

Abstract
Acrolein (2-propenal) is ubiquitously present in (cooked) foods and in the environment. It is formed from carbohydrates, vegetable oils and animal fats, amino acids during heating of foods, and by combustion of petroleum fuels and biodiesel. Chemical reactions responsible for release of acrolein include heat-induced dehydration of glycerol, retro-aldol cleavage of dehydrated carbohydrates, lipid peroxidation of polyunsaturated fatty acids, and Strecker degradation of methionine and threonine. Smoking of tobacco products equals or exceeds the total human exposure to acrolein from all other sources. The main endogenous sources of acrolein are myeloperoxidase-mediated degradation of threonine and amine oxidase-mediated degradation of spermine and spermidine, which may constitute a significant source of acrolein in situations of oxidative stress and inflammation. Acrolein is metabolized by conjugation with glutathione and excreted in the urine as mercapturic acid metabolites. Acrolein forms Michael adducts with ascorbic acid in vitro, but the biological relevance of this reaction is not clear. The biological effects of acrolein are a consequence of its reactivity towards biological nucleophiles such as guanine in DNA and cysteine, lysine, histidine, and arginine residues in critical regions of nuclear factors, proteases, and other proteins. Acrolein adduction disrupts the function of these biomacromolecules which may result in mutations, altered gene transcription, and modulation of apoptosis.
Keywords: Acrolein, Apoptosis, Cell signaling, Lipid peroxidation, Michael addition
1 Introduction
It has been known for centuries that a water-soluble material is obtained after treatment of fats with alkali to manufacture soap. The French chemist Chevreul (1823) [1], a pioneer of lipid chemistry, believed this sweet-tasting material to be an alcohol and named it ‘glycerin’ (glycerol). The thermal degradation product of glycerin was characterized as an aldehyde by Berzelius (1839) [2], who named it acrolein, which is a contraction of ‘acrid’ (referring to its pungent smell) and ‘oleum’ (oil or oil-like consistency). Redtenbacher (1843) [3] then demonstrated that acrolein can be prepared from glycerin by distillation in the presence of dehydrating agents such as phosphorous pentoxide. He remarked that ,in a very highly diluted condition the smell [of acrolein] is not altogether unpleasant, being somewhat ethereal, but a few drops of acrolein brought into a room soon bring the company to tears’ (quote from Roscoe and Schorlemmer’s A Treatise on Chemistry [4]). In today’s world, acrolein (2-propenal) is an important intermediate for the industrial-scale production of acrylic acid and plastics, which is not discussed in this review. Instead, we focus on the various exogenous and endogenous sources of acrolein and its fate in the human body: how is it eliminated and how does it exert biological and/or toxic effects?
2 Sources and human exposure
The sources of acrolein that are most relevant to human exposure and toxicity can be grouped into dietary, endogenous, and environmental sources. In the 1991 landmark review published by Esterbauer and colleagues [5], the ubiquitous presence of acrolein is attributed to incomplete combustion of petrol, wood, and plastic, to smoking of tobacco products, frying of foods in oils, endogenous lipid peroxidation, and to endogenous polyamine metabolism. Not many new sources of acrolein have been identified since publication of Esterbauer’s review. However, new insights into Maillard-type chemistry and the function of myeloperoxidase have led to the recognition of methionine and threonine as precursors of acrolein [6, 7]. We will briefly discuss the main chemical sources of acrolein and attempt to determine their relative contribution to the total exposure to humans.
2.1 Carbohydrates as a source of acrolein
Heating or baking of carbohydrate-containing foods results in the formation of reactive carbohydrate intermediates that can undergo carbon-carbon cleavage or react with amino acid residues in proteins. For example, heating of glucose may result in loss of the hydroxyl group at position 4 through dehydration, which would yield the appropriate β-hydroxy ketone moiety for release of the acrolein precursor, hydroxy acetone, via retro aldol cleavage of the 3,4-bond (Fig. 1). In support of this fragmentation pathway, thermal degradation studies of the Amadori product resulting from the reaction of single 13C-labeled glucose isotopomers with alanine yielded hydroxy acetone that contained carbon atoms 4, 5, and 6 of glucose [8]. A more prominent pathway produced hydroxy acetone from the C1-C3 fragment of glucose, which the authors rationalized by generation of 1-deoxyglucosone from the Amadori product followed by 3,5-enolization and retro aldol cleavage of the 3,4-bond (Fig. 1A).
Figure 1.

Formation of acrolein from glucose via hydroxy acetone (a). RA, retro aldol-cleavage.
How is acrolein formed from hydroxy acetone? The immediate precursor of acrolein should be 2-hydroxy propanal, which gives acrolein by (heat-induced) dehydration. It has been shown by Fourier-transform infrared spectroscopy that hydroxy acetone can undergo 2,3-enolization to produce the enediol (species b in Fig. 1B), which exists in equilibrium with 2-hydroxy propanal (c) [9]. The enolization process was promoted by increasing the temperature under acidic conditions [9]. Collectively, these studies lend support to the thermal formation of acrolein from glucose as shown in Fig. 1: glucose → deoxyglucosone → hydroxy acetone → 2-hydroxy propanal → acrolein.
The interaction between glucose and lipid peroxidation products is intuitively relevant to chronic inflammatory diseases such as atherosclerosis and diabetes. In a simple in vitro model system, Medina-Navarro and co-workers [10] showed increased formation of conjugated dienes (hydroperoxides) from linoleic and arachidonic acid and increased formation of acrolein (low micromolar concentrations) at increasing concentrations of glucose (range 0-15 mM). The incubations were performed with a fixed concentration of fatty acid (2.5 mM) in buffer solutions at pH 8.0 for periods up to 6 h. One interpretation of the results is that lipid peroxidation promotes the formation of deoxyglucosones, which can break down into acrolein by mechanisms shown in Fig. 1.
2.2 Tobacco
Acrolein exposure through cigarette smoke is generally considered to make up a large proportion of total human exposure. This is readily recognized by the observation that the main urinary metabolite of acrolein, 3-hydroxypropyl mercapturic acid, is found at about twice the level in urine of smokers compared to nonsmokers [11]. In the same study, the levels of the acrolein metabolite showed a median decrease of 78% after 4 wk of smoking abstinence (P < 0.0001).
What is the source of acrolein in cigarettes? In the manufacturing process of cigarettes, glycerol is added to tobacco in the range of about 1-5% by weight to maintain moisture and to absorb added flavors. In a toxicological evaluation study of glycerol as a cigarette ingredient, it was found that addition of glycerol at rates of 10 g or 15 g per 100 g tobacco led to a significant 9% increase of acrolein in the smoke (67 and 69 μg/cigarette, respectively), compared to the glycerol addition rate of 5 g per 100 g tobacco (60 μg acrolein/cigarette) and the control cigarettes (56 μg acrolein/cigarette) [12]. From these data it can be calculated that each gram of glycerol added to 100 g of tobacco increases the acrolein emission per cigarette by about 0.9 μg (r2 = 0.96). Thus, the glycerol-related acrolein emission is small compared to the total emission from cigarettes with no added glycerol (56 μg acrolein/cigarette). We will demonstrate in the following paragraph that carbohydrates in tobacco represent a far more important source of smoke-borne acrolein.
Sugars are normally present in tobacco at levels up to 20% by weight, depending on the method of curing. In addition, tobacco manufacturers add sugar-containing products to tobacco, such as corn syrup, sugar cane molasses, honey, and fruit juices to mask the harsh taste and irritability of tobacco smoke and to achieve brand recognition [13]. In a study of the effect of sugar levels in tobacco on smoke composition, an excellent correlation was found between fructose or saccharose levels in the range 0-16% w/w and the smoke components, formaldehyde, acetaldehyde, and acrolein (r2 values 0.90-0.98). Smoke-borne acrolein increased in the range 3.7-7.9 μg/cigarette per weight percent of sugar added, compared to control cigarettes that produced 118 μg of acrolein per cigarette [13, 14]. For example, addition of 16% sucrose to cigarettes led to an increase from 118 μg to 215 μg acrolein/cigarette[14], indicating that carbohydrates are a major source of acrolein in cigarettes. Because cigarettes also produce formaldehyde and acetaldehyde (11 and 578 μg/cigarette in the control cigarettes of the above study, respectively), it is conceivable that a portion of the acrolein is formed by aldol condensation of formaldehyde and acetaldehyde during the pyrolysis process.
2.3 Lipids as a source of acrolein
An obvious source of acrolein is the glycerol component of triacyl and diacyl glycerides, but can acrolein also be formed from fatty acids, e.g., by lipid peroxidation? While the mechanism of formation of 4-hydroxynonenal from linoleic acid has been studied in great detail [15, 16], there is little or no experimental evidence, published in the literature, that supports the proposed mechanism presented in Esterbauer’s 1991 review article [5] (Fig. 2). In fact, some authors have questioned the formation of acrolein from polyunsaturated fatty acids (PUFAs) by lipid peroxidation processes [17, 18]. On the other hand, there are many published studies reporting the formation of acrolein from fatty acids by oxidative degradation, some of which will be mentioned here. Examination of Esterbauer’s mechanism tells us that acrolein (consisting of an aldehyde and an olefin moiety) is unlikely to be formed from the fatty acid alkyl terminus (no PUFA has an olefin terminus) or the carboxy terminus (reduction of the R-COOH group to R-CHO will not take place in an oxidative environment). Esterbauer’s proposed mechanism is attractive because acrolein originates from the center of the aliphatic chain. There are two main reactions that result in carbon-carbon bond cleavage of lipid hydroperoxides: β-cleavage of the corresponding alkoxy radical, giving an aldehyde and an alkyl fragment, and Hock-fragmentation which yields two aldehyde fragments (mechanism shown in, e.g., Yin and Porter [19]). Thus, release of acrolein from a precursor lipid hydroperoxide should involve at least one β-cleavage fragment to account for the alkyl terminus, while the aldehyde moiety may arise either via β-cleavage of an alkoxy radical intermediate (shown in Fig. 2) or via Hock-cleavage of a hydroperoxide.
Figure 2.
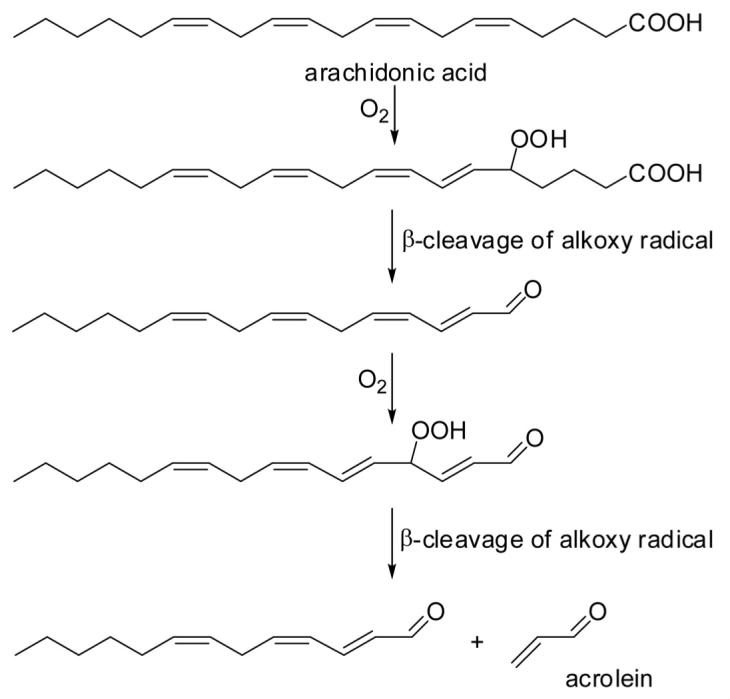
Proposed formation of acrolein from arachidonic acid according to Esterbauer and co-workers [5].
To determine the origin of acrolein released from lipids, one should distinguish between studies conducted with oils (di- or triglycerides) and studies conducted with fatty acids or their methyl esters. The difference is clearly demonstrated by Pederson and co-workers [20], who compared the heat-induced release of acrolein (and other volatiles) from rapeseed oil, rapeseed fatty acid methyl esters, and petroleum-derived diesel fuel. Although they did not quantify the volatiles, examination of the GC-MS chromatograms shows that rapeseed oil produced at least 102 times more acrolein than the mixture of fatty acid methyl esters in the reactor cell kept at 550°C, demonstrating again the significance of glycerol as a precursor of acrolein.
Small amounts of acrolein were detected upon Fe2+/H2O2-mediated oxidation of PUFAs and their ethyl esters (0.8-13 nmol/mg), but no detectable amounts of acrolein were generated from the mono-unsaturated fatty acid, oleic acid, or its ethyl ester [21]. A detailed and thorough study conducted by Pan and co-workers [22] indicates that acrolein can be released from the ethyl ester of the n-3 PUFA, all-cis-7,10,13,16,19-docosapentaenoic acid, by autoxidation at 50°C. GC and GC-MS analyses of the autoxidation mixture at 48 h of reaction yielded acrolein as a major degradation product, representing 8.1% of the total of volatiles by GC peak area. Other volatiles that were identified include 1-penten-3-ol (1.8%), 1-octene-3-ol (1.1%), 1-penten-3-one (4.3%), 1-octen-3-one (1.5%), and 1,5-octadien-3-one (3.3%). It is conceivable that these 1-alken-3-ols and 1-alken-3-ones were formed from their corresponding 3-hydroperoxy-1-alkenes, which could also yield acrolein by β-cleavage of alkoxy radical intermediates (Fig. 3). This would be analogous to the formation of 4-hydroxy-2-nonenal and 4-oxo-2-nonenal from a common hydroperoxy precursor, i.e., 4-hydroperoxy-2-nonenal [23, 24]. In an autoxidation study with cod liver oil stored under air at 25°C for up to 14 wk, an increase in the production of acrolein (from 46 ng/g to 152 ng/g oil), 1-penten-3-one, and 1-penten-3-ol was measured in the headspace after 2 wk of storage [25]. The addition of tocopherol concentrate (800 mg/kg oil) or a combination of tocopherol concentrate and ascorbyl palmitate (200 mg/kg oil) inhibited the formation of all three volatiles upon storage. Interestingly, the co-addition of ascorbyl palmitate decreased the level of acrolein in the headspace, but not the levels of 1-penten-3-one or 1-penten-3-ol. One interpretation of the results is that the formation of the three compounds is facilitated by a lipid peroxidation process as shown in Fig. 3 and that the addition of both antioxidants inhibits this process. The greater decrease of acrolein levels in the headspace of the oil treated with ascorbyl palmitate could be the result of a Michael addition reaction of the ascorbyl moiety with acrolein. Michael reactions between acrolein and ascorbate are well documented in the literature [26] and will be discussed in some detail in Section 3.2.
Figure 3.
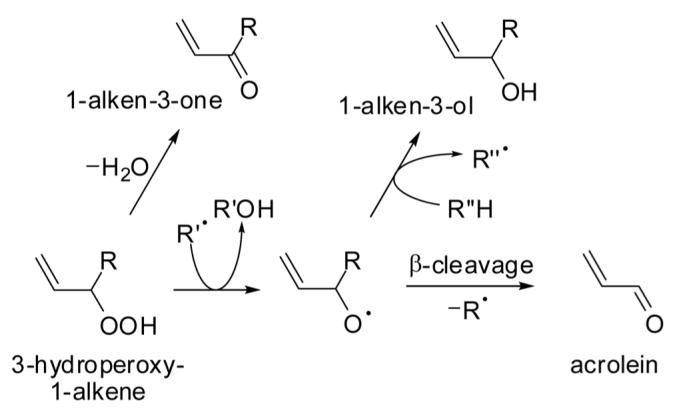
Hypothetical formation of acrolein, 1-alken-3-ones, and 1-alken-3-ols from 3-hydroperoxy-1-alkenes (authors’ interpretation of results obtained by Pan and co-workers [22]).
Umano and Shibamoto [27] determined the release of acrolein from cooking oils and beef fat heated at temperatures in the range 180-320°C for up to 6 h. They observed an increase in acrolein formation with time and temperature, as expected, but they also found a negative correlation between acrolein production and iodine values of the oils (a measure of the degree of unsaturation of the fatty acid moieties). The formation of glycerol requires water for hydrolysis of the triglycerides, which seems to be the limiting factor for acrolein generation from triglycerides. To explain the negative correlation, the authors hypothesized that fatty acid residues with a higher degree of unsaturation are more susceptible to water addition, thereby consuming water that would otherwise be available for ester hydrolysis. They also measured a substantially greater formation of acrolein when the oils and fats were heated in an air atmosphere compared to nitrogen (e.g., 120 g of corn oil released 54 mg of acrolein in N2 and 81 mg in O2 atmosphere at 300°C for 2 h), suggesting that free radical mechanisms contribute to acrolein formation. They proposed a mechanism involving a series of homolytic cleavages of ROC(O)R’ bonds as an alternative route to acrolein. The same corn oil produced only 5.4 mg of acrolein when heated at 280°C for 2 h, demonstrating that temperature is a major factor in acrolein formation from hot oils and fats. The same trend was observed in a study conducted by Fullana and co-workers [28]: increasing the temperature from 180°C to 240°C resulted in an increase in the generation of acrolein from 53 mg to 240 mg per liter of canola (rape-seed) oil. Olive oil yielded only 9 mg acrolein at 180°C and 34 mg at 240°C, while these numbers were 9 and 24 mg for extra virgin olive oil. The apparent ,resistance’ of olive oil (with a relatively low PUFA content) to generate acrolein in this study is not consistent with Umano and Shibamoto’s results, who found that olive oil was a ,high-acrolein’ producer [27]. Li and co-workers [29] compared the liberation of acrolein from diacylglycerol-rich and triacyl-rich oils derived from soybean and rapeseed oils during potato frying, but found no significant differences between the two types of oils (generation of acrolein was in the range 5.7-9.7 mg per kg of oil heated for 3 h at 180°C). The authors did not investigate the contribution of the potatoes and potato-derived water to the formation of acrolein.
From the studies discussed above, it seems that cooking with oil at temperatures of 180°C generates substantial amounts of acrolein (5-250 mg/kg oil) that is released into the atmosphere. Recently, the total emission of acrolein from commercial kitchens in Hong Kong was estimated at 7.7 tons per year, which far exceeds the annual emission of acrolein from vehicles in that city (1.8 tons/year) [30]. The authors note that the emissions from domestic kitchens and 26% of nonclassifiable commercial kitchens were not included in the estimates. Regardless of the accuracy of the estimates, cooking in highly urbanized areas is a major source of acrolein in the atmosphere. Epidemiological findings indicate that emission of acrolein from wok cooking rather than tobacco smoking is linked to the high incidence of lung cancer in Chinese women [31]. A study conducted by Shields and co-workers [32] determined the acrolein emissions from wok cooking to be highest for unrefined Chinese rapeseed oil and lowest for peanut oil. Acrolein emissions were reduced at lower temperatures, as expected, but also reduced when the antioxidant butylated hydroxyanisole was added to the cooking oil, suggesting that lipid peroxidation processes are involved in the formation of acrolein. Experiments with individual fatty acids revealed that heating of linolenic acid produced the greatest amounts of acrolein and the most mutagenic volatiles, measured in a Salmonella mutation assay. Taken together, these studies suggest that the acrolein-related health problems can be mitigated by better ventilation of cooking areas and by lowering cooking temperatures.
According to Uchida and co-workers [33], acrolein is produced when low-density lipoprotein (LDL) is oxidized and it forms a bis-adduct with lysine residues (called Nε-(3-formyl-3,4-dehydropiperidino)lysine or FDP-lysine, see Fig. 10 for structure) of LDL-protein. Their evidence is based on detection of FDP-lysine in Cu2+-mediated oxidation of LDL, by using an immunological assay that employs a monoclonal antibody specific to FDP-lysine. The FDP-lysine adduct was prepared chemically and fully characterized by spectroscopic means (NMR and MS). In separate experiments, free acrolein was detected by ELISA when arachidonic acid and other PUFAs were autoxidized in an iron/ascorbate-mediated free radical generating system. The authors concluded that ,acrolein is not just a pollutant but also a lipid peroxidation product that could be ubiquitously generated in biological systems [33].
Is lipid peroxidation a major source of acrolein in vivo? In urine of nonsmoking, healthy individuals, the predominant metabolite of the lipid peroxidation product, 4-hydroxy-2-nonenal (HNE), is the mercapturic acid derivative of 1,4-dihydroxynonene (DHN-MA, 2-acetamido-3-(1,4-dihydroxynonan-3-ylthio)propanoic acid). DHN-MA was found in human urine at concentrations of about 2.7 μg/L (8.4 nM) [34]. The analogous acrolein metabolite (S-(3-hydroxypropyl)mercapturic acid, HPMA) was reported to be present in the urine of a nonsmoker at a concentration of 422 μg/L (1.9 μM) [11]. Roethig et al. [35] determined the excretion of HPMA for nonsmoking subjects to be about 250 μg/24 h, which corresponds to about 0.7 μM if the urine volume was 1.6 L/day. These data suggest that there is 102-fold greater production of acrolein metabolites than HNE metabolites in humans. It is generally recognized that degradation of lipid hydroperoxides derived from linoleic or arachidonic acid yields far more HNE than acrolein [18, 36], and therefore it seems that lipid peroxidation is not likely to represent a major source of acrolein in humans. More prominent endogenous sources of acrolein are amino acids and polyamines (see Sections 2.5 and 2.6).
2.4 Biodiesel
If heating or combustion of PUFAs produces acrolein, should we be concerned about acrolein emissions when using biodiesel as an energy source? ,Biodiesel’ is most often defined as ,the mono alkyl esters of long chain fatty acids derived from renewable vegetable oils or animal fats for use in compression ignition (diesel) engines [37].’ Commercially available biodiesel is a mixture of fatty acid methyl or ethyl esters, produced by transesterification of triglycerides, and should not be a significant source of glycerol when properly refined. Indeed, acrolein emissions from biodiesel combustion are related to fuel quality and glycerol content [38]. In the United States, soybean oil is the primary raw material for the manufacture of biodiesel (75 million gallons in 2005), while rapeseed oil is mostly used in Europe for this purpose [37]. In general, biodiesel has undetectable SO2 and lower polyaromatic hydrocarbons emissions, but slightly higher nitrogen oxides (NOx) emissions than petroleum diesel [39]. In a comparison of a 20% biodiesel-petroleum diesel (B20) blend with petroleum diesel, no statistical difference was measured between acrolein emissions from a bus engine, commonly used in Italy [40]. Studies conducted under the auspices of the U.S. Environmental Protection Agency showed a consistent trend towards reduction in the acrolein emissions from biodiesel-petroleum diesel blends, but the differences were not statistically significant [39]. These data indicate that substitution of petroleum-based diesel with biodiesel does not significantly alter acrolein combustion emissions. Swanson and colleagues cautiously concluded that ,biodiesel exhaust emissions are less likely to present any risk to human health relative to petroleum diesel emissions,’ based on their review of the medical literature [37].
2.5 Amino acids as a source of acrolein
2.5.1 Methionine
Mottram and colleagues [6] demonstrated that the formation of acrylamide from asparagine and methionine requires a carbonyl compound, suggesting the involvement of a Schiff base. The Schiff base formed from methionine and 2,3-butanedione would produce ammonia and 3-(methylthio)propanal (methional) via Strecker degradation upon release of the carbonyl compound as proposed by Mottram and co-workers [6]. This mechanism is similar to the well-known formation of methional from methionine treated with ninhydrin, which converts the amino acid to the corresponding imino acid that subsequently breaks down into methional, carbon dioxide, and ammonia by hydrolysis [41-43]. Methional can form methanethiol and acrolein via retro Michael cleavage. Alternatively, decomposition of the oxidation product of methional (methional sulfoxide) would yield acrolein and methylsulfenic acid, which readily forms a disulfide with thiols (Fig. 4). Indeed, methanethiol, dimethyl sulfide, and acrolein were identified as volatile products from the reaction of methionine with ninhydrin [43]. The proposed mechanism in Fig. 4 would also explain the formation of acrolein from methionine and dehydroascorbic acid (a dicarbonyl compound) or ascorbic acid, by heating or boiling in aqueous solutions in the presence of atmospheric oxygen [44-46]. Interestingly, methional is the primary flavor compound of baked and cooked potatoes. In an effort to improve flavor retention during potato processing, Di and co-workers [47] developed a transgenic potato with sixfold higher concentrations of soluble methionine in the tubers compared to wild-type potatoes. The observed increase in methional formation during baking correlated with the soluble methionine levels in the tubers. Acrolein production was not measured in this study. Little is known about human exposure to acrolein through consumption of baked or cooked potatoes. It is conceivable that a fraction of the methional produced during cooking of potatoes is converted into acrolein after potato consumption by S-oxygenation and dissociation of the resulting sulfoxide (see Section 3).
Figure 4.

Formation of acrolein from methionine. The first three reaction steps are collectively known as the Strecker degradation of an α-amino acid.
2.5.2 Threonine
Threonine has been identified as an endogenous source of acrolein by Anderson and colleagues [7]. The conversion of threonine into acrolein was found to be mediated by myeloperoxidase present in human neutrophils (Fig. 5). Myeloperoxidase plays a role in killing bacteria and other pathogens by forming hypochlorous acid (HOCl) from chloride and hydrogen peroxide (H2O2). Acrolein production by myeloperoxidase in vitro requires both H2O2 and chloride ions, which indicates that a chlorinated intermediate is necessary to form acrolein and its precursor 2-hydroxypropanal (Fig. 5). Treatment of threonine with HOCl also generated acrolein and 2-hydroxypropanal. In a murine model of acute myocardial infarction, myeloperoxidase was found to be a major enzymatic source of acrolein, as demonstrated by a dramatic increase of acrolein in ischemic cardiac tissue obtained from wild-type mice as compared to myeloperoxi-dase-deficient (MPO null) mice [48]. The authors also observed that the MPO-null mice showed better cardiac function 24 days after the ischemic injury. These findings suggest that MPO-mediated oxidation of threonine can yield acrolein under conditions of acute oxidative stress in humans such as myocardial infarction and stroke.
Figure 5.

Proposed reaction pathway for the formation of acrolein from threonine mediated by myeloperoxidase (MPO) (adapted from Anderson et al. [7]).
Acrolein can also be formed from threonine during heat treatment of foods via Strecker degradation. In this case, 2-hydroxypropanal is the Strecker aldehyde [49] which forms acrolein by loss of a water molecule (cf. Fig. 1).
2.6 Polyamines
The physiological role of polyamines, such as spermine and spermidine, is to assist in the regulation of cell proliferation and differentiation. Polyamines, which are derived from arginine (ornithine) and decarboxylated S-adenosylmethionine [50], occur in all cells [51] and can reach high micromolar concentrations in brain tissue [52]. Acrolein is formed endogenously from polyamines by copper-dependent amine oxidases (plasma/serum amine oxidase, diamine oxidase, and semicarbazide-sensitive amine oxidase) and by FAD-dependent polyamine oxidases, monoamine oxidase, and spermine/polyamine oxidase [53]. 3-Aminopropanal appears to be the main metabolite of polyamine catabolism mediated by FAD-dependent enzymes, which can dissociate into acrolein and ammonia [53]. There has been some dispute as to whether spermine is metabolized by copper-dependent plasma amine oxidase at the primary amino termini or at the secondary amine sites [54-56]. Both pathways would yield spermidine, acrolein, and 3-aminopropanal via an intermediate imine species followed by hydrolysis and addition/elimination reactions. The group of Sayre [56] ended the dispute by showing that homospermine is exclusively metabolized at the primary amino groups, yielding aldehydes that do not spontaneously release acrolein via retro-Michael cleavage (which caused the confusion in previous studies). Thus, spermine is oxidatively deaminated by plasma amine oxidase to give spermidine and acrolein. Spermidine can undergo a second oxidative deamination, yielding putrescine and acrolein (Fig. 6). It was also shown by Lee and Sayre that the formation of 3-aminopropanal can be explained by Michael-type addition of ammonia to acrolein [56].
Figure 6.
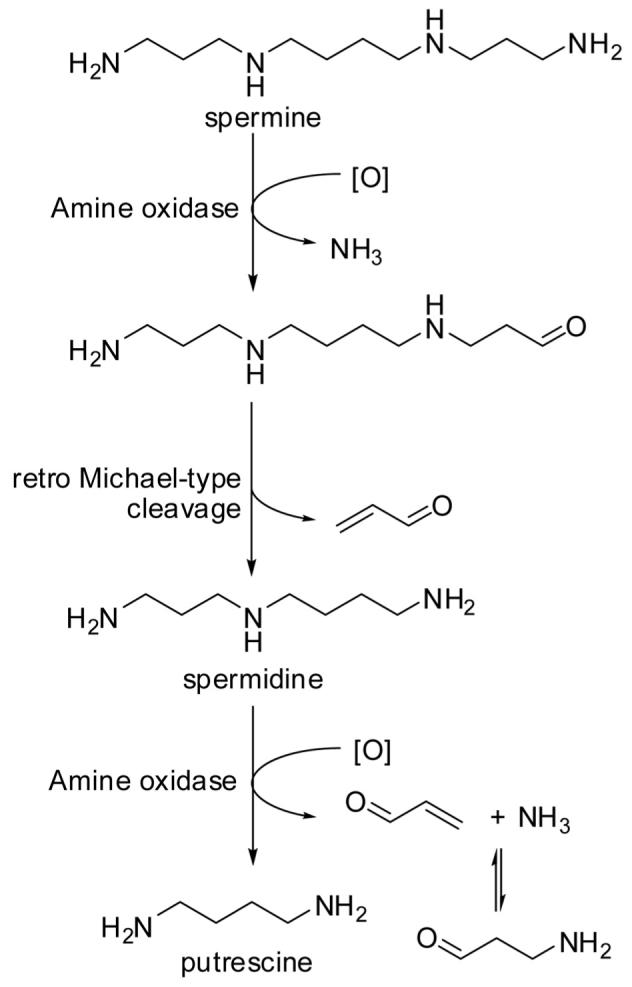
Amine oxidase-mediated catabolism of spermine yields acrolein and 3-aminopropanal.
The amine oxidase activity in plasma of patients with chronic renal failure was significantly enhanced compared to normal subjects (P < 0.01) as was the protein-bound acrolein levels in plasma (1.42 μM versus 0.53 μM, P < 0.01). FDP-lysine (see [33]) levels were also enhanced in these patients (170 μM versus 31.2 μM, P < 0.001) [57]. These data suggest that polyamine catabolism may be a significant source of acrolein compared to lipid peroxidation.
3 Metabolic fate
3.1 Glutathione conjugation with acrolein
Acrolein is soluble in water, in alcohol, and in diethyl ether, and therefore it can travel across membranes by passive diffusion. The main pathway for elimination of acrolein is conjugation with glutathione (GSH) in the liver, followed by enzymatic cleavage of the γ-glutamic acid and glycine residues, respectively, in the liver and in the kidney [58, 59], and N-acetylation of the resultant cysteine conjugate to form S-(3-oxopropyl)-N-acetylcysteine (OPMA) in the kidney. Reduction of this aldehyde yields S-(3-hydroxypropyl)-N-acetylcysteine (HPMA), the main metabolite of acrolein found in urine [11, 60], while oxidation of the aldehyde group produces S-carboxyethyl-N-acetylcysteine (carboxyethyl mercapturic acid, CEMA) (Fig. 7). Oxidation of acrolein may precede conjugation with GSH, thus forming acrylic acid. Enzyme-mediated epoxidation of acrolein produces glycidaldehyde, an instable intermediate that can react with water to yield glyceraldehyde [61] or it can form a conjugate with GSH [62]. Additional metabolites identified in the 0-24-h urine after oral administration of [2,3-14C]acrolein to rats (2.5 mg/kg) include 3-hydroxpropionic acid (6-30% relative abundance), malonic acid (trace), and N-acetyl-S-(2-carboxy-2-hydroxyethyl)-cysteine (8-18%), an end-product metabolite of GSH addition to glycidaldehyde [62] (Fig. 7). The feces in the [2,3-14C]acrolein metabolism study were also examined, but revealed no discrete metabolites: the radioactivity resided in a fraction with a molecular weight range of 2000-20000 Da [62]. Interestingly, a large proportion of the radioactivity was retained in oxalic acid (16-33%) when acrolein was administered orally, but no radioactive oxalic acid could be detected after intravenous injection, suggesting that it is formed in the gastrointestinal tract by the gut microflora [62].
Figure 7.

Metabolism of acrolein.
It has been pointed out by Esterbauer and others [5] that acrolein reacts 110-150 times faster with GSH than HNE. While GSH conjugation of acrolein can proceed without catalyst, the same enzyme that mediates the formation of the GSH-HNE conjugate also catalyzes the formation of the acrolein conjugate, i.e., glutathione-S-transferase isoenzyme A4-4 (GSTA4-4). When GSTA4-4 was overexpressed in mouse pancreatic islet endothelial (MS1) cells, the concentration of acrolein producing 50% reduction in cell viability (LC50 measured by MTT absorbance) increased from about 5 μM for the wild-type cells to about 38 μM for the transfected cells overexpressing GSTA4-4. In addition, the GSTA4-transfected cells showed a significantly greater growth rate than the wild-type cells, indicating that acrolein is detoxified by GSTA4-mediated conjugation with GSH [63]. Acrolein is also a substrate for GST A1-1, GST M1-1, GST P1-1 [64], and GST T1 [65].
Some individuals may be more susceptible to acrolein toxicity due to impaired GST activity. Palma and co-workers [66] found that smokers carrying the GSTM1-null genotype showed a significantly higher frequency of micronuclei (a measure of DNA damage) compared with GSTM1-positive smokers, while no such association was found in nonsmokers. Because acrolein is a substrate for GST M1-1 [64], smoking individuals with the GSTM1-null genotype may be particularly susceptible to the genotoxic and immunosuppressive effects of acrolein. A GST polymorphism does also exist for GST P1-1 in humans: four allelic variants for this GST isoenzyme have been identified, one of which has a significantly lower catalytic efficiency in GSH conjugation of acrolein [67].
The mercapturic acid of acrolein, OPMA, was shown to be more toxic than its homolog, S-(4-oxobutyl)-N-acetylcysteine, in human lung adenoma A549 cells [68]. The authors of this study concluded that the toxicity of OPMA is not due to OPMA itself, but due to release of acrolein by retro-Michael cleavage (β-elimination), which is not possible for the homolog. They also demonstrated that OPMA-S-oxide is more toxic than OPMA when cell proliferation was used as an index of toxicity (IC50 of 22 μM versus 83 μM). One explanation for this finding is that OPMA-S-oxide releases acrolein more facilely than OPMA due to the polarizing effect of the oxygen atom in the sulfoxide (see Fig. 7). Similar results were obtained by Hashmi and colleagues [69], who observed release of acrolein from OPMA-S-oxide but not from OPMA under basic conditions. While both conjugates showed cytotoxicity in isolated rat renal proximal tubular cells, cytotoxicity was reduced for OPMA but not for its S-oxide when the cells were treated with methimazole, an inhibitor of flavin-containing monooxygenase (FMO). The authors suggested that FMO-catalyzed S-oxygenation (bioactivation) of OPMA contributes to its cytotoxicity in the kidney [69], which would offer an explanation for the observed nephrotoxicity in rats given acrolein-GSH adduct intravenously [70] and possibly for acrolein’s suspected involvement in cyclophosphamide-related urinary bladder cancer (acrolein is a metabolite of cyclophosphamide) [71]. In general, S-oxygenation can be mediated by cytochrome P450s and by FMOs. One FMO isoenzyme, FMO2, is highly expressed in the lung of rabbits and can account for 10% or more of the total microsomal protein fraction [72]. Humans show genetic polymorphism in the expression of FMO2 in lung [73]. Although it is not known whether the GSH conjugate of acrolein is a substrate for FMO2, it is conceivable that the GSH conjugate is bioactivated in the lung, which could be relevant to acrolein exposure through cigarette smoke.
As mentioned above, HPMA is the major urinary metabolite of acrolein. When allylamine was administered to rats by gavage (5-150 mg/kg), 44-48% of the dose was converted to acrolein by oxidative deamination and found in the urine as HPMA during the first 24 h. A small amount (3% of the dose) was excreted in the urine as HPMA in the second 24-h period. Oral administration of acrolein to rats resulted in 79% recovery as HPMA in the 0-24-h urine [74]. In another study, the sum of HPMA and CEMA excreted in the urine during the first 24 h after intraperitoneal administration of acrolein to rats (0.5-2.0 mg/kg) accounted for 29.2 ± 6.5% of the dose. CEMAwas estimated to represent less than 10% of the total amount of mercapturic acids recovered in the urine [75]. In the same study, HPMA and CEMA were also found in the urine of rats exposed to acrolein by inhalation, but at lower recovery rates [75]. Unlike experiments conducted with liver tissue preparations from rats, lung tissue preparations failed to show conversion of acrolein into acrylic acid, suggesting that acrolein metabolism may be different in the liver and the lung [61].
In humans, the mean levels of HPMA in first-void morning urine are about 4.0 nmol/mg creatinine for smokers and 0.7 nmol/mg creatinine for nonsmokers [11]. The average concentration of 3-HPMA in the urine of the smoker subjects was 1095 ng/mL urine, which translates into HPMA excretion of about 1.7-2 mg/day [11] (= 7.7-9 μmol). For comparison, the exposure to acrolein from one cigarette is about 60 μg or 1.1 μmol (see Section 2.2).
3.2 Ascorbate adduction to acrolein
The reaction of ascorbic acid (vitamin C) with acrolein was first described by Fodor and colleagues in 1983 [26, 76]. It is classified as a Michael addition reaction, in which ascorbate (an enolate) plays the role of carbon nucleophile and acrolein that of Michael acceptor (Fig. 8). The Michael adduct undergoes further cyclization to form a 5,5,5-tricyclic spiro-compound in aqueous solvents and a 5,5,6-tricyclic system in anhydrous solvents [77]. The different outcomes of the cyclization reaction are most likely due to chelation (hydrogen-bonding) between OH-2 of the ascorbyl moiety with the lactone-carbonyl in anhydrous solvents, which leaves only OH-3 available for hemi-acetalization with the aldehyde functionality (pathway b in Fig. 8). In aqueous solvents, the lactone-carbonyl is H-bonded with solvent water molecules and OH-2 can now form the hemi-acetal with the aldehyde (pathway a). The reactions proceed in a stereo-specific manner and the structures of the products have been determined by X-ray diffraction and NMR spectroscopy [78, 79].
Figure 8.

Michael addition of ascorbate to acrolein: ,ascorbylation of acrolein’. DMF, dimethylformamide.
What is the biological relevance of this reaction, which we have termed ,nucleophilic ascorbylation’ (to distinguish it from the well-known (electrophilic) ascorbylation of proteins in which dehydroascorbic acid plays the role of electrophile, see, e.g., [80])? Can ascorbylation of acrolein compete with GSH conjugation? It is readily recognized that GSH is a ,better’ nucleophile than ascorbate, that is, GSH will react faster with acrolein than ascorbate. In other words, the outcome of the competition between both nucleophiles will show the acrolein-GSH adduct as the kinetic product, especially if the conjugation reaction is catalyzed by GST enzymes. As shown above, the acrolein-GSH adduct formation is reversible and acrolein can be released. Calculations conducted in our group indicate that ascorbylation of acrolein is thermodynamically favored over GSH adduction: the reaction AscH + GS-ACR → AscACR + GSH has a ΔG of -11 kcal/mol. In view of the fact that ascorbic acid and GSH both occur in cells at similar concentrations in the low millimolar range [81], ascorbylation could have biological relevance. Vitamin C-adequate cells exposed to acrolein show a dramatic decrease in the cellular level of vitamin C, and yet the (non-enzymatic) reaction of ascorbate with acrolein has often been ignored as a mechanism for the decrease of acrolein cytotoxicity or for the observed protection against acrolein cytotoxicity by ascorbic acid (see, e.g., [82-84]). For instance, Arai and colleagues [83] suggested that ,ascorbate can play and important role in preventing atherogenesis by suppressing the modification of apoE and VLDL by acrolein.’ Did adduction of ascorbic acid (1 mM) to acrolein (400 μM) contribute to the observed protection? Numerous acrolein exposure studies using cultured human cells have been conducted in the absence of ascorbic acid (human cells cannot synthesize ascorbic acid), which does not represent normal physiology and conclusions drawn from those studies as to acrolein toxicity may not be relevant to the in vivo situation.
4 Interaction of acrolein with biomolecular targets
4.1 Adduction of acrolein to DNA
The reaction of deoxyguanosine (dG) with acrolein to form 1,N2-propanodeoxyguanosine (PdG) adducts in vitro was first described by the Hecht laboratory [85]. Adduct formation can be initiated by Michael-type addition of either N-1 or N2 of dG to C-3 of acrolein to yield two regioisomers, α- and γ-hydroxy-PdG, each of which exist as a pair of stereo-isomers (Fig. 9). The γ-hydroxy isomer was formed as a major and the α-hydroxy isomer as a minor product after incubation of acrolein with dG or calf thymus DNA [85]. Both isomeric adducts were shown to induce base substitutions with G→C transversions predominating. α-Hydroxy-PdG gave rise to a higher miscoding frequency when incorporated into DNA of human xeroderma pigmentosum A cells [86, 87]. HydroxyPdG adducts have been detected in liver tissue DNA of humans and rodents without carcinogen treatment [88]. The levels of the γ-hydroxy-PdG adduct were about threefold higher in oral tissue obtained from smokers compared to nonsmokers (P = 0.003) [89].
Figure 9.

Adduction of deoxyguanosine to acrolein.
Feng and colleagues [90] reported that acrolein exposure to human lung cells produces a pattern of DNA damage in the p53 tumor suppressor gene which appeared to be similar to the p53 pattern of mutations found in lung cancer. They also observed that acrolein reduces the DNA repair capacity for damage induced by benzo[a]pyrene diol epoxide, a metabolite of the well-known cigarette procarcinogen, benzo[a]pyrene. In view of the much greater abundance of acrolein in cigarette smoke compared to benzo[a]pyrene (60 μg versus 1.5-15 ng per cigarette [91]), the authors suggest that acrolein may represent ,a major etiological agent for cigarette smoke-related lung cancer [90].’ This is a potentially important finding, because it opens avenues for decreasing the probability of cancer development in smokers by decreasing acrolein release from cigarettes through altering the amounts and composition of additives to cigarettes (see Section 2.2 and Hecht’s [91] commentary on the paper by Feng et al.).
4.2 Acrolein adduction to amino acids and cross-linking of proteins
Acrolein-induced toxicity has previously been reviewed by Beauchamp et al. [92] and by Kehrer and Biswal [93]. Numerous studies indicate that acrolein-induced toxic effects can at least to some extent be rationalized by depletion of cellular GSH [93]. In addition, acrolein exerts its biological effects through reaction with nucleophilic sites in proteins, i.e., the sulfhydryl group of cysteine, the imidazole moiety of histidine, and the ε-amino group of lysine, thereby impairing protein function. It prefers to form Michael-type adducts with cysteine residues due to its highly polarizable, conjugated π-electron system [94]. Similar to adduction of the guanidine moiety of DNA (Fig. 9), acrolein can form propano adducts with the guanidine group of arginine [95] (Fig. 10). Acrolein has been reported to form a bis-adduct with lysine residues, named Nε-(3-formyl-3,4-dehydropiperidino)lysine (FDP-lysine) [33]. The electrophilic α,β-unsaturated carbonyl moiety is retained in FDP-lysine, allowing it to react further with sulfhydryl compounds, such as GSH [96], or other nucleophilic sites in proteins under formation of cross-links [97]. FDP-lysine has been detected immunochemically in brain tissue from Alzheimer’s disease patients, particularly in neurofibrillary tangles of the microtubule-associated protein Tau [98]. Acrolein-modified α-synuclein has been detected by immunostaining using anti-acrolein monoclonal antibody, and subsequent in vitro experiments indicated that acrolein modification of α-synuclein promotes the formation of protein aggregates presumable via FDP-lysine-mediated cross-links [99]. There is a large number of potential protein targets that have redox- and/or electrophile-sensitive sites. Some examples are outlined in the following sections.
Figure 10.

Adduction of acrolein to amino acid residues in proteins.
4.3 Acrolein as a modulator of stress-mediated gene activation
Many transcription factors are regulated by redox-dependent mechanisms and utilize cysteine residues as ,redox switches.’ One toxicologically important example is the oxidative stress-sensing transcription factor, nuclear factor-κB (NF-κB). NF-κB is critically involved in the expression of more than 400 genes that play a role in the regulation of antioxidant defense, apoptosis, and inflammatory and immunological responses [100, 101]. The five members of the NF-κB family of proteins are p50/p105 (NF-κB1), p52/p100 (NF-κB2), c-Rel, RelB, and p65 (RelA). These proteins are characterized by their Rel homology domains, which control DNA binding, dimerization, and interactions with inhibitory factors known as Iκ-B proteins [101]. In its inactive state, NF-κB is bound to the inhibitor protein I-κB and resides in the cytoplasm. Activation of the transcription factor is mediated by phosphorylation of I-κB. Disassembly of the I-κB/NF-κB complex leads to inactivation of I-κB via ubiquitination and subsequent rapid degradation by the 26S proteosome. The release of NF-κB permits its translocation to the nucleus, where it can regulate the expression of specific genes [101].
Recently, Lambert et al. [102] reported on the immune-suppressing effects of cigarette smoke. Using human T-cells, they determined the effects of acrolein and related aldehydes on the production of proinflammatory cytokines, including interleukin-2 (IL-2), IL-10, granulocyte-macrophage colony-stimulating factor, interferon-γ, and tumor necrosis factor α (TNF-α). Acrolein inhibited the production of proinflammatory cytokines with an IC50 of 3 μM, which could be physiologically relevant in view of the amounts of acrolein generated by a single cigarette (60 μg = 1.1 μmol). The saturated aldehydes, acetaldehyde, propanal, and butanal, were inactive, presumably due to their inability to form Michael-type adducts with cysteine residues in NF-κB [102]. NF-κB-DNA binding was decreased by acrolein after mitogenic stimulation of T cells. In addition, these authors showed that acrolein was very effective in inhibiting the binding of NF-κB1 (p50 subunit) to the IL-2 promoter using a chromatin immunoprecipitation assay.
How does acrolein affect NF-κB-mediated gene activation? Using MS/MS, Lambert et al. [102] identified acrolein-modified amino acid residues in the p50 subunit, the DNA-binding domain of NFκB, after exposure of the protein to acrolein, i. e., Cys-61, Cys-87, Cys-118, Cys-123, Cys-261, Cys-272, Arg-230, and His-306. The in vitro experiment also revealed acrolein adduction to Arg-307, forming six-membered heterocycle. From previous mutation studies in combination with thiol labeling approaches, it was known that Cys-61 is critical for the DNA-binding activity of p50 [103, 104]. Analysis of the crystal structure of the NF-κB (p50/p65 heterodimer) bound to DNA supports a role of Arg-307 in mediating binding of p50 to DNA [105]. Crotonaldehyde exposure resulted in a similar pattern of adduction with modification of Cys-61 predominating. Taken together, Lambert and colleagues concluded that acrolein and other 2-alkenals found in cigarette smoke can disrupt physiological regulation of gene expression by direct modification of the DNA-binding domain of a transcription factor and, as a consequence, they may cause immunosuppression upon exposure via cigarette smoke.
4.4 Acrolein as an inducer of the Keap1-Nrf2-dependent ARE/EpRE signaling pathway
Nuclear erythroid-2 related factor 2 (Nrf2) is a member of the ,basic leucine zipper’-type transcription factor family. Nrf2 regulates the cellular redox homeostasis and orchestrates the enzymatic protection machinery against oxidative injuries and electrophile stress [106]. Nrf2 binds to and activates the ,antioxidant response element (ARE),’ also referred to as the ,electrophile response element (EpRE)’ [107]. Many ARE-dependent genes are involved in (i) synthesis of antioxidant defense enzymes (e.g., γ-glutamyl cysteine synthetase modifier subunit, γ-glutamyl cysteine synthetase catalytic subunit, heme oxygenase-1 (HO-1), superoxide dismutase, GSH reductase, GSH peroxidase, thioredoxin, thioredoxin reductase, peroxiredoxins, and cysteine/glutamate transporter), (ii) xenobiotic metabolism (e.g., NADP[H] quinone oxidoreductase 1, GSTs, and UDP-glucuronosyltransferase), and (iii) synthesis of xenobiotic efflux pump proteins (e.g., MRP1 and MRP2) [108]. Acrolein has been reported to induce transcriptional activation of ARE-dependent genes, resulting in the upregulation of phase II enzymes and cytoprotection against electrophile stress. For example, Wu et al. [109] demonstrated increased expression of the antioxidant enzyme HO-1 in endothelial cells that were exposed to acrolein. Exposure of human lung epithelial (A549) cells to acrolein (150 fmol/cell for 1 h) first depleted 80% of the intracellular GSH pool and then increased the transcription of γ-glutamylcysteine synthetase at 6-12 h post-treatment, resulting in normalization of GSH levels. Acrolein treatment also activated the transcription of phase II genes, as indicated by an increase in mRNA coding for NAD(P)H:quinone oxidoreductase [110].
How does acrolein induce transcription of ARE-driven genes and expression of stress-response proteins, such as phase II biotransformation enzymes and antioxidant proteins? In the cytoplasm, Nrf2 is inactive when bound to Kelch-like ECH-associated protein 1 (Keap1). Nrf2 activity is controlled by binding to and dissociation from Keap1, and by proteosomal degradation [111]. Under conditions of oxidant or electrophile stress, Keap1 loses affinity for Nrf2, thus promoting translocation of Nrf2 from the cytoplasm into the nucleus and induction of gene expression related to oxidant defense and electrophile detoxification [106, 112]. Human Keap1 is structurally organized into five major domains: the N-terminal domain (amino acids 1-60); the “Broad complex, tramtrack and bric-a-brac” BTB domain (amino acids 61-179); the central intervening IVR domain (amino acids 180-314); a double glycine-rich domain comprising six Kelch repeat motifs (amino acids 315-359, 361-410, 412-457, 459-504, 506-551, and 553-598); and a C-terminal domain (amino acids 599-624). The BTB and IVR domains are required for the redox- and electrophile-sensitive regulation of Nrf2 through a series of reactive cysteines [113-115]. Human Keap1 has 27 cysteine residues (while rodent Keap1 has 25), nine of which are predicted to be particularly reactive due to their location adjacent to basic amino acids. Especially neighboring lysine residues can decrease the pKa of the thiol and so increase the nucleophilicity of the thiol [106]. Moreover, cysteine residues coordinated with Zn2+ in Keap1 are particularly active as redox sensors [116].
Several research groups have studied the reactivity of the cysteine residues as Michael-type donors in murine and human Keap1-Nrf2 systems. It appeared that the reactivities of the cysteines in human Keap1 are different from the corresponding cysteines in mouse Keap1 [112-115, 117]. Despite some uncertainty regarding the exact order of the reactivity of the cysteine residues towards different electrophiles, Cys-151 (located in the BTB domain) was identified as the most reactive nucleophilic site in human Keap1 [113, 117]. It is conceivable that multiple cysteines in Keap1 can be modified by different inducers and it is likely that sites of sulfhydryl modification may vary among the different chemical compound classes and across species [118]. Furthermore, modification experiments of diverse Keap1-Nrf2 complexes indicated that sulfhydryl alkylation by different Michael acceptors did not lead per se to Keap1-Nrf2 complex dissociation and Nrf2 translocation. To account for these unexpected findings, Mesecar and colleagues [113] recently proposed an alternative model for Keap1-Nrf2-mediated cell signaling. In their model, nuclear accumulation of Nrf2 is achieved via the following events: (i) alkylation of Cys-151 by electrophiles leads to disruption of the homodimerization site and to conformational changes in the BTB domain of Keap1, which would result in altered accessibility of the site of ubiquitination in Nrf2 and thus decreased proteosomal degradation, and (ii) increased ubiquitination and turnover of Keap1. Both events would lead to elevated levels of active Nrf2 and Nrf2 nuclear accumulation. Although the details of the molecular mechanisms of the Nrf2-mediated response to electrophile stress remain to be elucidated, it seems somewhat provocative to suggest that, at low concentration, acrolein may actually exert cytoprotective effects by inducing phase II enzymes via activation of Nrf2 [119].
4.5 Acrolein adduction to phosphatases
Electrophile-mediated stress not only affects the transcription of many genes but also interferes with redox-sensitive signal transduction pathways, because many constituents of these pathways have reactive cysteine residues. The consequences of the reaction of acrolein with functionally critical protein thiol residues of protein tyrosine phosphatases (PTPs) have only recently been recognized.
All of the known PTPs contain a strictly conserved sequence motif at the active site, C(X)5R(S/T). The cysteine thiol of the active site attacks the phosphate group of phosphorylated tyrosine (pY), thereby forming a transient phosphocysteinyl enzyme intermediate [120, 121]. This mechanism has been well established for PTP1B, the archetypal member of the PTP family of enzymes. In vitro exposure of the catalytic subunit of human PTP1B (a.a. 1-322) to acrolein revealed that acrolein is a potent time-dependent inactivator of PTP1B. The concentration of acrolein required to achieve a half-maximal rate of inactivation, KI, was determined to be 2.3 ± 0.6 × 10-4 M and the maximum rate of inactivation at saturating concentrations of acrolein, kinact, was 0.02 ± 0.005 s-1. The apparent second-order rate constant for inactivation of the enzyme by acrolein (kinact/KI = 87 M-1s-1) is comparable to that for hydrogen peroxide (10 M -1s-1) [122], a known endogenous regulator of PTP1B activity. These data suggest that inactivation of PTP1B by acrolein is a consequence of covalent modification of the enzyme. Indeed, the active site cysteine, Cys-215, was identified by MS/MS as the major site of acrolein adduction, while histidine-214 of the active site showed only little reactivity towards acrolein [123]. In conclusion, inactivation of PTPs through acrolein adduction to cysteine in the active site leads to disruption of normal cellular signaling cascades and should be considered as an important chemical mechanism contributing to the cellular toxicity of acrolein [124].
4.6 Acrolein induces necrotic and apoptotic cell death
Acrolein can either induce apoptosis [82, 125] or have an inhibitory effect on apoptotic pathways as observed for human neutrophils [126]. Acrolein induced apoptosis in human lung epithelial (HBE1) cells at 10-25-μM and in isolated human alveolar macrophages at 25-μM exposure, as indicated by DNA fragmentation after 24 h of exposure. Acrolein has also been reported to cause necrotic cell death when proB lymphoid cells were exposed to acrolein at concentrations greater than 10 μM in the culture medium lacking serum [127]. Averill-Bates and colleagues [125, 128] demonstrated that acrolein can induce apoptosis in Chinese hamster ovary (CHO) cells, either through the intrinsic pathway which involves cytochrome c release [125] or through the extrinsic pathway by activation of death receptors [128]. Differences in cell types, cell culture conditions, medium composition, and acrolein concentration may explain some of the variable findings.
In general, there are two principal pathways of xenobiotic-induced apoptosis. First, in the extrinsic pathway xenobiotics interact with so-called death receptors (e.g., Fas, also called Apo-1 or CD95) or tumor-necrosis factor receptor, TNF-R) to cause activation of the death-inducing signaling complex (DISC). DISC recruits the initiator proteases, i. e., caspase-2, -8, and -10. Caspases are dormant as inactive zymogens and require cleavage by other caspases in order to become catalytically active proteases. The initiator caspases will then activate the effector caspases-3, -6, and -7 to initiate the cell-death program leading to degradation of cellular targets. Second, in the intrinsic or mitochondria-mediated pathway, intracellular stress signals lead to release of cytochrome c from the mitochondrial intermembrane space into the cytosol, where it binds to the apoptotic protease activating factor-1 (Apaf-1). Binding of cytochrome c to Apaf-1 triggers the formation of the apoptosome (l1 MDa oligomeric, Apaf-1-containing complex) that catalyses activation of the apoptosome-bound procaspase-9. Caspase-9 then activates the effector caspase-3 resulting in the execution of the cell-death program [129]. The events that lead to release of cytochrome c are not well understood, but one possible key event seems to be the formation of the mitochondrial permeability transition pore (PTP), a multi-subunit protein channel that is permeable for solutes of less than 1500 Da [130, 131]. The opening of this Ca2+-dependent pore is thought to control the commitment of the cell to die through apoptotic mechanisms or to undergo necrosis [132, 133].
Kern and Kehrer [127] reported that in murine FL5.12 proB lymphocytes, acrolein at low levels (<10 μM) induces only very modest levels of apoptosis while causing almost exclusively necrosis at higher doses. How can acrolein direct cellular death pathways, apoptosis versus necrosis? Tanel and Averill-Bates [125] have observed during their studies on apoptotic pathways in CHO cells that procaspase-3 was proteolytically processed but that caspase-3 was not catalytically active. Finkelstein et al. [126] reported that exposure of human neutrophils to acrolein also prevented the activation of caspase-3. Why can acrolein inactive cas-pases? Caspases are members of the cysteine-aspartate-specific proteases and, as such, have a cysteine residue in their active center [129]. The mature caspase-3 exists as heterotetrameric protein complexes, in which two heterodimers (p12-p17), consisting of the small (p12) and large subunit (p17), interact to form a 12-stranded β-sheet that is sand-wiched by α-helices. In the heterotetrameric complex (p12-p17)2, the two heterodimers are oriented in a head-to-tail configuration. Accordingly, the active site of each heterodimer is at opposite sites of the caspase fold. The catalytically active center of caspase-3 comprises Cys-163 and His-121 located in the p17 subunit. The tetrahedral transition state is stabilized by hydrogen bonding with the backbone amide proton of Gly-122 [134]. Due to the spatially close histidine-residue, the cysteine sulfhydryl group is particularly reactive and, in turn, can be readily regulated by oxidative or nitrosative stress [135] or become inactivated by alkylation reactions with reactive electrophiles, such as acrolein.
In addition, several lines of evidence suggest that exposure of cells to electrophiles decreases the mitochondrial membrane potential and promotes pore opening. Studies conducted by Darley-Usmar and colleagues [136] have demonstrated that the electrophilic lipid peroxidation product, 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2), is capable of inducing apoptosis in liver mitochondria. According to the ,two hit’ mechanism proposed by Brookes et al. [132], electrophile stress potentiates opening of the PTP under conditions of elevated Ca2+ levels in the mitochondrial matrix. Using biotin-tagged 15d-PGJ2, Darley-Usmar and colleagues were able to identify several putative mitochondrial protein targets of 15d-PGJ2, two of which seem to be particularly relevant to the opening of the PTP, i.e., ANT (adenine nucleotide translocase) and ATP synthase [132]. ANT, a constituent of the PTP complex, plays a role as one of the key modulators of the pore opening process in which ANT thiols are proposed to serve as redox-sensitive target sites [130, 131]. Elevated Ca2+ levels in the mitochondrial matrix result in upregulation of ATP synthase and other components of the oxidative phosphorylation machinery in order to meet the increased ATP demand [133]. The concomitant enhanced respiratory chain activity may increase oxidative stress and production of acrolein. In our laboratory, we have used aldehyde/keto-specific probes in combination with MS to characterize protein targets of α,β-unsaturated aldehydes in cardiac mitochondria isolated from rats of different ages. Our studies repeatedly identified Cys-256 in ANT as a major site of acrolein adduction (Fig. 11) [137]. We also identified several sites in subunits of ATP synthase that were modified by acrolein: Cys-294 in ATPase α-chain, Cys-78 in ATP synthase γ-chain, Cys-239 in ATP synthase B chain, Cys-100 in ATP synthase D chain, and Cys-141 in ATP synthase O subunit (unpublished results). It seems possible that inactivation of ATP synthase by a wide range of electrophiles may interfere with apoptotic cell death pathways. Further investigations are needed to examine how modifications by acrolein modulate the function of proteins and cellular processes.
Figure 11.

MALDI-MS/MS spectrum of the HICAT-labeled acrolein-modified peptide from ADP/ATP translocase 1 (ADT1_RAT, a.a. 245-258) found in heart mitochondria from 24-month-old untreated rats. As precursor ion for the MS/MS experiment, the peptide ion with m/z 2116.0 was used. The fragment ion data indicate that Cys-256 was modified by acrolein and the resulting protein carbonyl was subsequently tagged by the aldehyde/keto-specific probe HICAT (adapted from [137]).
5 Concluding remarks
What is the impact of acrolein exposure on human health? The most obvious single source of acrolein is tobacco smoke. Because there are many tobacco smoke constituents with biological effects detrimental to human health, it is often not possible to contribute a tobacco-related effect to acrolein. The study conducted by Lambert and colleagues [95] is significant, because it identified a link between acrolein and suppression of immune responses (a well-known effect of cigarette smoke) through inhibition of the NF-κB pathway (see Section 4.3). The acrolein concentrations used in this study (low micromolar range) may be reached in lung tissue after smoking several cigarettes. This finding may thus provide an explanation for the epidemiological observations that smoking or exposure to tobacco smoke increases the incidence of viral, bacterial, and mycobacterial diseases of the lung [138-141]. Similarly, cooking in poorly ventilated kitchens has been associated with respiratory illnesses, weakening of the immune system, and lung cancer in rural China [142]. It is conceivable that acrolein is co-responsible for these effects. As mentioned in Section 2.3, acrolein emissions from food cooking are far from negligible: the total acrolein emission from commercial kitchens in Hong Kong has been estimated at 7.7 tons per year (see [30]).
According to the U.S. Environmental Protection Agency, the main source of acrolein exposure to humans is the atmosphere, which contains 8.2-24.6 μg acrolein per m3 (mean 14.3 μg/m3) [143]. Acrolein concentrations in smoky indoor air can be much higher. Assuming that human air intake is 10.8 m3/24 h [144], acrolein exposure through atmospheric contact would amount to 154 μg or 2.75 μmol/24 h. This quantity of acrolein is roughly equal to the amount generated by smoking 2.5 cigarettes, which explains why there is a relatively high level of HPMA found in the urine of nonsmokers [11]. In view of the ever-increasing acrolein emissions into the environment, acrolein as a direct irritant may increasingly become a health hazard in individuals with respiratory diseases such as asthma [145].
Acknowledgments
The Stevens and Maier laboratories are supported by the National Institutes of Health (Grants R01HL081721, R01AG025372, S10RR022589, and P30ES000210)
Abbreviations
- ARE
antioxidant response element
- CEMA
carboxyethyl mercapturic acid
- dG
deoxyguanosine
- FDP-lysine
Nε-(3-formyl-3,4-dehydropiperidino)lysine
- FMO
flavin-containing monooxygenase
- GSH
glutathione
- GSTA4-4
glutathione-S-transferase isoenzyme A4-4
- HNE
4-hydroxy-2-nonenal
- HPMA
S-(3-hydroxypropyl)mercapturic acid (syn. S-(3-hydroxypropyl)-N-acetylcysteine)
- Keap1
Kelch-like ECH-associated protein 1
- LDL
low-density lipoprotein
- MPO
myeloperoxidase
- NF-κB
nuclear factor-κB
- Nrf2
nuclear erythroid-2 related factor 2
- OPMA
S-(3-oxopropyl)mercapturic acid (syn. S-(3-oxopropyl)-N-acetylcysteine)
- PdG
1,N2-propanodeoxyguanosine
- PTPs
protein tyrosine phosphatases
- PUFAs
polyunsaturated fatty acids
Footnotes
The authors have declared no conflict of interest.
6 References
- [1].Chevreul E. Rechereches chimiques sur les corps gras d’origine animale. Imprimerie Nationale; Paris: 1823. [Google Scholar]
- [2].Berzelius J. Lehrbuch der Chemie. Dresden and Leipzig; 1839. [Google Scholar]
- [3].Redtenbacher J. Ueber die Zerlegungsprodukte des Glycerinoxydes durch trockene Destillation. Ann. Chem. Pharm. 1843;47:113–148. [Google Scholar]
- [4].Roscoe H, Schorlemmer C. A Treatise on Chemistry. Volume III: The Chemistry of the Hydrocarbons and Their Derivatives. D. Appleton and Company; New York: 1884. [Google Scholar]
- [5].Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radical Biol. Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- [6].Mottram DS, Wedzicha BL, Dodson AT. Acrylamide is formed in the Maillard reaction. Nature. 2002;419:448–449. doi: 10.1038/419448a. [DOI] [PubMed] [Google Scholar]
- [7].Anderson MM, Hazen SL, Hsu FF, Heinecke JW. Human neutrophils employ the myeloperoxidase-hydrogen peroxide-chloride system to convert hydroxy-amino acids into glycolaldehyde, 2-hydroxypropanal, and acrolein. A mechanism for the generation of highly reactive alpha-hydroxy and alpha,beta-unsaturated aldehydes by phagocytes at sites of inflammation. J. Clin. Invest. 1997;99:424–432. doi: 10.1172/JCI119176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yaylayan VA, Keyhani A. Origin of carbohydrate degradation products in L-Alanine/D-[(13)C]glucose model systems. J. Agric. Food Chem. 2000;48:2415–2419. doi: 10.1021/jf000004n. [DOI] [PubMed] [Google Scholar]
- [9].Yaylayan VA, Harty-Majors S, Ismail AA. Monitoring carbonyl-amine reaction and enolization of 1-hydroxy-2-propanone (Acetol) by FTIR spectroscopy. J. Agric. Food Chem. 1999;47:2335–2340. doi: 10.1021/jf9812836. [DOI] [PubMed] [Google Scholar]
- [10].Medina-Navarro R, Duran-Reyes G, Diaz-Flores M, Kumate Rodriguez J, Hicks JJ. Glucose autoxidation produce acrolein from lipid peroxidation in vitro. Clin. Chim. Acta; Int. J. Clin. Chem. 2003;337:183–185. doi: 10.1016/j.cccn.2003.07.005. [DOI] [PubMed] [Google Scholar]
- [11].Carmella SG, Chen M, Zhang Y, Zhang S, et al. Quantitation of acrolein-derived (3-hydroxypropyl)mercapturic acid in human urine by liquid chromatography-atmospheric pressure chemical ionization tandem mass spectrometry: effects of cigarette smoking. Chem. Res. Toxicol. 2007;20:986–990. doi: 10.1021/tx700075y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Carmines EL, Gaworski CL. Toxicological evaluation of glycerin as a cigarette ingredient. Food. Chem. Toxicol. 2005;43:1521–1539. doi: 10.1016/j.fct.2005.04.010. [DOI] [PubMed] [Google Scholar]
- [13].Talhout R, Opperhuizen A, van Amsterdam JG. Sugars as tobacco ingredient: Effects on mainstream smoke composition. Food. Chem. Toxicol. 2006;44:1789–1798. doi: 10.1016/j.fct.2006.06.016. [DOI] [PubMed] [Google Scholar]
- [14].Shelar G, Bernasek P, Furin O, Tobaccodocuments.org Sugar/Nicotine Study. 1992 http://tobaccodocuments.org/product_design/510697389-7410.html.
- [15].Schneider C, Tallman KA, Porter NA, Brash AR. Two distinct pathways of formation of 4-hydroxynonenal. Mechanisms of nonenzymatic transformation of the 9- and 13-hydroperoxides of linoleic acid to 4-hydroxyalkenals. J. Biol. Chem. 2001;276:20831–20838. doi: 10.1074/jbc.M101821200. [DOI] [PubMed] [Google Scholar]
- [16].Schneider C, Porter NA, Brash AR. Autoxidative transformation of chiral omega6 hydroxy linoleic and arachidonic acids to chiral 4-hydroxy-2E-nonenal. Chem. Res. Toxicol. 2004;17:937–941. doi: 10.1021/tx049913n. [DOI] [PubMed] [Google Scholar]
- [17].Spiteller G. Peroxyl radicals: inductors of neurodegenerative and other inflammatory diseases. Their origin and how they transform cholesterol, phospholipids, plasmalogens, polyunsaturated fatty acids, sugars, and proteins into deleterious products. Free Radical Biol. Med. 2006;41:362–387. doi: 10.1016/j.freeradbiomed.2006.03.013. [DOI] [PubMed] [Google Scholar]
- [18].Spiteller P, Kern W, Reiner J, Spiteller G. Aldehydic lipid peroxidation products derived from linoleic acid. Biochim. Biophys. Acta. 2001;1531:188–208. doi: 10.1016/s1388-1981(01)00100-7. [DOI] [PubMed] [Google Scholar]
- [19].Yin H, Porter NA. New insights regarding the autoxidation of polyunsaturated fatty acids. Antiox. Redox Signaling. 2005;7:170–184. doi: 10.1089/ars.2005.7.170. [DOI] [PubMed] [Google Scholar]
- [20].Pederson J, Ingemarsson A, Olsson J. Oxidation of rapeseed oil, rapeseed methyl ester (RME) and diesel fuel studied with GC/MS. Chemosphere. 1999;38:2467–2474. [Google Scholar]
- [21].Tamura H, Kitta K, Shibamoto T. Formation of reactive aldehydes from fatty acids in a Fe2+/H2O2 oxidation system. J. Agric. Food Chem. 1991;39:439–442. [Google Scholar]
- [22].Pan X, Kaneko H, Ushio H, Ohshima T. Oxidation of all-cis-7,10,10,13,16,19-docosapentaenoic acid ethyl ester. Hydroperoxide distribution and volatile characterization. Eur. J. Lipid Sci. Technol. 2005;107:228–238. [Google Scholar]
- [23].Lee SH, Blair IA. Characterization of 4-oxo-2-nonenal as a novel product of lipid peroxidation. Chem. Res. Toxicol. 2000;13:698–702. doi: 10.1021/tx000101a. [DOI] [PubMed] [Google Scholar]
- [24].Blair IA. Lipid hydroperoxide-mediated DNA damage. Exp. Gerontol. 2001;36:1473–1481. doi: 10.1016/s0531-5565(01)00133-4. [DOI] [PubMed] [Google Scholar]
- [25].Olsen E, Vogt G, Saarem K, Greibrokk T, Nilsson A. Autoxidation of cod liver oil with tocopherol and ascorbyl palmitate. JAOCS. 2005;82:97–103. [Google Scholar]
- [26].Fodor G, Arnold R, Mohacsi T, Karle I, Flippen-Anderson J. A new role for l-ascorbic acid: Michael donor to alpha,beta-unsaturated carbonyl compounds. Tetrahedron. 1983;39:2137–2145. [Google Scholar]
- [27].Umano K, Shibamoto T. Analysis of acrolein from heated cooking oils and beef fat. J. Agric. Food Chem. 1987;35:909–912. [Google Scholar]
- [28].Fullana A, Carbonell-Barrachina AA, Sidhu S. Comparison of volatile aldehydes present in the cooking fumes of extra virgin olive, olive, and canola oils. J. Agric. Food Chem. 2004;52:5207–5214. doi: 10.1021/jf035241f. [DOI] [PubMed] [Google Scholar]
- [29].Li C, Kimura F, Endo Y, Maruyama C, Fujimoto K. Deterioration of diacylglycerol- and triacylglycerol-rich oils during frying of potatoes. Eur. J. Lipid Sci. Technol. 2005;107:173–179. [Google Scholar]
- [30].Ho S, Yu J, Chu K, Yeung L. Carbonyl emissions from commercial cooking sources in Hong Kong. J. Air Waste Management Assoc. 2006;56:1091–1098. doi: 10.1080/10473289.2006.10464532. [DOI] [PubMed] [Google Scholar]
- [31].Gao YT, Blot WJ, Zheng W, Ershow AG, et al. Lung cancer among Chinese women. Int. J. Cancer. 1987;40:604–609. doi: 10.1002/ijc.2910400505. [DOI] [PubMed] [Google Scholar]
- [32].Shields PG, Xu GX, Blot WJ, Fraumeni JF, Jr., et al. Mutagens from heated Chinese and U.S. cooking oils. J. Natl. Cancer Inst. 1995;87:836–841. doi: 10.1093/jnci/87.11.836. [DOI] [PubMed] [Google Scholar]
- [33].Uchida K, Kanematsu M, Morimitsu Y, Osawa T, et al. Acrolein is a product of lipid peroxidation reaction. Formation of free acrolein and its conjugate with lysine residues in oxidized low density lipoproteins. J. Biol. Chem. 1998;273:16058–16066. doi: 10.1074/jbc.273.26.16058. [DOI] [PubMed] [Google Scholar]
- [34].Alary J, Debrauwer L, Fernandez Y, Cravedi JP, et al. 1,4-Dihydroxynonene mercapturic acid, the major end metabolite of exogenous 4-hydroxy-2-nonenal, is a physiological component of rat and human urine. Chem. Res. Toxicol. 1998;11:130–135. doi: 10.1021/tx970139w. [DOI] [PubMed] [Google Scholar]
- [35].Roethig HJ, Zedler BK, Kinser RD, Feng S, et al. Short-term clinical exposure evaluation of a second-generation electrically heated cigarette smoking system. J. Clin. Pharmacol. 2007;47:518–530. doi: 10.1177/0091270006297686. [DOI] [PubMed] [Google Scholar]
- [36].Spiteller G. Lipid peroxidation in aging and age-dependent diseases. Exp. Gerontol. 2001;36:1425–1457. doi: 10.1016/s0531-5565(01)00131-0. [DOI] [PubMed] [Google Scholar]
- [37].Swanson K, Madden M, Ghio A. Biodiesel exhaust: The need for health effects research. Environ. Health Perspect. 2007;115:496–499. doi: 10.1289/ehp.9631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Graboski M, McCormick R. Combustion of fat and vegetable oil derived fuels in diesel engines. Progr. Energy Combust. Sci. 1998;24:125–164. [Google Scholar]
- [39].Anonymous, United States Environmental Protection Agency A Comprehensive Analysis of Biodiesel Impacts on Exhaust Emissions, Draft Technical Report. 2002 www.epa.gov/otaq/models/analysis/biodsl/p02001.pdf.
- [40].Turrio-Baldassarri L, Battistelli CL, Conti L, Crebelli R, et al. Emission comparison of urban bus engine fueled with diesel oil and ,biodiesel’ blend. Sci. Total Environ. 2004;327:147–162. doi: 10.1016/j.scitotenv.2003.10.033. [DOI] [PubMed] [Google Scholar]
- [41].Hunter I, Potter E. Microdetermination of volatile aldehydes. Anal. Chem. 1958;30:293–295. [Google Scholar]
- [42].Roth H, Blaschke G. Pharmazeutische Analytik. Thieme; Stuttgart and New York: 1981. [Google Scholar]
- [43].Ballance P. Production of volatile compounds related to the flavor of foods from the Strecker degradation of methionine. J. Sci. Food Agric. 1961;12:532–536. [Google Scholar]
- [44].Ishizuka S, Sakurai H, Ishii K. Reaction products produced from the reaction of dehydroascorbic acid with methionine. Nihon Daigaku Nojuigakubu Gijutsu Kenkyu Hokuku. 1979;36:111–120. CAN 190:202457. [Google Scholar]
- [45].Alarcon R. Formation of acrolein from various amino acids and polyamines under degradation at 100 C. Environ. Res. 1976;12:317–326. doi: 10.1016/0013-9351(76)90041-4. [DOI] [PubMed] [Google Scholar]
- [46].Self R, Rolley H, Joyce A. Some volatile compounds from cooked potatoes. J. Sci. Food Agric. 1963;14:8–14. [Google Scholar]
- [47].Di R, Kim J, Martin MN, Leustek T, et al. Enhancement of the primary flavor compound methional in potato by increasing the level of soluble methionine. J. Agric. Food Chem. 2003;51:5695–5702. doi: 10.1021/jf030148c. [DOI] [PubMed] [Google Scholar]
- [48].Vasilyev N, Williams T, Brennan ML, Unzek S, et al. Myeloperoxidase-generated oxidants modulate left ventricular remodeling but not infarct size after myocardial infarction. Circulation. 2005;112:2812–2820. doi: 10.1161/CIRCULATIONAHA.105.542340. [DOI] [PubMed] [Google Scholar]
- [49].Li J, Ho CT. Generation of aldehydes from Maillard reactions of glucose and amino acids. Special Publication-Royal Society of Chemistry. 2005;300:213–218. [Google Scholar]
- [50].Loefller G, Petrides P. Biochemie und Pathobiochemie. Springer; Berlin: 1998. [Google Scholar]
- [51].Hoet PH, Nemery B. Polyamines in the lung: polyamine uptake and polyamine-linked pathological or toxicological conditions. Am. J. Physiol. 2000;278:L417–433. doi: 10.1152/ajplung.2000.278.3.L417. [DOI] [PubMed] [Google Scholar]
- [52].Shaw GG, Pateman AJ. The regional distribution of the polyamines spermidine and spermine in brain. J. Neurochem. 1973;20:1225–1230. doi: 10.1111/j.1471-4159.1973.tb00091.x. [DOI] [PubMed] [Google Scholar]
- [53].O’Brien PJ, Siraki AG, Shangari N. Aldehyde sources, metabolism, molecular toxicity mechanisms, and possible effects on human health. Crit. Rev. Toxicol. 2005;35:609–662. doi: 10.1080/10408440591002183. [DOI] [PubMed] [Google Scholar]
- [54].Tabor CW, Tabor H, Bachrach U. Identification of the aminoaldehydes produced by the oxidation of spermine and spermidine with purified plasma amine oxidase. J. Biol. Chem. 1964;239:2194–2203. [PubMed] [Google Scholar]
- [55].Houen G, Bock K, Jensen A. HPLC and NMR investigation of the serum amine oxidase catalyzed oxidation of polyamines. Acta Chem. Scand. 1994;48:52–60. doi: 10.3891/acta.chem.scand.48-0052. [DOI] [PubMed] [Google Scholar]
- [56].Lee Y, Sayre LM. Reaffirmation that metabolism of polyamines by bovine plasma amine oxidase occurs strictly at the primary amino termini. J. Biol. Chem. 1998;273:19490–19494. doi: 10.1074/jbc.273.31.19490. [DOI] [PubMed] [Google Scholar]
- [57].Sakata K, Kashiwagi K, Sharmin S, Ueda S, Igarashi K. Acrolein produced from polyamines as one of the uraemic toxins. Biochem. Soc. Trans. 2003;31:371–374. doi: 10.1042/bst0310371. [DOI] [PubMed] [Google Scholar]
- [58].Lieberman MW, Barrios R, Carter BZ, Habib GM, et al. gamma-Glutamyl transpeptidase. What does the organization and expression of a multipromoter gene tell us about its functions? Am. J. Pathol. 1995;147:1175–1185. [PMC free article] [PubMed] [Google Scholar]
- [59].Josch C, Klotz LO, Sies H. Identification of cytosolic leucyl aminopeptidase (EC 3.4.11.1) as the major cysteinylglycine-hydrolysing activity in rat liver. Biol. Chem. 2003;384:213–218. doi: 10.1515/BC.2003.023. [DOI] [PubMed] [Google Scholar]
- [60].Kaye CM. Biosynthesis of mercapturic acids from allyl alcohol, allyl esters and acrolein. Biochem. J. 1973;134:1093–1101. doi: 10.1042/bj1341093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Patel JM, Wood JC, Leibman KC. The biotransformation of allyl alcohol and acrolein in rat liver and lung preparations. Drug Metab. Dispos. 1980;8:305–308. [PubMed] [Google Scholar]
- [62].Parent RA, Paust DE, Schrimpf MK, Talaat RE, et al. Metabolism and distribution of [2,3-14C]acrolein in Sprague-Dawley rats. II. Identification of urinary and fecal metabolites. Toxicol. Sci. 1998;43:110–120. doi: 10.1006/toxs.1998.2462. [DOI] [PubMed] [Google Scholar]
- [63].Yang Y, Yang Y, Trent MB, He N, et al. Glutathione-S-transferase A4-4 modulates oxidative stress in endothelium: possible role in human atherosclerosis. Atherosclerosis. 2004;173:211–221. doi: 10.1016/j.atherosclerosis.2003.12.023. [DOI] [PubMed] [Google Scholar]
- [64].Berhane K, Widersten M, Engstrom A, Kozarich JW, Mannervik B. Detoxication of base propenals and other alpha, beta-unsaturated aldehyde products of radical reactions and lipid peroxidation by human glutathione transferases. Proc. Natl. Acad. Sci. USA. 1994;91:1480–1484. doi: 10.1073/pnas.91.4.1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Lemke TL, Williams DA. Foye’s Principles of Medicinal Chemistry. Lippincott Williams and Wilkens, Wolters Kluwer; Baltimore and Philadelphia: 2008. pp. 253–326. [Google Scholar]
- [66].Palma S, Cornetta T, Padua L, Cozzi R, et al. Influence of glutathione S-transferase polymorphisms on genotoxic effects induced by tobacco smoke. Mutat. Res. 2007;633:1–12. doi: 10.1016/j.mrgentox.2007.03.014. [DOI] [PubMed] [Google Scholar]
- [67].Pal A, Hu X, Zimniak P, Singh SV. Catalytic efficiencies of allelic variants of human glutathione S-transferase Pi in the glutathione conjugation of alpha, beta-unsaturated aldehydes. Cancer Lett. 2000;154:39–43. doi: 10.1016/s0304-3835(00)00390-6. [DOI] [PubMed] [Google Scholar]
- [68].Park SB, Osterloh JD, Vamvakas S, Hashmi M, et al. Flavin-containing monooxygenase-dependent stereoselective S-oxygenation and cytotoxicity of cysteine S-conjugates and mercapturates. Chem. Res. Toxicol. 1992;5:193–201. doi: 10.1021/tx00026a008. [DOI] [PubMed] [Google Scholar]
- [69].Hashmi M, Vamvakas S, Anders MW. Bioactivation mechanism of S-(3-oxopropyl)-N-acetyl-L-cysteine, the mercapturic acid of acrolein. Chem. Res. Toxicol. 1992;5:360–365. doi: 10.1021/tx00027a007. [DOI] [PubMed] [Google Scholar]
- [70].Horvath JJ, Witmer CM, Witz G. Nephrotoxicity of the 1:1 acrolein-glutathione adduct in the rat. Toxicol. Appl. Pharmacol. 1992;117:200–207. doi: 10.1016/0041-008x(92)90238-n. [DOI] [PubMed] [Google Scholar]
- [71].Cannon J, Linke CA, Cos LR. Cyclophosphamide-associated carcinoma of urothelium: modalities for prevention. Urology. 1991;38:413–416. doi: 10.1016/0090-4295(91)80228-y. [DOI] [PubMed] [Google Scholar]
- [72].Williams DE, Hale SE, Muerhoff AS, Masters BS. Rabbit lung flavin-containing monooxygenase. Purification, characterization, and induction during pregnancy. Mol. Pharmacol. 1985;28:381–390. [PubMed] [Google Scholar]
- [73].Whetstine JR, Yueh MF, McCarver DG, Williams DE, et al. Ethnic differences in human flavin-containing monooxygenase 2 (FMO2) polymorphisms: detection of expressed protein in African-Americans. Toxicol. Appl. Pharmacol. 2000;168:216–224. doi: 10.1006/taap.2000.9050. [DOI] [PubMed] [Google Scholar]
- [74].Sanduja R, Ansari GA, Boor PJ. 3-Hydroxypropylmer-capturic acid: a biologic marker of exposure to allylic and related compounds. J. Appl. Toxicol. 1989;9:235–238. doi: 10.1002/jat.2550090406. [DOI] [PubMed] [Google Scholar]
- [75].Linhart I, Frantik E, Vodickova L, Vosmanska M, et al. Biotransformation of acrolein in rat: excretion of mercapturic acids after inhalation and intraperitoneal injection. Toxicol. Appl. Pharmacol. 1996;136:155–160. doi: 10.1006/taap.1996.0019. [DOI] [PubMed] [Google Scholar]
- [76].Roth H, Eger K, Troschuetz R. Pharmazeutische Chemie II: Arzneistoffanalyse: Reaktivitaet, Stabilitaet, Analytik. Georg Thieme Verlag; Stuttgart and New York: 1985. [Google Scholar]
- [77].Eger K, Folkers G, Zimmerman W, Schmidt R, Hiller W. Unexpected formation of a heterocyclic spiran in the Michael reaction of ascorbic acid with acrolein. J. Chem. Res. Synopses. 1987;9:277. [Google Scholar]
- [78].Eger K, Schmidt M, Albert K, Schmid J. Formation of heterocyclic products in Michael reactions of ascorbic acid with alpha, beta-unsaturated compounds. 3. Structural analysis of Michael adducts of ascorbic acid by carbon-13 NMR spectroscopy in the solid state. J. Heterocycl. Chem. 1992;29:1225–1228. [Google Scholar]
- [79].Eger K, Schmidt R. Heterocyclizations of ascorbic acid with alpha,beta-unsaturated aldehydes and ketones. II. Michael reactions of ascorbic acid with acrolein derivatives. Archiv der Pharmazie. 1989;322:127–132. [Google Scholar]
- [80].Saxena P, Saxena AK, Cui XL, Obrenovich M, et al. Transition metal-catalyzed oxidation of ascorbate in human cataract extracts: possible role of advanced glycation end products. Invest. Ophthalmol. Vis. Sci. 2000;41:1473–1481. [PubMed] [Google Scholar]
- [81].Halliwell B, Gutteridge J. Free Radicals in Biology and Medicine. Oxford University Press; London: 1999. [Google Scholar]
- [82].Nardini M, Finkelstein EI, Reddy S, Valacchi G, et al. Acrolein-induced cytotoxicity in cultured human bronchial epithelial cells. Modulation by alpha-tocopherol and ascorbic acid. Toxicology. 2002;170:173–185. doi: 10.1016/s0300-483x(01)00540-6. [DOI] [PubMed] [Google Scholar]
- [83].Arai H, Uchida K, Nakamura K. Effect of ascorbate on acrolein modification of very low density lipoprotein and uptake of oxidized apolipoprotein e by hepatocytes. Biosci. Biotechnol. Biochem. 2005;69:1760–1762. doi: 10.1271/bbb.69.1760. [DOI] [PubMed] [Google Scholar]
- [84].Logan MP, Parker S, Shi R. Glutathione and ascorbic acid enhance recovery of Guinea pig spinal cord white matter following ischemia and acrolein exposure. Pathobiology. 2005;72:171–178. doi: 10.1159/000086786. [DOI] [PubMed] [Google Scholar]
- [85].Chung FL, Young R, Hecht SS. Formation of cyclic 1,N2-propanodeoxyguanosine adducts in DNA upon reaction with acrolein or crotonaldehyde. Cancer Res. 1984;44:990–995. [PubMed] [Google Scholar]
- [86].Stein S, Lao Y, Yang IY, Hecht SS, Moriya M. Genotoxicity of acetaldehyde- and crotonaldehyde-induced 1,N2-propanodeoxyguanosine DNA adducts in human cells. Mutat. Res. 2006;608:1–7. doi: 10.1016/j.mrgentox.2006.01.009. [DOI] [PubMed] [Google Scholar]
- [87].Yang IY, Chan G, Miller H, Huang Y, et al. Mutagenesis by acrolein-derived propanodeoxyguanosine adducts in human cells. Biochemistry. 2002;41:13826–13832. doi: 10.1021/bi0264723. [DOI] [PubMed] [Google Scholar]
- [88].Nath RG, Chung FL. Detection of exocyclic 1,N2-propanodeoxyguanosine adducts as common DNA lesions in rodents and humans. Proc. Natl. Acad. Sci. USA. 1994;91:7491–7495. doi: 10.1073/pnas.91.16.7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Nath RG, Ocando JE, Guttenplan JB, Chung FL. 1,N2-propanodeoxyguanosine adducts: potential new biomarkers of smoking-induced DNA damage in human oral tissue. Cancer Res. 1998;58:581–584. [PubMed] [Google Scholar]
- [90].Feng Z, Hu W, Hu Y, Tang MS. Acrolein is a major cigarette-related lung cancer agent: Preferential binding at p53 mutational hotspots and inhibition of DNA repair. Proc. Natl. Acad. Sci. USA. 2006;103:15404–15409. doi: 10.1073/pnas.0607031103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Hecht SS. Smoking and lung cancer-a new role for an old toxicant? Proc. Natl. Acad. Sci. USA. 2006;103:15725–15726. doi: 10.1073/pnas.0607811103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Beauchamp RO, Jr., Andjelkovich DA, Kligerman AD, Morgan KT, Heck HD. A critical review of the literature on acrolein toxicity. Crit. Rev. Toxicol. 1985;14:309–380. doi: 10.3109/10408448509037461. [DOI] [PubMed] [Google Scholar]
- [93].Kehrer JP, Biswal SS. The molecular effects of acrolein. Toxicol. Sci. 2000;57:6–15. doi: 10.1093/toxsci/57.1.6. [DOI] [PubMed] [Google Scholar]
- [94].Lopachin RM, Barber DS, Geohagen BC, Gavin T, et al. Structure-toxicity analysis of type-2 alkenes: in vitro neurotoxicity. Toxicol. Sci. 2007;95:136–146. doi: 10.1093/toxsci/kfl127. [DOI] [PubMed] [Google Scholar]
- [95].Lambert C, Li J, Jonscher K, Yang TC, et al. Acrolein inhibits cytokine gene expression by alkylating cysteine and arginine residues in the NF-kappaB1 DNA binding domain. J. Biol. Chem. 2007;282:19666–19675. doi: 10.1074/jbc.M611527200. [DOI] [PubMed] [Google Scholar]
- [96].Furuhata A, Nakamura M, Osawa T, Uchida K. Thiolation of protein-bound carcinogenic aldehyde. An electrophilic acrolein-lysine adduct that covalently binds to thiols. J. Biol. Chem. 2002;277:27919–27926. doi: 10.1074/jbc.M202794200. [DOI] [PubMed] [Google Scholar]
- [97].Kuhla B, Haase C, Flach K, Luth HJ, et al. Effect of pseudophosphorylation and cross-linking by lipid peroxidation and advanced glycation end product precursors on tau aggregation and filament formation. J. Biol. Chem. 2007;282:6984–6991. doi: 10.1074/jbc.M609521200. [DOI] [PubMed] [Google Scholar]
- [98].Calingasan NY, Uchida K, Gibson GE. Protein-bound acrolein: a novel marker of oxidative stress in Alzheimer’s disease. J. Neurochem. 1999;72:751–756. doi: 10.1046/j.1471-4159.1999.0720751.x. [DOI] [PubMed] [Google Scholar]
- [99].Shamoto-Nagai M, Maruyama W, Hashizume Y, Yoshida M, et al. In parkinsonian substantia nigra, alpha-synuclein is modified by acrolein, a lipid-peroxidation product, and accumulates in the dopamine neurons with inhibition of proteasome activity. J. Neural. Transm. 2007 doi: 10.1007/s00702-007-0789-2. DOI: 10.1007/s00702-007-0789-2. [DOI] [PubMed] [Google Scholar]
- [100].Baldwin AS., Jr. The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu. Rev. Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- [101].Orlowski RZ, Baldwin AS., Jr. NF-kappaB as a therapeutic target in cancer. Trends Mol. Med. 2002;8:385–389. doi: 10.1016/s1471-4914(02)02375-4. [DOI] [PubMed] [Google Scholar]
- [102].Lambert C, McCue J, Portas M, Ouyang Y, et al. Acrolein in cigarette smoke inhibits T-cell responses. J. Allergy Clin. Immunol. 2005;116:916–922. doi: 10.1016/j.jaci.2005.05.046. [DOI] [PubMed] [Google Scholar]
- [103].Matthews JR, Kaszubska W, Turcatti G, Wells TN, Hay RT. Role of cysteine62 in DNA recognition by the P50 subunit of NF-kappa B. Nucleic Acids Res. 1993;21:1727–1734. doi: 10.1093/nar/21.8.1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Nishi T, Shimizu N, Hiramoto M, Sato I, et al. Spatial redox regulation of a critical cysteine residue of NF-kappa B in vivo. J. Biol. Chem. 2002;277:44548–44556. doi: 10.1074/jbc.M202970200. [DOI] [PubMed] [Google Scholar]
- [105].Chen FE, Huang DB, Chen YQ, Ghosh G. Crystal structure of p50/p65 heterodimer of transcription factor NF-kappaB bound to DNA. Nature. 1998;391:410–413. doi: 10.1038/34956. [DOI] [PubMed] [Google Scholar]
- [106].Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Ann. Rev. Pharmacol. Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- [107].Levonen AL, Landar A, Ramachandran A, Ceaser EK, et al. Cellular mechanisms of redox cell signalling: role of cysteine modification in controlling antioxidant defences in response to electrophilic lipid oxidation products. Biochem. J. 2004;378:373–382. doi: 10.1042/BJ20031049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Singh A, Misra V, Thimmulappa RK, Lee H, et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006;3:e420. doi: 10.1371/journal.pmed.0030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Wu CC, Hsieh CW, Lai PH, Lin JB, et al. Upregulation of endothelial heme oxygenase-1 expression through the activation of the JNK pathway by sublethal concentrations of acrolein. Toxicol. Appl. Pharmacol. 2006;214:244–252. doi: 10.1016/j.taap.2005.12.013. [DOI] [PubMed] [Google Scholar]
- [110].Tirumalai R, Rajesh Kumar T, Mai KH, Biswal S. Acrolein causes transcriptional induction of phase II genes by activation of Nrf2 in human lung type II epithelial (A549) cells. Toxicol. Lett. 2002;132:27–36. doi: 10.1016/s0378-4274(02)00055-3. [DOI] [PubMed] [Google Scholar]
- [111].Kang MI, Kobayashi A, Wakabayashi N, Kim SG, Yamamoto M. Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proc. Natl. Acad. Sci. USA. 2004;101:2046–2051. doi: 10.1073/pnas.0308347100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Wakabayashi N, Dinkova-Kostova AT, Holtzclaw WD, Kang MI, et al. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc. Natl. Acad. Sci. USA. 2004;101:2040–2045. doi: 10.1073/pnas.0307301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Eggler AL, Liu G, Pezzuto JM, van Breemen RB, Mesecar AD. Modifying specific cysteines of the electrophile-sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proc. Natl. Acad. Sci. USA. 2005;102:10070–10075. doi: 10.1073/pnas.0502402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Hong F, Freeman ML, Liebler DC. Identification of sensor cysteines in human Keap1 modified by the cancer chemopreventive agent sulforaphane. Chem. Res. Toxicol. 2005;18:1917–1926. doi: 10.1021/tx0502138. [DOI] [PubMed] [Google Scholar]
- [115].Hong F, Sekhar KR, Freeman ML, Liebler DC. Specific patterns of electrophile adduction trigger Keap1 ubiquitination and Nrf2 activation. J. Biol. Chem. 2005;280:31768–31775. doi: 10.1074/jbc.M503346200. [DOI] [PubMed] [Google Scholar]
- [116].Dinkova-Kostova AT, Holtzclaw WD, Wakabayashi N. Keap1, the sensor for electrophiles and oxidants that regulates the phase 2 response, is a zinc metalloprotein. Biochemistry. 2005;44:6889–6899. doi: 10.1021/bi047434h. [DOI] [PubMed] [Google Scholar]
- [117].Sakurai T, Kanayama M, Shibata T, Itoh K, et al. Ebselen, a seleno-organic antioxidant, as an electrophile. Chem. Res. Toxicol. 2006;19:1196–1204. doi: 10.1021/tx0601105. [DOI] [PubMed] [Google Scholar]
- [118].Dinkova-Kostova AT, Holtzclaw WD, Kensler TW. The role of Keap1 in cellular protective responses. Chem. Res. Toxicol. 2005;18:1779–1791. doi: 10.1021/tx050217c. [DOI] [PubMed] [Google Scholar]
- [119].Satoh T, Okamoto SI, Cui J, Watanabe Y, et al. Activation of the Keap1/Nrf2 pathway for neuroprotection by electrophilic phase II inducers. Proc. Natl. Acad. Sci. USA. 2006;103:768–773. doi: 10.1073/pnas.0505723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Zhang ZY. Chemical and mechanistic approaches to the study of protein tyrosine phosphatases. Acc. Chem. Res. 2003;36:385–392. doi: 10.1021/ar020122r. [DOI] [PubMed] [Google Scholar]
- [121].Jackson MD, Denu JM. Molecular reactions of protein phosphatases-insights from structure and chemistry. Chem. Rev. 2001;101:2313–2340. doi: 10.1021/cr000247e. [DOI] [PubMed] [Google Scholar]
- [122].Denu JM, Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry. 1998;37:5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- [123].Seiner DR, Labutti JN, Gates KS. Kinetics and mechanism of protein tyrosine phosphatase 1B inactivation by acrolein. Chem. Res. Toxicol. 2007;20:1315–1320. doi: 10.1021/tx700213s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Takeuchi K, Kato M, Suzuki H, Akhand AA, et al. Acrolein induces activation of the epidermal growth factor receptor of human keratinocytes for cell death. J. Cell. Biochem. 2001;81:679–688. doi: 10.1002/jcb.1105. [DOI] [PubMed] [Google Scholar]
- [125].Tanel A, Averill-Bates DA. The aldehyde acrolein induces apoptosis via activation of the mitochondrial pathway. Biochim. Biophys. Acta. 2005;1743:255–267. doi: 10.1016/j.bbamcr.2004.11.007. [DOI] [PubMed] [Google Scholar]
- [126].Finkelstein EI, Nardini M, van der Vliet A. Inhibition of neutrophil apoptosis by acrolein: a mechanism of tobacco-related lung disease? Am. J. Physiol. 2001;281:L732–739. doi: 10.1152/ajplung.2001.281.3.L732. [DOI] [PubMed] [Google Scholar]
- [127].Kern JC, Kehrer JP. Acrolein-induced cell death: a caspase-influenced decision between apoptosis and oncosis/necrosis. Chem. Biol. Interact. 2002;139:79–95. doi: 10.1016/s0009-2797(01)00295-2. [DOI] [PubMed] [Google Scholar]
- [128].Tanel A, Averill-Bates DA. Activation of the death receptor pathway of apoptosis by the aldehyde acrolein. Free Radical Biol. Med. 2007;42:798–810. doi: 10.1016/j.freeradbiomed.2006.12.009. [DOI] [PubMed] [Google Scholar]
- [129].Lavrik IN, Golks A, Krammer PH. Caspases: pharmacological manipulation of cell death. J. Clin. Invest. 2005;115:2665–2672. doi: 10.1172/JCI26252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 1999;341(Pt 2):233–249. [PMC free article] [PubMed] [Google Scholar]
- [131].Crompton M, Virji S, Doyle V, Johnson N, Ward JM. The mitochondrial permeability transition pore. Biochem. Soc. Symp. 1999;66:167–179. doi: 10.1042/bss0660167. [DOI] [PubMed] [Google Scholar]
- [132].Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004;287:C817–833. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- [133].Nicotera P, Leist M, Ferrando-May E. Intracellular ATP, a switch in the decision between apoptosis and necrosis. Toxicol. Lett. 1998;102-103:139–142. doi: 10.1016/s0378-4274(98)00298-7. [DOI] [PubMed] [Google Scholar]
- [134].Mittl PR, Di Marco S, Krebs JF, Bai X, et al. Structure of recombinant human CPP32 in complex with the tetrapeptide acetyl-Asp-Val-Ala-Asp fluoromethyl ketone. J. Biol. Chem. 1997;272:6539–6547. doi: 10.1074/jbc.272.10.6539. [DOI] [PubMed] [Google Scholar]
- [135].Chandra J, Samali A, Orrenius S. Triggering and modulation of apoptosis by oxidative stress. Free Radical Biol. Med. 2000;29:323–333. doi: 10.1016/s0891-5849(00)00302-6. [DOI] [PubMed] [Google Scholar]
- [136].Landar A, Shiva S, Levonen AL, Oh JY, et al. Induction of the permeability transition and cytochrome c release by 15-deoxy-Delta 12,14-prostaglandin J2 in nitachondria. Biochem. J. 2006;394:185–195. doi: 10.1042/BJ20051259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Han B, Stevens JF, Maier CS. Design, synthesis, and application of a hydrazide-functionalized isotope-coded affinity tag for the quantification of oxylipid-protein conjugates. Anal. Chem. 2007;79:3342–3354. doi: 10.1021/ac062262a. [DOI] [PubMed] [Google Scholar]
- [138].Stratton K, Shetty P, Wallace R, Bondurant S. Clearing the smoke: the science base for tobacco harm reduction-executive summary. Tob. Control. 2001;10:189–195. doi: 10.1136/tc.10.2.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Altet MN, Alcaide J, Plans P, Taberner JL, et al. Passive smoking and risk of pulmonary tuberculosis in children immediately following infection. A case-control study. Tuber. Lung Dis. 1996;77:537–544. doi: 10.1016/s0962-8479(96)90052-0. [DOI] [PubMed] [Google Scholar]
- [140].Alcaide J, Altet MN, Plans P, Parron I, et al. Cigarette smoking as a risk factor for tuberculosis in young adults: a case-control study. Tuber. Lung Dis. 1996;77:112–116. doi: 10.1016/s0962-8479(96)90024-6. [DOI] [PubMed] [Google Scholar]
- [141].Miguez-Burbano MJ, Burbano X, Ashkin D, Pitchenik A, et al. Impact of tobacco use on the development of opportunistic respiratory infections in HIV seropositive patients on antiretroviral therapy. Addiction Biol. 2003;8:39–43. doi: 10.1080/1355621031000069864. [DOI] [PubMed] [Google Scholar]
- [142].Zhang JJ, Smith KR. Household air pollution from coal and biomass fuels in china: measurements, health impacts, and interventions. Environ. Health Perspect. 2007;115:848–855. doi: 10.1289/ehp.9479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].United States Environmental Protection Agency . Toxicological review of acrolein. Washington, DC: 2003. CAS Nr. 107-02-8. [Google Scholar]
- [144].Silbernagel S, Despopoulos A. Taschenatlas der Physiologie. Thieme; Stuttgart and New York: 1990. [Google Scholar]
- [145].Leikauf GD. Hazardous air pollutants and asthma. Environ. Health Perspect. 2002;110(Suppl 4):505–526. doi: 10.1289/ehp.02110s4505. [DOI] [PMC free article] [PubMed] [Google Scholar]