Kim, S. H., and Surh, Y. J. (2006) Carcinogenesis 27 1465-1474 [DOI] [PubMed] [Google Scholar]
- 34.Jobin, C., Bradham, C. A., Russo, M. P., Juma, B., Narula, A. S., Brenner, D. A., and Sartor, R. B. (1999) J. Immunol. 163 3474-3483 [PubMed] [Google Scholar]
- 35.Jijon, H., Allard, B., and Jobin, C. (2004) Cell. Signal. 16 1023-1032 [DOI] [PubMed] [Google Scholar]
- 36.Cross, R. K., and Wilson, K. T. (2003) Inflamm. Bowel Dis. 9 179-189 [DOI] [PubMed] [Google Scholar]
- 37.Vallance, B. A., Deng, W., De Grado, M., Chan, C., Jacobson, K., and Finlay, B. B. (2002) Infect. Immun. 70 6424-6435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jung, H. C., Eckmann, L., Yang, S. K., Panja, A., Fierer, J., Morzycka-Wroblewska, E., and Kagnoff, M. F. (1995) J. Clin. Investig. 95 55-65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sartor, R. B. (2006) Nat. Clin. Pract. Gastroenterol. Hepatol. 3 390-407 [DOI] [PubMed] [Google Scholar]
- 40.Elewaut, D., DiDonato, J. A., Kim, J. M., Truong, F., Eckmann, L., and Kagnoff, M. F. (1999) J. Immunol. 163 1457-1466 [PubMed] [Google Scholar]
- 41.Laskowitz, D. T., McKenna, S. E., Song, P., Wang, H., Durham, L., Yeung, N., Christensen, D., and Vitek, M. P. (2007) J. Neurotrauma 24 1093-1107 [DOI] [PubMed] [Google Scholar]
- 42.Wang, Y., Xiang, G. S., Kourouma, F., and Umar, S. (2006) Br. J. Pharmacol. 148 814-824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khan, M. A., Ma, C., Knodler, L. A., Valdez, Y., Rosenberger, C. M., Deng, W., Finlay, B. B., and Vallance, B. A. (2006) Infect. Immun. 74 2522-2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schreiber, S., Nikolaus, S., and Hampe, J. (1998) Gut 42 477-484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scheinman, R. I., Cogswell, P. C., Lofquist, A. K., and Baldwin, A. S., Jr. (1995) Science 270 283-286 [DOI] [PubMed] [Google Scholar]
- 46.Kopp, E., and Ghosh, S. (1994) Science 265 956-959 [DOI] [PubMed] [Google Scholar]
- 47.Wahl, C., Liptay, S., Adler, G., and Schmid, R. M. (1998) J. Clin. Investig. 101 1163-1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fichtner-Feigl, S., Fuss, I. J., Preiss, J. C., Strober, W., and Kitani, A. (2005) J. Clin. Investig. 115 3057-3071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fukata, M., Michelsen, K. S., Eri, R., Thomas, L. S., Hu, B., Lukasek, K., Nast, C. C., Lechago, J., Xu, R., Naiki, Y., Soliman, A., Arditi, M., and Abreu, M. T. (2005) Am. J. Physiol. Gastrointest. Liver. Physiol. 288 G1055-G1065 [DOI] [PubMed] [Google Scholar]
- 50.Greten, F. R., Eckmann, L., Greten, T. F., Park, J. M., Li, Z. W., Egan, L. J., Kagnoff, M. F., and Karin, M. (2004) Cell 118 285-296 [DOI] [PubMed] [Google Scholar]
- 51.Xiao, H., Gulen, M. F., Qin, J., Yao, J., Bulek, K., Kish, D., Altuntas, C. Z., Wald, D., Ma, C., Zhou, H., Tuohy, V. K., Fairchild, R. L., de la Motte, C., Cua, D., Vallance, B. A., and Li, X. (2007) Immunity 26 461-475 [DOI] [PubMed] [Google Scholar]