

Full Paper
Microwave and fluorous technologies are employed to increase reaction speed and reduce separation time in solution-phase parallel synthesis of a substituted 3-aminoimidazo[1,2-a]pyridine and 3-aminoimidazo[1,2-a]pyrazine library. Multicomponent reactions of perfluorooctanesulfonyl-tagged benzaldehydes with 2-aminopyridines (or 2-aminopyrazines) and isocyanides are followed by Pd-catalyzed cross-coupling reactions with boronic acids or thiols to form biaryls or aryl sulfides. Reaction mixtures are purified by fluorous solid-phase extraction (F-SPE) or crystallization.
Introduction
Microwave irradiation has been widely used as a powerful and easily controllable heating source for organic reactions. Reactions conducted under microwave usually have short reaction times, high yields, and better selectivities.1 The coupling of microwave-assisted reactions with phase-tag-based separations, such as solid-phase2 or fluorous synthesis,3 has great potential to further improve the efficiency of organic synthesis. Fluorous synthesis is a new solution-phase technology that has favorable homogeneous reaction kinetics and solid-phase type separation.4 Perfluoroalkyl groups employed in fluorous synthesis have good physical and chemical properties including high stability at elevated temperature, low ability to absorb microwave, good solubility in many common organic solvents. Limitations associated with solid-phase synthesis such as slower heterogeneous reactions, resin swelling in different solvents, requirement of large excess reagents to complete the reaction, and difficulty to analyze resin-bound intermediates are not concerns in fluorous synthesis.
Multicomponent reactions (MCRs) are powerful tools to assemble molecules with high diversity.5 Performing additional reactions after MCRs can increase the molecular complexity and improve drug-like characteristics of the initial products.6 We recently reported the use of perfluorooctylsulfonates as an alternative to halogen atoms and triflate groups for Suzuki and other cross-coupling reactions in the formation C-C, C-S, and C-H bonds.7 In this paper, perfluorooctanesulfonyl benzaldehydes are used for the synthesis of 3-aminoimidazo[1,2-a]pyridine and 3-aminoimidazo[1,2-a]pyrazine derivatives 4.
Imidazo[1,2-a]pyridines/pyrazines are biologically interesting compounds which possess antifungal, antibacterial, cytoprotective, cardiac stimulating, and benzodiazepine receptor antagonistic properties.8 The Blackburn and other groups have reported solution-phase syntheses of 4 by 3-component condensation reactions involving 2-aminopyridines/2-aminopyrazines, aldehydes, and isonitriles (Scheme 1A).9 The MCRs completed in 48–72 hours at ambient temperature and purifications were conducted by resin capture and release.9a The Blackburn group also developed a solid-phase synthesis with polymer-supported aldehydes to simplify the purification of condensation products (Scheme 1B).10 We report here a new method that incorporates microwave irradiation and fluorous tagging strategy11 to speed up reaction and separation processes (Scheme 1C). The post-condensation modification was carried out by Pd-catalyzed cross-coupling reactions to cleave the fluorous tag and introduce an additional diversity point (YR4). In the fluorous approach, the perfluoroalkanesulfonyl tag has three functions: as a phenol protecting group for the condensation reaction, as a phase tag for purification of reaction mixture, and as an activating group for the cross-coupling reaction. The final products have four diversity points (R1 to R4) with additional variations of X (= N or CH) and Y (= nothing or S).
Results and Discussion
Method development
Our initial effort was focused on testing the feasibility of MCR with fluorous aldehyde 1. Following our previous work on perfluoroalkylsulfonates,7 we made fluorous benzaldehyde 1 by a reaction of perfluorooctanesulfonyl fluoride with 3-hydroxy-4-methoxybenzaldehyde. The fluorous benzaldehyde 1 could be purified by flash chromatography or fluorous solid-phase extraction (F-SPE). It was then mixed with 2-aminopyridine and cyclohexylisonitrile for MCR (Scheme 2). The mixture was heated under microwave irradiation at 150 °C for 10 min in 3:1 CH2Cl2/MeOH. The desired product 4d was purified by F-SPE or precipitation. The resulting 4d was subjected to the Suzuki coupling reaction with 4-methoxyboronic acid under microwave irradiation at 130 °C for 20 min using Pd(dppf)Cl2 (dppf = 1,1’-bis(diphenylphosphino)ferrocene) as a catalyst, K2CO3 as a base, and 4:4:1 acetone/toluene/H2O as a co-solvent. The final product 5d was isolated from the reaction mixture by F-SPE. After the development of a fluorous synthetic approach, we then explored the scope and limitations of this method by synthesize a demonstration library.
3-Component condensation reactions
An array of 60 (4 x 5 x 3) MCRs were conducted through parallel synthesis by using five 2-aminopyridines/2-aminopyrazines, four aldehydes, and three isonitriles (Scheme 3). Since microwave reaction of 60 condensation reactions in sequential would take more than 10 h under the conditions described in Scheme 2, we decide to run parallel reactions by converting microwave heating to conventional heating. We found that at 60 °C using MeOH as the solvent, the MCR had 90% conversion after 6 h. When temperature increased to 80 °C with EtOH as the solvent, the reaction completed in 2 h. Products 4 could be precipitated out directly from the reaction mixture at room temperature with purities greater than 85%, F-SPE was not needed. The yields of sixty MCRs are summarized in Table 1. Due to their high solubility in EtOH four compounds 4{2,1,3}, 4{2,2,1}, 4 {3,2,1}, 4{2,2,3} did not precipitate out from the reaction mixture.
Table 1.
Yields of compounds 4{1–4,1–5,1–3}
| 4 | yield | 4 | yield | 4 | yield | 4 | yield | 4 | yield |
|---|---|---|---|---|---|---|---|---|---|
| {1,1,1} | 38% | {1,2,1} | 40% | {1,3,1} | 53% | {1,4,1} | 103% | {1,5,1} | 58% |
| {2,1,1} | 29% | {2,2,1} | 0% | {2,3,1} | 58% | {2,4,1} | 70% | {2,5,1} | 55% |
| {3,1,1} | 40% | {3,2,1} | 0% | {3,3,1} | 48% | {3,4,1} | 66% | {3,5,1} | 31% |
| {4,1,1} | 39% | {4,2,1} | 22% | {4,3,1} | 69% | {4,4,1} | 48% | {4,5,1} | 26% |
| {1,1,2} | 48% | {1,2,2} | 26% | {1,3,2} | 68% | {1,4,2} | 48% | {1,5,2} | 56% |
| {2,1,2} | 34% | {2,2,2} | 32% | {2,3,2} | 31% | {2,4,2} | 82% | {2,5,2} | 75% |
| {3,1,2} | 66% | {3,2,2} | 39% | {3,3,2} | 49% | {3,4,2} | 36% | {3,5,2} | 35% |
| {4,1,2} | 63% | {4,2,2} | 54% | {4,3,2} | 43% | {4,4,2} | 53% | {4,5,2} | 51% |
| {1,1,3} | 35% | {1,2,3} | 27% | {1,3,3} | 36% | {1,4,3} | 65% | {1,5,3} | 49% |
| {2,1,3} | 0% | {2,2,3} | 0% | {2,3,3} | 51% | {2,4,3} | 73% | {2,5,3} | 55% |
| {3,1,3} | 39% | {3,2,3} | 47% | {3,3,3} | 38% | {3,4,3} | 61% | {3,5,3} | 34% |
| {4,1,3} | 30% | {4,2,3} | 41% | {4,3,3} | 33% | {4,4,3} | 36% | {4,5,3} | 56% |
Cross coupling reactions
To evaluate cross-coupling reactions, some representative compounds 4{1–4,1–5,1–3} were randomly selected for two kinds of cross-coupling reactions. Sixteen analogs of 4 were reacted with two boronic acids 6{1–2} to form biaryl compounds 5 (R4Y = R4), while another sixteen were coupled with two thiols 6{3–4} to form aryl sulfides 5 (R4Y = R4S) (Scheme 4).
Under the conditions described in Scheme 2, byproducts imine 7 and subsequent hydrolyzed product 8 were observed from some of the reaction mixtures (Scheme 5). The formation of imine 7 was indicated by the color change from orange (imine) to light purple (aniline) when the imine was treated with Na(OAc)3BH and acetic acid in CH2Cl2. LC-MS analysis also suggested the formation of 7 and 8 .12
We found the formation of 7 and 8 could be suppressed in the absence of water. However, the conversion of 4 was low because the Suzuki coupling requires OH- to generate the trihydroxyborate anion.13 Better results were obtained by using dimethyl ethylene glycol ether (DME) as the solvent with 2–3 drops of water under microwave irradiation at 150 °C for 10 min. The reaction mixtures were purified by F-SPE to remove the fluorous components followed by silica gel SPE with 20:80 EtOAc/hexane to remove the less polar byproducts. The product was collected by elution with 50:50 EtOAc/hexane, while polar impurities remained on the cartridge. The yields and purities of the Suzuki coupling products are summarized in Table 2. Reactions with 3-chlorobenzeneboronic acid 6{1} gave better yield than those with the more electron-deficient 4-acetylbenzeneboronic acid 6{2}.
Table 2.
Yields and purities for Suzuki coupling products 5{1–4,1–5,1–3,1–2}
| 5 | yield | purity | 5 | yield | purity |
|---|---|---|---|---|---|
| {1,1,1,1} | 49% | 58% | {2,1,2,2} | 34% | 86% |
| {1,1,3,1} | 41% | 89% | {3,2,3,2} | 17% | 45% |
| {4,2,2,1} | 27% | 88% | {1,3,1,2} | 12% | 20% |
| {2,3,2,1} | 58% | 91% | {3,3,2,2} | 25% | 75% |
| {1,4,1,1} | 24% | 75% | {3,3,3,2} | 16% | 68% |
| {2,4,1,1} | 41% | 86% | {4,4,2,2} | 26% | 79% |
| {3,4,1,1} | 42% | 76% | {1,5,2,2} | 10% | 21% |
| {1,5,3,1} | 18% | 81% | {3,5,2,2} | 8% | 35% |
The cross-coupling reactions of 4 with thiols were conducted under microwave irradiation at 180 °C for 5 min using Pd(dppf)Cl2 as a catalyst, Cs2CO3 as a base, and anhydrous DMF as a solvent. The reaction mixtures were purified by F-SPE followed by silica gel SPE (Table 3). Structures of some representative cross-coupling products 5 are listed in Scheme 6.
Table 3.
Yields and purities for aryl sulfide products 5{1–4,1–5,1–3,3–4}
| 5 | Yield | purity | 5 | yield | purity |
|---|---|---|---|---|---|
| {2,1,1,3} | 30% | 86% | {3,2,2,4} | 30% | 46% |
| {2,2,2,3} | 28% | 93% | {4,2,3,4} | 41% | 77% |
| {1,3,2,3} | 8% | 80% | {2,1,3,4} | 29% | 88% |
| {4,3,3,3} | 65% | 73% | {1,3,3,4} | 22% | 75% |
| {1,4,2,3} | 4% | 73% | {4,4,1,4} | 45% | 54% |
| {1,5,1,3} | 8% | 42% | {2,4,2,4} | 17% | 64% |
| {4,5,2,3} | 37% | 52% | {4,1,3,4} | 15% | 76% |
| {2,5,3,3} | 16% | 75% | {4,5,3,4} | 48% | 52% |
Conclusions
A fluorous synthetic route to 3-aminoimidazo[1,2-a]pyridines and 3-aminoimidazo[1,2-a]pyrazines derivatives has been developed by conducting MCRs followed by post-condensation reactions. The microwave heating has been applied to both steps. The MCRs were established under microwave heating and later converted to conventional heating for quick parallel library synthesis. The post-condensation reactions were conducted by microwave-assisted Pd-catalyzed cross-coupling reactions of fluorous sulfonates with boronic acids to form biaryls or with thiols to form aryl sulfides. At each reaction step, the reaction mixtures were analyzed by LC-MS or TLC, which is important for the reactions which have low conversion or producing byproducts. The F-SPE purification of the reaction mixtures from MCRs was unnecessary because recrystallization of the product proved to be an easier choice. This modification reflects the compatibility of fluorous technology with other purification techniques. Because of competitive reactions and different reactivity of substrates, the yields for both MCRs and coupling reactions were relatively low, which also affected the purities of intermediates 4 and products 5 after F-SPE and recrystallization.
Experimental Section
General
Perfluorooctanesulfonyl fluoride, building blocks 1, 2, 3, and reagents were obtained from commercial sources. FluoroFlash® SPE cartridges were obtained from Fluorous Technologies, Inc. SPE purification was conducted in parallel on a 2 x 12 SPE manifold available from Supelco and Fisher. Microwave reactions were conducted sequentially on a CEM Explorer® single mode microwave reactor. NMR spectra were obtained on a Bruker AC-270 spectrometer (270 MHz). CDCl3 was used as the solvent unless otherwise specified. LC-MS spectra were obtained on an Agilent 1100 system. Purities were calculated based on UV254 absorption. APCI was used as ion source for all MS.
General SPE procedures for F-SPE
14 A mixture containing fluorous and non-fluorous compounds in a minimum amount of solvent (< 20% of cartridge volume) was loaded onto a FluoroFlash® cartridge preconditioned with 80:20 MeOH/H2O. The cartridge was eluted with 80:20 MeOH/H2O (2–5 cartridge volume) for the non-fluorous fraction followed by same amount of MeOH for the fluorous fraction. The loading of a SPE cartridge was around 1–5%. The SPE was conducted on a standard 2 x 12 SPE manifold. The vacuum was controlled to allow the solvent level moves about 1 inch/min. The cartridge can be reused by washing thoroughly with acetone or THF.
General procedure for the preparation of perfluorooctanesulfonyl benzaldehyde 1{1–4}
To a solution of a hydroxybenzaldehyde (110 mmol) in DMF (30 mL) was added K2CO3 powder (135 mmol) at room temperature. The reaction mixture was stirred for 10 minutes before perfluorooctanesulfonyl fluoride (91 mmol) was added neat. The mixture was heated at 70 °C for 2–8 hours. The cooled reaction mixture was filtered and the solid was rinsed with EtOAc. The filtrate was washed with water, dried over MgSO4 and concentrated. The residue was dissolved in DCM (10 mL), loaded onto Si gel (500 g) and eluted with EtOAc/hexane (20%, 2x500 mL; 30%, 500 mL). The fractions were combined and concentrated to give the product as a white solid.
Analytical data for 1{1}: 1H NMR (ppm) δ 7.48 (d, J = 8.6 Hz, 2H), 8.01 (d, J = 8.6 Hz, 2H), 10.05 (s, 1H).
Analytical data for 1{2}: 1H NMR (ppm) δ 7.57 (dd, J = 8.1, 1.6 Hz, 1H), 7.68 (t, J = 8.1 Hz, 1H), 7.81, (s, 1H), 7.94 (dd, J = 7.7, 1.0 Hz, 1H), 10.05 (s, 1H).
Analytical data for 1{3}: 1H NMR (ppm) δ 4.02 (s, 3H), 7.43 (d, J = 8.1 Hz, 1H), 7.53 (dd, J = 8.1, 1.6 Hz, 1H), 7.58 (d, J = 1.6 Hz, 1 H), 10.00 (s, 1H).
Analytical data for 1{4}: 1H NMR (ppm) δ 4.01 (s, 3H), 7.18 (d, J = 8.4 Hz, 1H), 7.76 (s, 1H), 7.88 (d, J = 8.4 Hz, 1 H), 9.88 (s, 1H).
General Procedure for preparation of 4{1–4,1–5,1–3}
Method A, microwave conditions: Fluorous benzaldehyde 1 (1.0 equiv.), 2-aminopyridine or pyrazine (1.1 equiv.), isonitrile (1.2 equiv.) and Sc(OTf)3 (0.05 equiv.) in 3:1 DCM/MeOH were heated under microwaves to 150 °C in 2 min ramp time. The reaction was maintained at this temperature for 10 min. After cooling to room temperature, the white precipitate was filtered, washed with cold MeOH and dried under vacuum to afford 4. Method B, thermo conditions: Fluorous benzaldehyde 1 (1.0 equiv.), 2-aminopyridine or pyrazine (1.1 equiv.), isonitrile (1.2 equiv.) and Sc(OTf)3 (0.05 equiv.) in EtOH were heated to 80 °C for 2 h. The cooled reaction mixture was diluted with hexane and filtered. The solid was washed with hexane and dried to afford 4.
Analytical data for 4{1,1,1}: 1H NMR (ppm) δ 3.42 (t, J = 5.9 Hz, 1H), 4.21 (d, J = 5.9 Hz, 2H), 6.79 (t, J = 6.7 Hz, 1H), 7.18 (dd, J = 6.8, 6.6 Hz, 1H), 7.22–7.40 (m, 7H), 7.57 (d, J = 6.8 Hz, 1H), 7.95 (d, J = 6.6 Hz, 1H), 8.08 (d, J = 8.7 Hz, 2 H); MS 798 (M+H).
Analytical data for 4{4,1,2} (4d): 1H NMR (ppm) δ 1.04–1.16 (m, 5H), 1.52–1.90 (m, 5H), 2.90–3.10 (m, 2H), 3.98 (s, 3H), 6.83 (t, J = 6.7 Hz, 1H), 7.08–7.26 (m, 2H), 7.56 (d, J = 9.0 Hz, 1H), 8.01–8.20 (m, 3H); MS 820 (M+H).
Analytical data for 4{1,1,3}: 1H NMR (ppm) δ 0.93 (t, J = 7.2 Hz, 3H), 1.35–1.55 (m, 2H), 1.55–1.70 (m, 2H), 3.06 (q, J = 6.7 Hz, 2H), 3.22–3.48 (m, 1H), 6.92 (t, J = 6.7 Hz, 1H), 7.22–7.33 (m, 1H), 7.37 (d, J = 8.9 Hz, 2H), 7.66 (d, J = 8.9 Hz, 1H), 8.13 (d, J = 6.7 Hz, 1H), 8.17 (d, J = 8.9 Hz, 2 H); MS 764 (M+H).
Analytical data for 4{1,3,2}: 1H NMR (ppm) δ 1.00–1.38 (m, 5H), 1.50–1.90 (m, 5H), 2.43 (s, 3H), 2.85–3.05 (m, 1H), 3.30–3.60 (m, 1H), 6.78 (d, J = 7.0 Hz, 1H), 7.35 (d, J = 8.6 Hz, 2H), 7.47–7.59 (br, 1H), 8.08 (d, J = 7.0 Hz, 1H), 8.25 (d, J = 8.6 Hz, 2H); MS 804 (M+H).
Analytical data for 4{1,3,3}: 1H NMR (ppm) δ 0.93 (t, J = 7.3 Hz, 3H), 1.35–1.55 (m, 2H), 1.55–1.70 (m, 2H), 2.41 (s, 3H), 3.04 (q, J = 6.7 Hz, 2H), 3.11–3.18 (m, 1H), 6.70 (dd, J = 6.7, 1.3 Hz, 1H), 7.34 (s, 1H), 7.35 (d, J = 8.6 Hz, 2H), 7.95 (d, J = 7.0 Hz, 1H), 8.13 (d, J = 8.6 Hz, 2H); MS 778 (M+H).
Analytical data for 4{2,1,1}: 1H NMR (ppm) δ 3.85–4.15 (br, 1H), 4.21 (d, J = 5.7 Hz, 2H), 6.92 (t, J = 6.7 Hz, 1H), 7.18–7.33 (m, 6H), 7.33–7.44 (m, 1H), 7.57 (t, J = 8.1 Hz, 1H), 7.78–7.95 (m, 1H), 7.98–8.08 (m, 2 H), 8.18 (d, J = 8.1 Hz, 1H); MS 798 (M+H).
Analytical data for 4{2,1,2}: 1H NMR (ppm) δ 1.05–1.38 (m, 5H), 1.52–1.92 (m, 5H), 2.90–3.08 (m, 1H), 3.15–3.30 (m, 1H), 6.91 (t, J = 6.7 Hz, 1H), 7.20–7.35 (m, 2H), 7.55 (t, J = 8.1 Hz, 1H), 7.69 (d, J = 9.0 Hz, 1H), 8.12–8.32 (m, 2H), 8.23 (d, J = 8.1 Hz, 1H); MS 790 (M+H).
Analytical data for 4{2,3,2}: 1H NMR (ppm) δ 1.07–1.38 (m, 5H), 1.52–1.90 (m, 5H), 2.42 (s, 3H), 2.90–3.08 (m, 1H), 3.08–3.17 (m, 1H), 6.71 (dd, J = 6.9, 1.1 Hz, 1H), 7.21 (dd, J = 8.1, 2.4 Hz, 1H), 7.39 (s, 1H), 7.53 (t, J = 8.1 Hz, 1H), 8.01 (d, J = 6.9 Hz, 1H), 8.13(d, J = 2.2 Hz, 1H), 8.20 (d, J = 8.1 Hz, 1H); MS 804 (M+H).
Analytical data for 4{4,2,3}: 1H NMR (ppm) δ 0.92 (t, J = 7.3 Hz, 3H), 1.35–1.53 (m, 2H), 1.53–1.70 (m, 2H), 3.10–3.30 (m, 3H), 4.00 (s, 3H), 7.19 (d, J = 8.7 Hz, 1H), 7.86 (d, J = 4.6 Hz, 1H), 7.94 (d, J = 1.8 Hz, 1H), 8.05 (dd, J = 8.7, 1.9 Hz, 1H), 8.20–8.32 (m, 1H), 8.97 (s, 1H); MS 795 (M+H).
Analytical data for 4{4,4,2}: 1H NMR (ppm) δ 1.05–1.38 (m, 5H), 1.52–1.90 (m, 5H), 2.39 (s, 3H), 2.98–3.08 (m, 1H), 3.20–3.40 (m, 1H), 3.98 (s, 3H), 7.09–7.22 (m, 2H), 7.65 (d, J = 9.2 Hz, 1H), 7.94 (s, 1H), 8.11 (d, J = 1.8 Hz, 1H), 8.20 (dd, J = 8.7, 1.8 Hz, 1H); MS 834 (M+H).
General procedure for preparation of biaryls 5{1–4,1–5,1–3,1–2}
Method A
A mixture of 4 (0.2 mmol), boronic acid (0.19 mmol), K2CO3 (0.4 mmol), Pd(dppf)Cl2 (10 mol%) in 4:4:1 acetone/tolune/H2O (2 mL) was heated to 130 °C in 5 min under microwave irradiation. The reaction was maintained at this temperature for 20 min and cooled to room temperature. The mixture was loaded onto a FluoroFlash® SPE cartridge (5 g) and the cartridge was eluted with 80:20 MeOH/H2O (3x10 mL) and then acetone (2x10 mL). The MeOH/H2O fractions were combined and concentrated to afford the desired product.
Analytical data for 5d: 1H NMR (ppm) δ 1.05–1.38 (m, 5H), 1.51–1.94 (m, 5H), 2.98–3.15 (m, 2H), 3.87 (s, 3H), 3.89 (s, 3H), 6.78 (t, J = 6.8 Hz, 1H), 6.98 (d, J = 8.4 Hz, 2H), 7.02–7.20 (m, 2H), 7.49–7.62 (m, 3H), 7.98–8.04 (m, 2H), 8.13 (d, J = 6.6 Hz, 1H); MS 428 (M+H).
Method B
To a mixture of 4 (0.2 mmol), boronic acid (0.19 mmol), K2CO3 (0.4 mmol), Pd(dppf)Cl2 (10 mol%) in DME (2 mL) was added 2 drops of water. The mixture was heated to 150 °C in 2 minutes under microwave irradiation. The reaction was maintained at this temperature for 10 minutes and cooled to room temperature. The mixture was diluted with 80:20 MeOH/H2O (5 mL) and loaded onto a FluoroFlash® SPE cartridge (5 g). The cartridge was eluted with 80:20 MeOH/H2O (3x10 mL) and then acetone (2x10 mL). The MeOH/H2O fractions were combined and concentrated. The residue was dissolved in DCM (0.5 mL) and loaded onto a Si-SPE cartridge (2 g). The cartridge was eluted with EtOAc/hexane (20% 10 mL then 50% 3x10 mL). The 50% fractions were combined and concentrated to afford the desired product. For pyrazines, the gradient was 50% (10 mL) followed by 100% (3x10 mL). The 100% fractions were combined and concentrated to give the desired product.
Analytical data for 5{2,3,2,1}: 1H NMR (ppm) δ 1.03–1.38 (m, 5H), 1.51–1.94 (m, 5H), 2.38 (s, 3H), 2.94–3.13 (m, 1H), 3.13–3.45 (m, 1H), 6.63 (dd, J = 6.9, 0.8 Hz, 1H), 7.27–7.45 (m, 3H), 7.50–7.63 (m, 3H), 7.67 (s, 1H), 8.00 (d, J = 7.6 Hz, 1H), 8.02–8.13 (m, 1H), 8.27 (t, J = 0.8 Hz, 1H); MS 416 (M+H), 418 (M+2+H).
Analytical data for 5{1,1,3,1}: 1H NMR (ppm) δ 0.95 (t, J = 7.2 Hz, 3H), 1.35–1.56 (m, 2H), 1.56–1.74 (m, 2H), 3.08 (t, J = 7.0 Hz, 2H), 3.15–3.38 (m, 1H), 6.84 (td, J = 7.0, 0.9 Hz, 1H), 7.14 (dd, J = 7.0, 1.2 Hz, 1H), 7.30–7.45 (m, 2H), 7.53 (dd, J = 4.9, 0.9 Hz, 1H), 7.56–7.72 (m, 4H), 8.05–8.15 (m, 3H); MS 376 (M+H), 378 (M+2+H).
Analytical data for 5{2,1,2,2}: 1H NMR (ppm) δ 1.03–1.38 (m, 5H), 1.51–1.94 (m, 5H), 2.65 (s, 3H), 2.94–3.09 (m, 1H), 3.09–3.30 (m, 1H), 6.82 (t, J = 6.7 Hz, 1H), 7.17 (td, J = 6.7, 1.0 Hz, 1H), 7.50–7.63 (m, 3H), 7.77 (d, J = 8.4 Hz, 2H), 8.03 (d, J = 8.4 Hz, 2H), 8.09–8.12 (m, 1H), 8.14 (d, J = 6.7 Hz, 1H) 8.37 (s, 1H); MS 410 (M+H).
Analytical data for 5{4,4,2,2}: 1H NMR (ppm) δ 1.05–1.38 (m, 5H), 1.51–1.94 (m, 5H), 2.35 (s, 3H), 2.65 (s, 3H), 2.85–3.15 (m, 2H), 3.88 (s, 3H), 7.00 (dd, J = 9.1, 1.4 Hz, 1H), 7.07 (d, J = 9.3 Hz, 1H), 7.46 (d, J = 9.1 Hz, 1H), 7.71 (d, J = 8.4 Hz, 2H), 7.88 (s, 1H), 8.01 (d, J = 8.4 Hz, 2H), 8.05–8.15 (m, 2H); MS 454 (M+H).
General procedure for preparation of sulfides 5{1–4,1–5,1–3,3–4}
A mixture of 4 (0.2 mmol), thiol or thiophenol (0.19 mmol), Cs2CO3 (0.4 mmol), and Pd(dppf)Cl2 (10 mol%) in DMF (2 mL) was heated under microwaves to 180 °C in 2 min ramp time. The reaction was maintained at this temperature for 5 min and cooled to room temperature. The mixture was diluted with 80:20 MeOH/H2O (5 mL) and loaded onto a FluoroFlash® SPE cartridge (5 g). The cartridge was eluted with 80:20 MeOH/H2O (3x10 mL) and then acetone (2x10 mL). The MeOH/H2O fractions were combined and concentrated. The residue was dissolved in DCM (0.5 mL) and loaded on a Si-SPE cartridge (2 g). The cartridge was eluted with EtOAc/hexane (20%, 10 mL; 50%, 3x10 mL). The 50% fractions were combined and concentrated to afford the desired product. For pyrazines, the gradient was 50% (10 mL) followed by 100% (3x10 mL). 100% fractions were combined and concentrated to afford the desired product.
Analytical data for 5{2,1,1,3}: 1H NMR (ppm) δ 1.03–1.38 (m, 5H), 1.51–1.94 (m, 5H), 3.10–3.28 (m, 1H), 3.50–4.10 (br, 1H), 4.20 (s, 2H), 6.79 (td, J = 6.7, 0.9 Hz, 1H), 7.10–7.25 (m, 1H), 7.28–7.45 (m, 7H), 7.62 (d, J = 9.1 Hz, 1H), 7.80–7.88 (m, 1H), 7.97–8.10 (m, 2H); MS 414 (M+H).
Analytical data for 5{1,3,2,3}: 1H NMR (ppm) δ 1.05–1.51 (m, 10H), 1.51–1.93 (m, 8H), 1.93–2.13 (m, 2H), 2.40 (s, 3H), 2.83–3.10 (m, 2H), 3.10–3.30 (m, 1H), 6.65 (dd, J = 7.0, 1.4 Hz, 1H), 7.30–7.40 (m, 1H), 7.44 (d, J = 8.4 Hz, 2H), 7.91–8.06 (m, 3H); MS 420 (M+H).
Analytical data for 5{1,3,3,4}: 1H NMR (ppm) δ 0.92 (t, J = 7.3 Hz, 3H), 1.33–1.51 (m, 2H), 1.51–1.70 (m, 2H), 2.38 (s, 3H), 2.85–3.08 (m, 3H), 3.83 (s, 3H), 6.64 (dd, J = 7.0, 1.3 Hz, 1H), 6.92 (d, J = 8.6 Hz, 2H), 7.23 (d, J = 8.4 Hz, 2H), 7.31 (s, 1H), 7.44 (d, J = 8.6 Hz, 2H), 7.86 (d, J = 8.4 Hz, 2H), 7.92 (d, J = 6.7 Hz, 1H); MS 418 (M+H).
Analytical data for 5{4,2,3,4}: 1H NMR (ppm) δ 0.90 (t, J = 7.3 Hz, 3H), 1.25–1.51 (m, 4H), 2.65–2.83 (m, 2H), 2.83–3.00 (m, 1H), 3.83 (s, 3H), 3.97 (s, 3H), 6.88–7.03 (m, 3H), 7.33 (d, J = 2.1 Hz, 1H), 7.50 (d, J = 8.6 Hz, 2H), 7.74–7.83 (m, 2H), 7.86 (d, J = 1.1 Hz, 1H), 8.91 (d, J = 1.3 Hz, 1H); MS 435 (M+H).
Scheme 1.
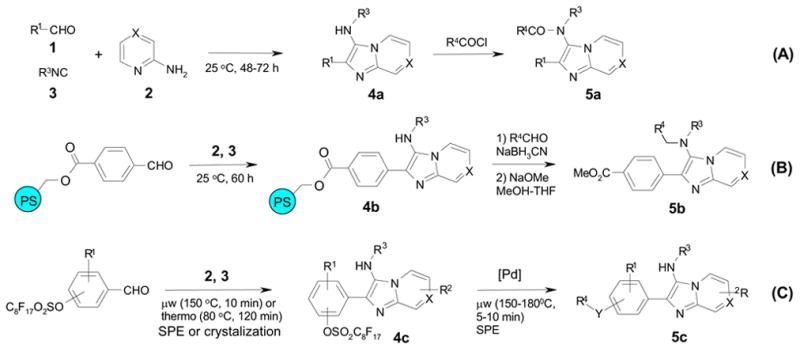
Solution-phase (A), solid-phase (B), and fluorous (C) syntheses of imidazo[1,2-a]pyridine/pyrazine derivatives by multicomponent and post-condensation reactions
Scheme 2.
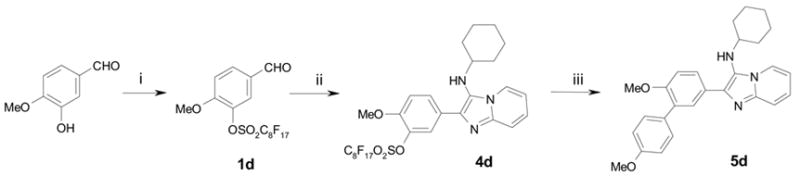
Microwave-assisted fluorous synthesis of imidazo[1,2-a]pyrazine 4d followed by detagging with Suzuki coupling reaction for 5. i) C8F17SO2F (0.9 equiv), K2CO3 (2.0 equiv), DMF, 70 °C, 5 h, 90% yield; ii) 2-aminopyridine (1.1 equiv), cyclohexylisonitrile (1.2 equiv), Sc(OTf)3 (0.05 equiv), 3:1 CH2Cl2/MeOH, microwave (150 w, 150 °C, 10 min), 76%; iii) 4-methoxyboronic acid (0.95 equiv), Pd(pddf)Cl2 (0.1 equiv), K2CO3 (2.0 equiv), 4:4:1 acetone/toluene/H2O, microwave (150 w, 130 °C, 20 min), F-SPE eluted with 80:20 MeOH-H2O then MeOH, 75% yield (85% purity by LC-MS, 254 nm)
Scheme 3.
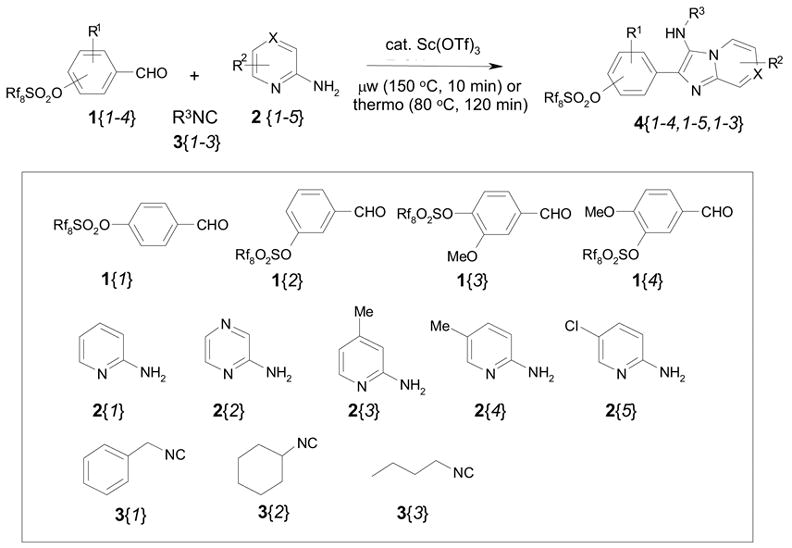
Parallel synthesis of 60 imidazo[1,2-a]pyridine and imidazo[1,2-a]pyrazine analogs
Scheme 4.
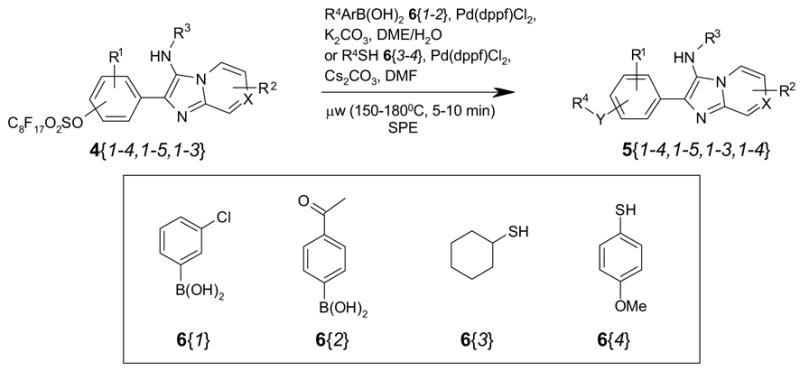
Parallel cross-coupling reactions for 5{1–4,1–5,1–3,1–2}
Scheme 5.
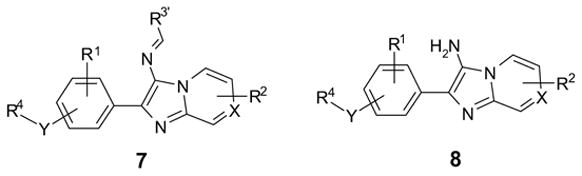
Possible byproducts 7 and 8 found in the mixture of coupling reactions
Scheme 6.
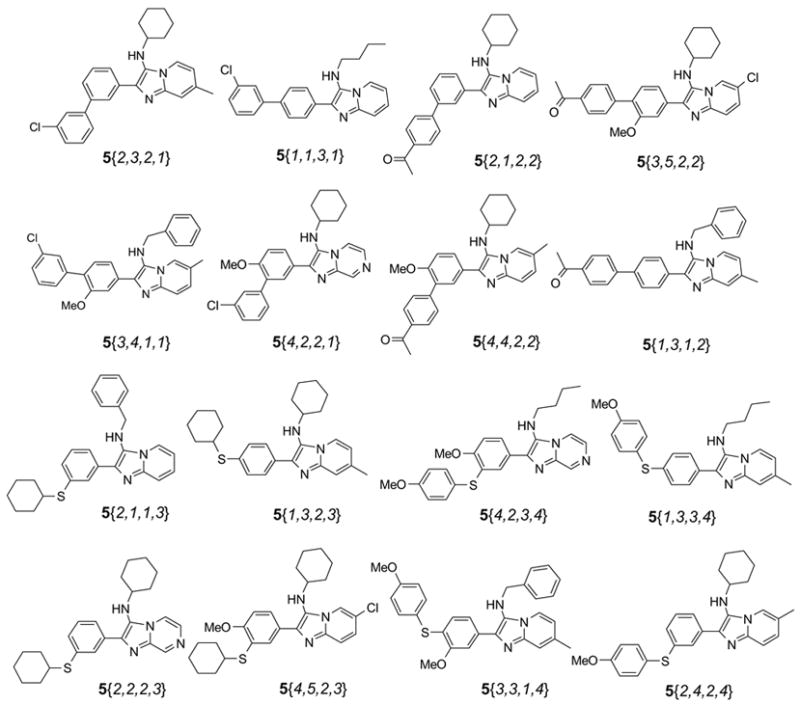
Representative structures of cross-coupling products 5
Acknowledgments
This work was supported by National Institutes of General Medical Sciences SBIR funding (2R44GM062717-02). Thanks to Prof. Dennis Curran and Dr. Sivaraman Dadapani for help with the manuscript.
Abbreviations
- MCR
multicomponent reaction
- SPE
solid-phase extraction
- F-SPE
fluorous solid-phase extraction
- LC-MS
liquid chromatography-mass spectrometry
- 1H NMR
proton nuclear magnetic resonance,
- DMF
N,N-dimethylformamide
- THF
tetrahydrofuran
- DME
dimethyl ethylene glycol ether
- dppf
1,1’-bis(dipenylphosphino)ferrocene
- OTf
trifluoromethanesulfonyl
- DCM
dichloromethane
References and Notes
- 1.Recent reviews on microwave synthesis: Loupy A, editor. Microwaves in Organic Synthesis. Wiley-VCH; Weinheim: 2002. ; b) Hayes BL. Microwave Synthesis: Chemistry at the Speed of Light. CEM Publishing; Matthews, NC: 2002. [Google Scholar]; c) Larhed M, Moberg C, Hallberg A. Acc Chem Res. 2002;35:717–727. doi: 10.1021/ar010074v. [DOI] [PubMed] [Google Scholar]; d) Wathey B, Tierney J, Lidström P, Westman J. Drug Discovery Today. 2002;7:373–380. doi: 10.1016/s1359-6446(02)02178-5. [DOI] [PubMed] [Google Scholar]; e) Kappe CO. Curr Opin Chem Biol. 2002;6:314–320. doi: 10.1016/s1367-5931(02)00306-x. [DOI] [PubMed] [Google Scholar]; f) Larhed M, Hallberg A. Drug Discovery Today. 2001;6:406–416. doi: 10.1016/s1359-6446(01)01735-4. [DOI] [PubMed] [Google Scholar]; g) Lidström P, Tierney J, Wathey B, Westman J. Tetrahedron. 2001;57:9225–9283. [Google Scholar]; h) Varma RS. Pure Appl Chem. 2001;73:193–198. [Google Scholar]
- 2.a) Lidström P, Westman J, Lewis A. Comb Chem High Throughput Screening. 2002;5:441–448. doi: 10.2174/1386207023330147. [DOI] [PubMed] [Google Scholar]; b) Kappe CO. Am Lab. 2001;33:13–19. [Google Scholar]; c) Larhed M, Lindeberg G, Hallberg A. Tetrahedron Lett. 1996;37:8219–8222. [Google Scholar]
- 3.a) Larhed M, Hoshino M, Hadida S, Curran DP, Hallberg A. J Org Chem. 1997;62:5583–5587. [Google Scholar]; b) Olofsson K, Kim SY, Larhed M, Curran DP, Hallberg A. J Org Chem. 1999;64:4539–4541. [Google Scholar]
- 4.a) Gladysz JA, Curran DP, Horvath IT, editors. The Handbook of Fluorous Chemistry. Wiley-VCH; Weinheim: 2004. [Google Scholar]; b) Zhang W. Chem Rev. 2004;104:2531–2556. doi: 10.1021/cr030600r. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhang W. Tetrahedron. 2003;59:4475–4489. [Google Scholar]; d) Pozzi G, Shepperson I. Coord Chem Rev. 2003;242:115–124. [Google Scholar]; e) Tzschucke CC, Markert C, Bannwarth W, Roller S, Hebel A. Angew Chem Int Ed. 2002;41:3964–4000. doi: 10.1002/1521-3773(20021104)41:21<3964::AID-ANIE3964>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]; f) Yoshida J, Itami K. Chem Rev. 2002;102:3693–3716. doi: 10.1021/cr0103524. [DOI] [PubMed] [Google Scholar]; g) Dobbs AP, Kimberley MR. J Fluorine Chem. 2002;118:3–17. [Google Scholar]; h) Curran DP. Synlett. 2001:1488–1496. [Google Scholar]; i) Curran DP. In: Stimulating Concepts in Chemistry. Stoddard F, Reinhoudt D, Shibasaki M, editors. Wiley-VCH; New York: 2000. pp. 25–37. [Google Scholar]; j) Curran DP. Angew Chem, Int Ed Eng. 1998;37:1174–1196. doi: 10.1002/(SICI)1521-3773(19980518)37:9<1174::AID-ANIE1174>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 5.Recent reviews on MCRs: Zhu J. Eur J Org Chem. 2003:1133–1144.; b) Pulici M, Cervi G, Martina K, Quartieri F. Combin Chem High Throughput Syn. 2003;6:693–727. doi: 10.2174/138620703771981241. [DOI] [PubMed] [Google Scholar]; c) von Wangelin AJ, Neumann H, Gordes D, Kaus S, Strubing D, Beller M. Chem Eur J. 2003;9:4286–4294. doi: 10.1002/chem.200305048. [DOI] [PubMed] [Google Scholar]; d) Nair V, Rajesh C, Vinod AU, Bindu S, Sreekanth AR, Mathen JS, Balagopal LB. Acc Chem Res. 2003;36:899–907. doi: 10.1021/ar020258p. [DOI] [PubMed] [Google Scholar]; e) Orru RVA, de Greef M. Synthesis. 2003:1471–1499. [Google Scholar]; f) Litvinov VP. Russian Chem Rev. 2003;72:69–85. [Google Scholar]; g) Ugi I. Pure Appl Chem. 2001;73:187–191. [Google Scholar]; h) Ugi I, Hech S. Combin Chem High Throughput Syn. 2001;4:1–34. doi: 10.2174/1386207013331291. [DOI] [PubMed] [Google Scholar]; i) Domling A, Ugi I. Angew Chem Int Ed. 2000;39:3168–3210. doi: 10.1002/1521-3773(20000915)39:18<3168::aid-anie3168>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 6.a) Hulme C, Gore C. Curr Med Chem. 2003;10:51–80. doi: 10.2174/0929867033368600. [DOI] [PubMed] [Google Scholar]; b) Hulme C, Nixey T. Curr Opin Drug Discov Dev. 2003;6:921–929. [PubMed] [Google Scholar]; c) Armstrong RW, Combs AP, Tempest P, Brown SD, Keating TA. Acc Chem Res. 1996;29:123–131. [Google Scholar]
- 7.a) Zhang W, Chen CHT, Lu Y, Nagashima T. Org Lett. 2004;6:1473–1476. doi: 10.1021/ol0496428. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang W, Nagashima T, Lu Y, Chen CHT. Tetrahedron Lett. 2004;45:4611–4613. [Google Scholar]; c) Zhang W, Lu Y, Chen CHT. Mol Diversity. 2003;7:199–202. doi: 10.1023/b:modi.0000006825.12186.5f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Trapani G, Franco M, Ricciardi L, Latrofa A, Genchi G, Sanna E, Tuveri F, Cagetti E, Biggio G, Liso G. J Med Chem. 1997;40:3109–3118. doi: 10.1021/jm970112+. [DOI] [PubMed] [Google Scholar]; b) Rival Y, Grassy G, Michel G. Chem Pharm Bull. 1992;40:1170–1176. doi: 10.1248/cpb.40.1170. [DOI] [PubMed] [Google Scholar]; c) Starrett JE, Montzka TA, Crosswell AR, Cavanagh RL. J Med Chem. 1989;32:2204–2210. doi: 10.1021/jm00129a028. [DOI] [PubMed] [Google Scholar]; d) Sabalayrolles C, Cros GH, Milhavet JC, Rechenq E, Chapat JP, Boucard M, Serrano JJ, McNeill JH. J Med Chem. 1984;27:206–212. doi: 10.1021/jm00368a018. [DOI] [PubMed] [Google Scholar]; e) Fisher MH, Lusi A. J Med Chem. 1972;15:982–985. doi: 10.1021/jm00279a026. [DOI] [PubMed] [Google Scholar]
- 9.Blackburn C, Guan B, Fleming P, Shiosaki K, Tsai S. Tetrahedron Lett. 1998;39:3635–3638.Blackburn C. Tetrahedron Lett. 1998;39:5469–5299.see also Groebke K, Weber L, Fridolin M. Synlett. 1998:661–663.Bienayme H, Bouzid K. Angew Chem, Int Ed Engl. 1998;37:2234–2237. doi: 10.1002/(SICI)1521-3773(19980904)37:16<2234::AID-ANIE2234>3.0.CO;2-R.
- 10.Blackburn C, Guan B. Tetrahedron Lett. 2000;41:1495–1500. [Google Scholar]
- 11.Reviews of fluorous protecting groups and tags: Zhang W. Speciality Chem Magazine. 2004;24(5):30–32.; Zhang W. In: The Handbook of Fluorous Chemistry. Gladysz JA, Curran DP, Horvath IT, editors. Wiley-VCH; Weinheim: 2004. pp. 222–236. [Google Scholar]
- 12.Isolation of compounds 7 and 8 as well as their structure characterization have been attempted.
- 13.a) Miyaura N, Suzuki A. Chem Rev. 1995;95:2457–2483. [Google Scholar]; b) Farina V, Krishnamurthy V, Scott WJ. Organic Reactions. Wiley; New York: 1997. [Google Scholar]
- 14.For more information on F-SPE, log on to: http://fluorous.com/download/an_spe.pdf