

Abstract
We report here a method for proteomics pattern discovery by utilizing a self-organizing map approach to analyze data obtained from a novel multiplex iTRAQ™ proteomics method. Through the application of this technique, we were able to delineate the early molecular events preceding dorsal root ganglia neurite outgrowth induced by either nerve growth factor (NGF) or an immunophilin ligand, JNJ460. Following pattern analysis we discovered that each neurotrophic agent promoted mostly distinct increases in protein expression with few overlapping patterns. In the NGF-treated group, proteins possessing “biosynthesis function” (p < 0.002) and “ribosome localization” (p < 0.0003) were increased, while proteins promoting “organogenesis” (p < 0.004) and related “signal transduction” (p < 0.008) functions were notably increased in the JNJ460-treated group. This study suggests that the properties of neurite outgrowth triggered by NGF and JNJ460 can be distinguished at the proteome level. Multiplexed proteomics analysis, along with pattern discovery bioinformatics tools, has the capability to differentiate subtle neuroproteomics patterns.
Keywords: iTRAQ, Nerve growth factor, Immunophilin ligand, Mass spectrometry, SOM analysis, Proteomic pattern
1. Introduction
We describe here a proteomic pattern discovery method using the iTRAQ™ proteomics approach in combination with self-organizing map (SOM) data analysis routine. Effective implementation of SOM has enabled us to ascertain proteomic patterns in regenerating dorsal root ganglia (DRGs) treated with either nerve growth factor (NGF) or JNJ460, an immunophilin ligand. NGF and immunophilin ligands are among distinct classes of molecules known to mediate axonal outgrowth and nerve regeneration. JNJ460 is a non-immunosuppressive derivative of FK506, an immunosuppressant macrolide drug isolated from the bacterium Streptomyces tsukubaensis. FK506 is used clinically to prevent allograft rejection in organ transplantations (Schreiber and Crabtree, 1992). In addition to its role in organ transplantation, FK506 is also recognized as having a nerve regenerative property independent of its immunosuppressive effects. FK506 accelerates functional recovery of injured nerves by increasing the rate of axonal regeneration in rat sciatic nerve and DRG, as well as decreases infarct size in ischemic stoke models (Hamilton and Steiner, 1998). Over the past decade, several non-immunosuppressive FK506 derivatives have been developed, including JNJ460 and GPI1046. These molecules retain the neuroregenerative capabilities of FK506, but do so without immunosuppression, since they lack the calcineurin effecter domain (Armistead et al., 1995).
NGF promotes the survival and differentiation of subsets of neurons in both the peripheral and central nervous systems (Yuen et al., 1996). It exerts its action by binding to TrkA, a receptor tyrosine kinase that initiates signal pathways important for differentiation, including the Ras/Raf/MAP kinase pathway (Kaplan, 1998). Previous studies have shown that the immunophilin ligands function to potentiate signals from neurotrophins and prime neurons to better respond to neurotrophic factors (Tanaka et al., 2003). However, it remains unclear how immunophilin ligands cooperate with neurotrophic factors to promote neurite outgrowth.
In order to understand the early response mechanisms of JNJ460, we utilized iTRAQ-based method to compare DRG proteomic changes among NGF, JNJ460 treatments and the controls. The iTRAQ labeling reagents consist of a balance groups, and a peptide reactive group which labels all the primary amines (i.e. peptide N-termini and/or lysine side chains), and a reporter group capable of differentiating up to four samples. The peptide quantification is based on the relative peak areas of the four reporter ions, m/z 114.1, 115.1, 116.1 and 117.1, produced following tandem mass spectrometry fragmentation of iTRAQ-labeled peptides (DeSouza et al., 2005). This technology has been successfully applied to elucidate biochemical pathways involved in reperfusion stress (Hirsch et al., in press), to discover cancer markers (DeSouza et al., 2005), to characterize dynamic phosphorylation events in the epidermal growth factor receptor signaling network (Zhang et al., 2005) and to evaluate diurnal peptide variation in parotid saliva (Hardt et al., 2005).
Since we needed to simultaneously compare protein expression among three groups, a pattern recognition analysis routine is more preferable to conventional binary comparison methods. SOM is a clustering method based on machine learning using artificial neural networks. Through reiterative learning, this algorithm produces a two-dimensional (or 3D) grid, in which similar records appear close to each other and dissimilar records appear more distant (Tamayo et al., 1999). Unlike Bayesian-based clustering method, SOM does not require prior knowledge of the data grouping characteristics. In addition, SOM is easy to implement, computationally efficient and scalable, which makes it amenable for gene expression analysis. It has been used for cancer classification, signal transduction pathway studies as well as pharmacological differentiation of drug treatments (Golub et al., 1999; Ideker et al., 2001). Although commonly used in cDNA microarray studies, more recent applications of SOM to proteomics studies allow one to analyze datasets with multiplex trend data points such as time course and dose response (Zhang et al., 2005).
In the present study, we used SOM to analyze the early responsive proteins elevated by two agents that promote neurite outgrowth, NGF and JNJ460. Interestingly, we observed divergent proteomic patterns of neurite outgrowth. JNJ460 treatment resulted in the increase of proteins involved in “organogenesis” and related “signal transduction” events. By comparison, predominant increase of ribosomal proteins and proteins with “biosynthesis” activities were observed in NGF treatment. This proteomic pattern analysis method should be applicable for other neuroproteomic discoveries.
2. Materials and methods
2.1. Tissue collection, culture and protein extraction
All animal experiments were conducted under the direction of IACUC-approved protocols. DRGs were dissected from P1 to P3 C57Bl/6 mice as previously described (Birge et al., 2004). In control group, 110 DRGs were pooled and placed in ice-cold phosphate buffer solution (PBS, pH 7.4) containing 2% glucose. The tissues were transferred onto sterile polyornithine–laminin tissue culture plates in Dulbecco’s modified Eagle media (Cellgro, Herndon, VA, USA) supplemented with 10% fetal calf serum, 2% glucose and 1% penicillin–streptomycin and incubated at 37 °C for 4 h. In the two treatment groups, DRGs were cultured with media supplemented with either 1 μM JNJ460 or 100 ng/ml NGF. After 4-h incubation, DRGs were collected via centrifugation at 1000 × g for 1 min. The supernatants were discarded and the DRGs were washed twice with PBS and stored at −80 °C. In order to minimize fluctuations in expression patterns, we performed five replicate experiments for the proteomic analysis. The pooled DRGs were homogenized in 200 μl of lysis buffer (pH 10.0) containing 25 mM triethylammonium bicarbonate, 20 mM Na2CO3 and 2 μl of protease inhibitor cocktail (Sigma, St Louis, MO, USA). After centrifugation at 19,000 × g for 30 min, the supernatants were adjusted to pH 7.5, with HEPES (100 mM). Protein concentration was measured via Bradford assay.
2.2. iTRAQ sample preparation
iTRAQ reagents were purchased from Applied Biosystems Inc. (ABI, Framingham, MA, USA). Seventy micrograms of each protein extract was denatured and alkylated as described according to manufacturer’s protocol (iTRAQ™ Reagents Chemistry Reference Guide, 2004). Each sample was then digested with 10 μg of trypsin (Promega, Madison, WI, USA) at 37 °C overnight. The peptides were labeled with the iTRAQ reagents as follows: duplicated controls with tags 114 and 117; JNJ460 with tag 115; NGF with tag 116. The labeled peptides were mixed in even ratios.
2.3. Two-dimensional liquid chromatography and tandem mass spectrometry (MS/MS)
The peptide mixture was resuspended in 0.5 ml of cation exchange mobile phase A containing 10 mM KH2PO4 and 20% acetonitrile (ACN) (pH 3.0). The peptides were separated on a BioCAD™ Perfusion Chromatography System (ABI) equipped with a polysulfoethyl A column (4.6 mm × 200 mm, 5 μm, 300 Å, Poly LC Inc., Columbia, MD, USA) plus an upstream guard column (4 mm × 10 mm). Mobile phase B consisted of 600 mM KCl, 10 mM KH2PO4 and 20% ACN (pH 3.0). The column was first washed isocratically with mobile phase A for 10 min at 1.0 ml/min to remove unbound materials. Bound peptides were then eluted with a 40 min linear gradient from 0 to 50% B, followed by a 10 min linear gradient from 50 to 100% B. Two minute fractions were collected and desalted via PepClean™ C18 spin columns (Pierce, Rockford, IL, USA). Peptides in each ion exchange fraction were further separated on an Ultimate™ Chromatography System equipped with a Probot matrix-assisted laser desorption ionization (MALDI) spotting device (Dionex, Sunnyvale, CA, USA). Peptides were captured onto a reversed phase 0.3 mm × 5 mm trapping column and resolved on a 0.1 mm × 150 mm capillary PepMap column (3 μm, 100 Å, C18, Dionex) with a 70 min gradient of solvent A (5% ACN, 0.1% trifluoroacetic acid, TFA) and solvent B (95% ACN, 0.1% TFA): 0–4 min, from 5 to 8% B, at 34 min, to 18% B, at 57 min, to 35% B and at 64 min to 95% B. The HPLC eluent was mixed in a 1:3 ratio with matrix (7 mg/ml α-cyano-4-hydroxycinnamic acid in 60% ACN, 5 mM ammonium monobasic phosphate and the internal calibrants, 50 fmol/μl each of GFP and ACTH, 18–39) through a 30 nl mixing tee, and spotted onto MALDI plates in a 18 × 18 spot array format. The peptides were analyzed on a 4700 Proteomics Analyzer tandem mass spectrometer (ABI) in a data-dependent fashion. MS spectra (m/z 800–3600) were acquired in positive ion mode with internal mass calibration. Eight most intense ions per spot were selected for subsequent MS/MS analysis in 1 keV mode. Each spectrum was averaged over 4000 laser shots.
2.4. Bioinformatics and self-organizing-map (SOM)
Peptide identification was performed by searching the MS/MS spectra against the SwissProt database (v. 46) using a local MASCOT search engine (v. 1.9) on a GPS (v. 3.5, ABI) server. The following search parameters were used: trypsin with one missed cleavage was selected; mass tolerance was 50 ppm for the precursors and 0.3 Da for the MS/MS ions; iTRAQ-labeled N-termini and lysines and MMTS-labeled cysteines were set as fixed modifications; oxidized methionines and iTRAQ-labeled tyrosines were set as variable modifications. Only peptides identified with confidence interval (C.I.) values of at least 95% were used for protein quantification and pattern analysis.
For quantitative analysis, isotopic carryover-corrected iTRAQ reporter ion peak areas (RPAs) were extracted from the raw data using the GPS server. Data normalization was performed using the median RPAs of all identified peptides assuming overall protein concentrations in each sample were comparable. Peptide expression ratios were calculated as JNJ460 (115.1) or NGF (116.1) RPA over the average of two control RPAs (114.1 or 117.1). The ratios were transformed into log2 numeric for subsequent analysis. A protein expression ratio was computed as the average of all corresponding peptides, with at least two peptides per protein. And expression range was reported as the protein expression ratio plus/minus the standard deviation of all corresponding peptides. An in-house perl script was written to automate all computational processes. Protein ratios presented in Tables 2 and 3 are anti-logged data for more convenient data interpretation.
Table 2.
Selected proteins upregulated in JNJ460-treated DRGs
| Accession numbera | Protein | Ratiob | Rangeb |
|---|---|---|---|
| GO function: organogenesis | |||
| P02772 | α-Fetoprotein | 1.4 | 1.6–1.3 |
| P68033 | Actin | 1.3 | 1.4–1.2 |
| P10637 | Microtubule-associated protein tau | 1.3 | 1.6–1.1 |
| Q62234 | Myomesin 1 | 1.3 | 1.4–1.2 |
| P13541 | Myosin heavy chain, fast skeletal muscle | 1.3 | 1.3–1.3 |
| P05977 | Myosin light chain 1 | 1.3 | 1.5–1.1 |
| Q9Z2H5 | Neuronal protein 4.1 | 1.3 | 1.4–1.2 |
| Q9QZ47 | Troponin T | 1.5 | 1.7–1.3 |
| GO function: signal transduction | |||
| Q9QYF9 | N-myc downstream regulated gene 3 protein (NDRG3) | 1.5 | 1.8–1.2 |
| Q61074 | Protein phosphatase 2C gamma isoform (PP2C) | 1.3 | 1.5–1.2 |
| Q99JI6 | Ras-related protein Rap-1b | 1.3 | 1.5–1.1 |
| P62137 | Serine/threonine-protein phosphatase PP1-α | 1.3 | 1.5–1.2 |
SwissProt accession number.
Protein expression ratios are anti-logged values from the average of all log2-based peptide expression ratios.
Table 3.
Selected proteins upregulated in NGF-treated DRGs
| Accession numbera | Protein | Ratio | Rangeb |
|---|---|---|---|
| GO function: ribosome | |||
| Q921I9 | Exosome complex exonuclease RRP41 | 1.3 | 1.5–1.1 |
| P62983 | 40S ribosomal protein S27a | 1.4 | 1.7–1.1 |
| P47955 | 60S acidic ribosomal protein P1 | 1.3 | 1.7–1.0 |
| GO function: biosynthesis | |||
| P63242 | Eukaryotic translation initiation factor 5A | 1.3 | 1.4–1.2 |
| Q8BXQ2 | GPI transamidase component PIG-T | 1.3 | 1.7–1.0 |
| Other functions | |||
| P97370 | Sodium/potassium-transporting ATPase 3-3 | 1.3 | 1.3–1.2 |
| Q9CPQ3 | Mitochondria import receptor TOM22 | 1.3 | 1.9–0.9 |
| P62761 | Neuronal visinin-like protein 1 (NVP-1) | 1.5 | 1.8–1.2 |
| Q9EQH2 | Adipocyte-derived leucine aminopeptidase | 1.3 | 1.4–1.3 |
SwissProt accession number.
Protein expression ratios are anti-logged values from the average of all log2-based peptide expression ratios.
To distinguish proteomic patterns, expression ratios from both NGF and JNJ460 treatments relative to the control average were analyzed using a SOM routine with Spotfire software package (v8.2, Somerville, MA, USA) with default settings (bubble neighborhood function with radius of 2.5 × 0; linear learning function with initial ratio at 0.05; 12,500 iteration was used to generate a 3 × 3 grid). The software initially mapped the “nodes” of the grid (group) into k-dimensional space (k = 3 for the three sets of ratio values compared) and then iteratively adjusted them for the best fit. With each iteration, a data point (a protein) will be randomly selected and the location of all the nodes will be adjusted in the k-dimensional space according to their distances to the selected data point, with the closest node moved the most whereas distant nodes moved less. After 12,500 iterations, nine distinct clusters were produced to yield the best cluster separation as well as preventing over-clustering. Increasing in clustering group number did not produce more distinct patterns while decreasing group number generated overlapping groups with little relevance for biological inference. Thus, we settled at 3 × 3 grid for this study. The protein functional “enrichments” in each treatment group were then analyzed by DAVID software tools (Dennis et al., 2003) based on gene ontology (GO) annotations. GO annotations are composed of three ontologies: biological process (B.P.), molecular function (M.F.) and cellular component (C.C.) (Ashburner et al., 2000). For each level 3 GO term, a hypergeometric test was used to compare the number of proteins in the group against all identified proteins, and a p-value was produced to indicate the enrichment significance for that term.
2.5. Western blotting
Western blotting was also used to validate selected protein expression changes. Triplicate experiments were carried out: 10 μg/lane of extracted proteins were probed with anti-troponin T (Research Diagnostics Inc., Flanders, NJ, USA), anti-eIF-5A (Pharmingen, San Diego, CA, USA) or anti-FKBP52 (BIOMOL Research Laboratories Inc. Plymouth Meeting, PA, USA), and secondary antibody-coupled signal was detected using ECL chemiluminescence method (Perkin-Elmer, Boston, MA, USA). Quantification was calculated with Quantity One software (v. 4.3.1, Bio-Rad, San Francisco, CA, USA).
3. Results
3.1. SOM clustery of iTRAQ quantified proteins into nine groups
DRGs from neonatal C57 Bl6 mice were surgically explanted and incubated in media supplemented with either (100 ng/ml) NGF or (1 μM) JNJ460 for 4 h in order to delineate differences in their regenerative paradigms. Searching all 26,051 MS/MS spectra against mouse proteins in SwissProt protein database led to the assignment of 5320 unique peptides from 985 proteins.
SOM analysis of the expression ratios from both treatments in comparison to the averaged control divided the identified proteins into nine groups (Fig. 1 and Supplementary Table 1). The groupings revealed divergent protein expression patterns following these treatments. For example, groups 2, 4 and 5 contained proteins that did not change significantly from either treatment. The proteins clustered in group 1 were slightly decreased while the ones in group 9 were increased following both treatments. GO functional analysis revealed that most of the proteins enriched in group 9 were related to cellular metabolism, indicating metabolic activation upon both pharmacological treatments (Supplementary Table 2). The proteins clustered in groups 3 and 6 contain NGF-specific elevated proteins, where the ones in groups 7 and 8 contain JNJ460-specific elevated proteins.
Fig. 1.
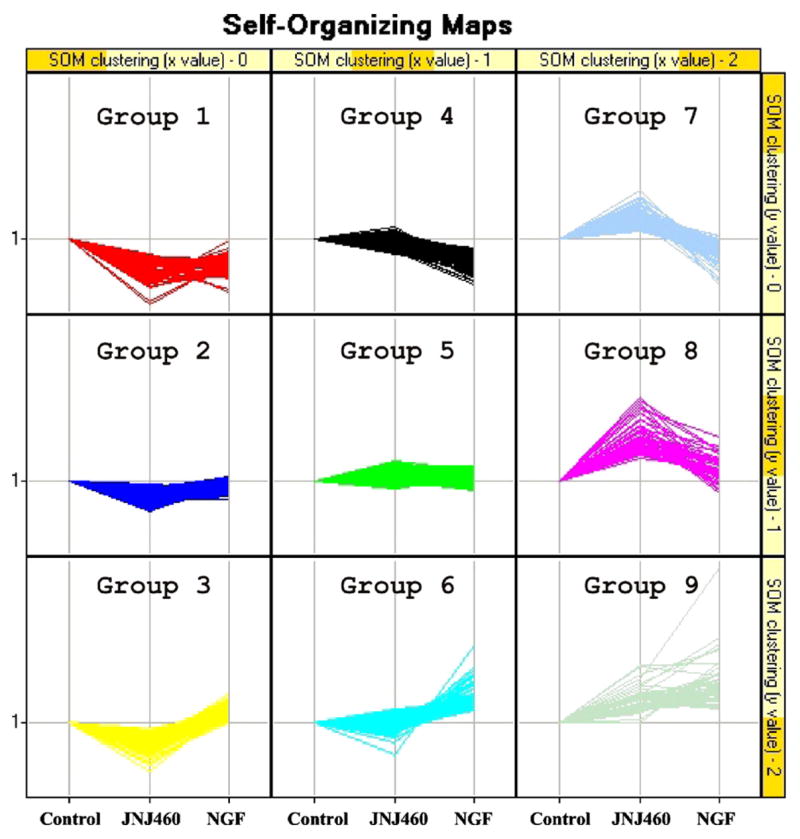
Proteomic clustering by self-organizing map. SOM analysis of proteins identified in iTRAQ experiments using Spotfire software produced a 3 × 3 cluster. The control is shown as the baseline, to which JNJ460 and NGF treatments are compared to. Groups 3 and 6 include proteins that are specifically upregulated upon NGF treatment; whereas groups 7 and 8 include proteins upregulated with JNJ460 treatment. Proteins showing similar trends between the two treatments are clustered in groups 1 and 9.
3.2. Divergent neurite outgrowth patterns from NGF and JNJ460 treatments
Functional comparison of proteins enriched in groups 6 and 8 describes the divergent patterns (Table 1). Besides cellular metabolism (p < 0.004), NGF activated both biosynthesis machinery (p < 0.002) and ribosome (p < 0.0002). By comparison, in addition to elevating stress response (p < 0.005), JNJ460 promoted both organogenesis (p < 0.004) and related signal transduction pathways (p < 0.008). Further analyses of the most dramatically changed proteins in each group provided more details to corroborate the divergent patterns.
Table 1.
Divergent DRG proteomic patterns from NGF and JNJ460 treatments
| Enriched function (GO term) | Protein count | p-Value | SOM group (treatment) | GO categorya |
|---|---|---|---|---|
| Ribosome | 23 | 0.0002 | 6 (NGF) | C.C. |
| Cellular metabolism | 109 | 0.0004 | 6 (NGF) | B.P. |
| Primary metabolism | 101 | 0.0014 | 6 (NGF) | B.P. |
| Biosynthesis | 44 | 0.0014 | 6 (NGF) | B.P. |
| Cytoplasm | 106 | 0.0020 | 6 (NGF) | C.C. |
| Ribonucleoprotein complex | 26 | 0.0028 | 6 (NGF) | C.C. |
| Response to external stimulus | 11 | 0.0025 | 8 (JNJ460) | B.P. |
| Organogenesis | 20 | 0.0039 | 8 (JNJ460) | B.P. |
| Response to biotic stimulus | 12 | 0.0046 | 8 (JNJ460) | B.P. |
| Response to stress | 14 | 0.0050 | 8 (JNJ460) | B.P. |
| Signal transduction | 19 | 0.0076 | 8 (JNJ460) | B.P. |
Definition: C.C., cellular component; B.P., biological process.
3.3. Distinct protein function enrichment in each treatment group
A unique feature in the proteome of JNJ460-treated DRGs relative to that of the control was the apparent increase of structural proteins with “organogenesis” properties (Table 2), including α-fetoprotein, actin, microtubule-associated protein tau, myomesin 1, myosin heavy chain, myosin light chain 1, neuronal protein 4.1 and troponin T (Fig. 2). In addition, signal transduction proteins activated by JNJ460 treatment included N-myc downstream regulated gene 3 protein, Ras-related protein Rap-1b and two phosphatases: protein phosphatase 2C and serine/threonine phosphatase PP1-α. By comparison, ganglia treated with NGF showed a different pattern with a major subset of proteins involved in ribosomal function and biosynthesis (Table 3). Ribosomal proteins with either RNA processing function (exosome complex exonuclease RRP41) or protein synthesis function (40S ribosomal protein S27a and 60S acidic ribosomal protein P1) were increased. The increase of translation eIF-5A corresponds with the growth promotion function of NGF (Fig. 2). In addition, GPI transamidase component PIG-T is involved in phosphatidylinositol-glycan biosynthesis. Two transport proteins were increased by NGF treatment, sodium/potassium-transporting ATPase β-3 is involved with the exchange of Na+ and K+ ions across the plasma membrane, while TOM22 is a receptor for nearly synthesized mitochondrial proteins. Additional NGF-specific elevated proteins included neuronal visin-like protein 1 and adipocyte-derived leucine aminopeptidase.
Fig. 2.
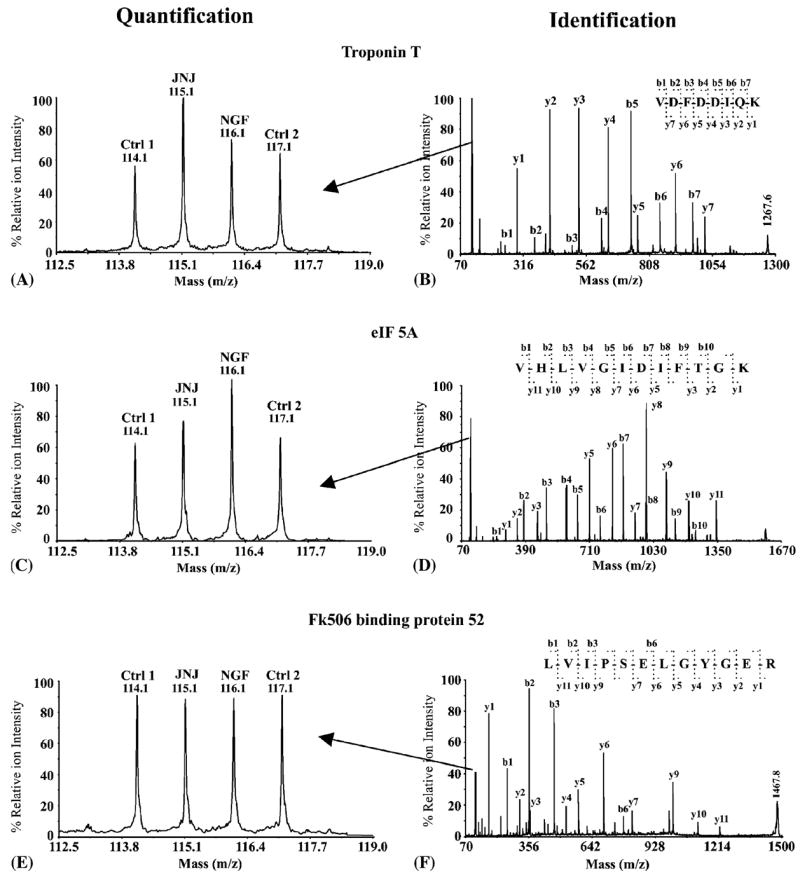
Examples of protein identification and quantification using the iTRAQ approach. Troponin T (A and B) or eukaryotic translation initiation factor 5A (C and D) was upregulated in either JNJ460 or NGF-treated DRGs, respectively; whereas FK506 binding protein 52 (E and F) was unchanged. Peptide sequences were deduced from the MS/MS spectra, (B, D and F) based on the observation of continuous series of either N-terminal (b series) or C-terminal (y series) ions. Quantification of peptides was based on the relative iTRAQ reporter peak areas (A, C and E).
3.4. Validation by Western blotting
The proteomic patterns revealed by SOM analysis of iTRAQ data were consistent with Western blot analysis of selected proteins. In Fig. 2, three representative MS/MS spectra of peptides from troponin T (elevated in JNJ460 treatment, A and B), eIF-5A (elevated in NGF treatment, C and D) and FKBP52 (unchanged, E and F) are shown. Similarly, Western blot analysis showed a comparable degree of protein expression changes elicited by either NGF or JNJ460 treatment (Fig. 3).
Fig. 3.
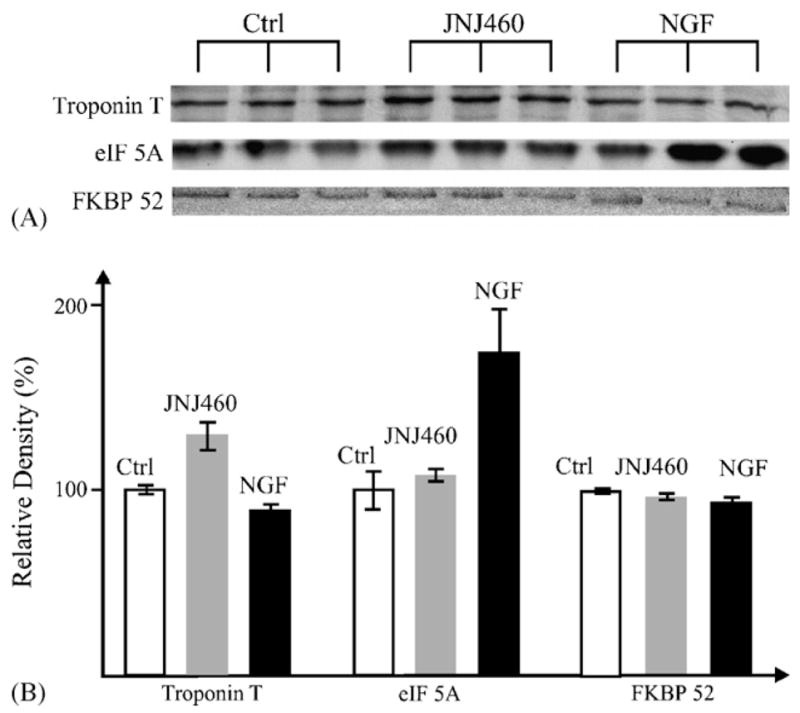
Western blot analysis of selected proteins. (A) DRG extracts (10 μg/lane) from the three treatments were blotted with specific antibodies and (B) protein expression levels were plotted based on the relative band densities measured using Quantity One software (Bio-Rad). FKBP52 is the control.
4. Discussion
In this report, we described a method utilizing the combination of SOM and iTRAQ-based approach for proteomic pattern discovery. The iTRAQ technology is a powerful tool for studying protein changes among up to four groups. However, due to both cost and significant mass spectrometry time demand, repetitive iTRAQ experiments are not always feasible. In addition, data validation is also not trivial. Comparing with RT-PCR used for verifying large scale microarray results, selecting specific antibodies for validating iTRAQ results by Western blotting can be both expensive and not always possible. SOM and other clustering methods provide alternative strategies. Instead of focusing on selected “significantly changed” proteins/genes, the expressions patterns of all data points are analyzed as a whole. SOM provides a snapshot of a dataset and reveals prominent patterns quickly. Successful application of SOM for iTRAQ data analysis has enabled us to discover divergent neurite outgrowth patterns instigated by JNJ460 and NGF.
In general, the quantitative changes revealed by iTRAQ analysis are accurate and are consistent with Western blotting, albeit the latter method usually reports more pronounced changes. For example, both troponin T and eIF-5A showed ~1.3–1.5-fold increases from iTRAQ analysis (Tables 2 and 3) and ~1.4–2.0-fold in Western blotting (Fig. 3). Earlier studies have shown that changes beyond 20% from the population mean reported by iTRAQ analysis could be considered significant based on variance and analytical noise analysis (Unwin et al., 2005, in press). Recent work by others (Keshamouni et al., 2006) and our group (Hu et al., in press) also found that ~20% changes observed in iTRAQ data were consistent with the corresponding Western blotting quantification of identical proteins. Quantification accuracy could be affected by many factors, including sample quantity, complexity, analytical hardware limitations, bioinformatic assignment of protein isoforms in addition to biological variations, which we have analyzed extensively (Hu et al., in press).
In our experimental design, four hour incubation time was chosen based on earlier microarray analysis of identical pharmacological treatments of isolated Schwann cells (Birge et al., 2004). In those experiments, the earliest mRNA induction events induced by JNJ460 occurred at 4 h, including the activation of a number of transcription factors and early response genes. Therefore, the likely proteomic changes during the same period could provide clues on the different growth features as results of either NGF or JNJ460 treatments. Longer incubation times could result in the characterization of mainly structural proteins that increase significantly during the process of neurite extension, which is usually observed after 1–2 days of either NGF or JNJ460 treatments (Birge et al., 2004). This would mask relatively subtle but important changes in the key regulatory proteins responsible for unraveling regeneration-initiating events due to JNJ460 treatment.
In the present analysis, we found numerous NGF induced proteins that play important roles in protein synthesis, including exosome complex exonuclease RRP41, 40S ribosomal protein S27a, 60S acidic ribosomal protein P1 and translation eIF-5A. Interestingly, ganglia treated with JNJ460 showed a cluster of proteins involved in organogenesis. Actin and myosin are cytoskeletal proteins and are important determinants of axonal regeneration (Luo, 2002). For example, we have observed here that both myosin heavy chain and its regulatory light chain were upregulated as a result of JNJ460 treatment, which is indicative of actomyosin remodeling. Other upregulated structure proteins are also important for actomyosin remodeling. Microtubule-associated protein tau binds to and stabilizes microtubules, is regulated directly by phosphorylation, and is important for axonal outgrowth and neuronal polarity (Stoothoff and Johnson, 2005). Further studies are required to fully understand how JNJ460 activates signaling pathways for neuritogenesis.
An important conclusion drawn from this study is that immunophilin ligands and neurotrophins have distinct initial mechanisms of action as evident by the global changes in the proteome patterns of regenerating neurons treated with these factors. Previous studies have shown that immunophilin ligands complement the action of neurotrophic factors and in part by enhancing the MAPK/JNK pathways (Price et al., 2003). On the other hand, earlier study has shown that JNJ460 induces neurogenesis indirectly in a Schwann cell dependent manner, but not when directly administered to PC12 cells (Birge et al., 2004). The data presented here further suggest that the early events associated with JNJ460 are functionally distinct to the effects of NGF, may offer a mechanism for the observed priming effects of JNJ460 on NGF.
In conclusion, we report here a method for proteomics pattern discovery. By combining the iTRAQ™ proteomics approach with self-organizing map method, we were able to delineate the early incipient molecular events preceding DRG neurite outgrowth induced by either NGF or an immunophilin ligand, JNJ460. Previous studies have shown that immunophilin ligands require low concentrations of other neurotrophic factors, such as NGF and therefore it was not ruled out that immunophilin ligands function indirectly, to lower the cellular threshold of neurotrophic factor sensitivity (Schreiber and Crabtree, 1992). We postulate that the mechanism for the lowered sensitivity to NGF may be the result of early actomysin remodeling activity of immunophilin ligands, and as such offer a possible mechanism for the observed synergistic actions of neurotrophic factors and immunophilin ligands. The proteomic pattern discovery method described here could provide an effective means to distinguish the pharmacological effects of other neurological agents.
Supplementary Material
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.jneumeth.2006.05.010.
Acknowledgments
This project is supported in part by an NIH grant, NS046593 to HL and a grant from the Robert Wood Johnson Research Foundation to RB. We dedicate this work to the loving memory of our colleague and dear friend, Dr. Longwen Deng.
References
- Armistead DM, Badia MC, Deininger DD, Duffy JP, Saunders JO, Tung RD, et al. Design, synthesis and structure of non-macrocyclic inhibitors of FKBP12, the major binding protein for the immunosuppressant FK506. Acta Crystallogr D Biol Crystallogr. 1995;51(Pt 4):522–8. doi: 10.1107/S0907444994014502. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat Genet. 2000;25(1):25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birge RB, Wadsworth S, Akakura R, Abeysinghe H, Kanojia R, MacIelag M, et al. A role for schwann cells in the neuroregenerative effects of a non-immunosuppressive fk506 derivative, jnj460. Neuroscience. 2004;124(2):351–66. doi: 10.1016/j.neuroscience.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, et al. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4(5):P3. [PubMed] [Google Scholar]
- DeSouza L, Diehl G, Rodrigues MJ, Guo J, Romaschin AD, Colgan TJ, et al. Search for cancer markers from endometrial tissues using differentially labeled tags iTRAQ and cICAT with multidimensional liquid chromatography and tandem mass spectrometry. J Proteome Res. 2005;4(2):377–86. doi: 10.1021/pr049821j. [DOI] [PubMed] [Google Scholar]
- Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286(5439):531–7. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- Hamilton GS, Steiner JP. Immunophilins: beyond immunosuppression. J Med Chem. 1998;41(26):5119–43. doi: 10.1021/jm980307x. [DOI] [PubMed] [Google Scholar]
- Hardt M, Witkowska HE, Webb S, Thomas LR, Dixon SE, Hall SC, et al. Assessing the effects of diurnal variation on the composition of human parotid saliva: quantitative analysis of native peptides using iTRAQ reagents. Anal Chem. 2005;77(15):4947–54. doi: 10.1021/ac050161r. [DOI] [PubMed] [Google Scholar]
- Hirsch J, Hanse KC, Choi S, Noh J, Hirose R, Roberts JP, et al. Warm ischemia induced alterations in oxidative and inflammatory proteins in hepatic Kupffer cells in rats. Mol Cell Proteomics. doi: 10.1074/mcp.M500320-MCP200. in press. [DOI] [PubMed] [Google Scholar]
- Hu J, Qian J, Borisov O, Pan S, Li Y, Liu T, et al. Optimized proteomic analysis of a mouse model of cerebellar dysfunction using amine-specific isobaric tag. Proteomics. doi: 10.1002/pmic.200600026. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ideker T, Thorsson V, Ranish JA, Christmas R, Buhler J, Eng JK, et al. Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science. 2001;292(5518):929–34. doi: 10.1126/science.292.5518.929. [DOI] [PubMed] [Google Scholar]
- Kaplan DR. Studying signal transduction in neuronal cells: the Trk/NGF system. Prog Brain Res. 1998;117:35–46. doi: 10.1016/s0079-6123(08)64005-4. [DOI] [PubMed] [Google Scholar]
- Keshamouni VG, Michailidis G, Grasso CS, Anthwal S, Strahler JR, Walker A, et al. Differential protein expression profiling by iTRAQ-2DLC-MS/MS of lung cancer cells undergoing epithelial–mesenchymal transition reveals a migratory/invasive phenotype. J Proteome Res. 2006;5:1143–54. doi: 10.1021/pr050455t. [DOI] [PubMed] [Google Scholar]
- Luo L. Actin cytoskeleton regulation in neuronal morphogenesis and structural plasticity. Annu Rev Cell Dev Biol. 2002;18:601–35. doi: 10.1146/annurev.cellbio.18.031802.150501. [DOI] [PubMed] [Google Scholar]
- Price RD, Yamaji T, Matsuoka N. FK506 potentiates NGF-induced neurite outgrowth via the Ras/Raf/MAP kinase pathway. Br J Pharmacol. 2003;140(5):825–9. doi: 10.1038/sj.bjp.0705522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber SL, Crabtree GR. The mechanism of action of cyclosporin A and FK506. Immunol Today. 1992;13(4):136–42. doi: 10.1016/0167-5699(92)90111-J. [DOI] [PubMed] [Google Scholar]
- Stoothoff WH, Johnson GV. Tau phosphorylation: physiological and pathological consequences. Biochim Biophys Acta. 2005;1739(2–3):280–97. doi: 10.1016/j.bbadis.2004.06.017. [DOI] [PubMed] [Google Scholar]
- Tamayo P, Slonim D, Mesirov J, Zhu Q, Kitareewan S, Dmitrovsky E, et al. Interpreting patterns of gene expression with self-organizing maps: methods and application to hematopoietic differentiation. Proc Natl Acad Sci USA. 1999;96(6):2907–12. doi: 10.1073/pnas.96.6.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Fujita N, Ogawa N. Immunosuppressive (FK506) and non-immunosuppressive (GPI1046) immunophilin ligands activate neurotrophic factors in the mouse brain. Brain Res. 2003;970(1–2):250–3. doi: 10.1016/s0006-8993(03)02434-x. [DOI] [PubMed] [Google Scholar]
- Unwin RD, Pierce A, Watson RB, Sternberg DW, Whetton AD. Quantitative proteomic analysis using isobaric protein tags enables rapid comparison of changes in transcript and protein levels in transformed cells. Mol Cell Proteomics. 2005;4(7):924–35. doi: 10.1074/mcp.M400193-MCP200. [DOI] [PubMed] [Google Scholar]
- Unwin RD, Smith DL, Blinco D, Wilson CL, Miller CJ, Evans CA, et al. Quantitative proteomics reveals post-translational control as a regulatory factor in primary hematopoietic stem cells. Blood. doi: 10.1182/blood-2005-12-4995. in press. [DOI] [PubMed] [Google Scholar]
- Yuen EC, Howe CL, Li Y, Holtzman DM, Mobley WC. Nerve growth factor and the neurotrophic factor hypothesis. Brain Dev. 1996;18(5):362–8. doi: 10.1016/0387-7604(96)00051-4. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wolf-Yadlin A, Ross PL, Pappin DJ, Rush J, Lauffenburger DA, et al. Time-resolved mass spectrometry of tyrosine phosphorylation sites in the epidermal growth factor receptor signaling network reveals dynamic modules. Mol Cell Proteomics. 2005;4(9):1240–50. doi: 10.1074/mcp.M500089-MCP200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.jneumeth.2006.05.010.