

Abstract
Autophagy is a highly conserved cellular process responsible for the degradation of long-lived proteins and organelles. Autophagy occurs at low levels under normal conditions, but is upregulated in response to stress such as nutrient deprivation, hypoxia, mitochondrial dysfunction, and infection. Upregulation of autophagy may be beneficial to the cell by recycling of proteins to generate free amino acids and fatty acids needed to maintain energy production, by removing damaged organelles, and by preventing accumulation of protein aggragates. In contrast, there is evidence that enhanced autophagy can contribute to cell death, possibly through excessive self-digestion. In the heart, autophagy is essential role for maintaining cellular homeostasis under normal conditions and increased autophagy can be seen in conditions of starvation, ischemia/reperfusion, and heart failure. However, the functional significance of autophagy in heart disease is unclear and controversial. Here, we review the literature and discuss the evidence that autophagy can have both beneficial and detrimental roles in the myocardium depending on the level of autophagy, and discuss potential mechanisms by which autophagy provides protection in cells.
Keywords: autophagy, cell death, mitochondria, Beclin 1, ischemia/reperfusion, heart failure
Introduction
Autophagy is an evolutionarily conserved process involved in the degradation of long-lived proteins and organelles. Cytoplasmic material is sequestered by an autophagosome and subsequently delivered to the lysosome where it is degraded by lysosomal proteases. There are three forms of autophagy; chaperone-mediated autophagy, microautophagy and macroautophagy. Macroautophagy is the most common form of autophagy in mammalian cells and will be herein referred to as autophagy. Autophagy occurs at low levels under normal conditions, but is also upregulated in response to stress such as nutrient deprivation, hypoxia, mitochondrial dysfunction, and infection [1–4]. Autophagy can promote cell survival by generating free amino acids and fatty acids required to maintain function during nutrient-limiting conditions, or by removing damaged organelles and intracellular pathogens. However, autophagy might also promote cell death through excessive self-digestion and degradation of essential cellular consituents. Recent studies have also reported interactions between the autophagic and apoptotic pathways [5, 6].
Autophagy is an important process in the heart. It occurs at low basal levels under normal conditions and is important for the turnover of organelles [7]. Many studies have demonstrated that autophagy is upregulated in the heart in response to stress such as ischemia/reperfusion [4, 8–12]. Increased numbers of autophagosomes are a prominent feature in many cardiovascular diseases such as cardiac hypertrophy and heart failure [13–18]. However, the functional role of the enhanced autophagy in the diseased heart is unclear and studies have yielded conflicting results regarding the role of autophagy in the heart. It is controversial whether the increased number of autophagosomes in dying cells contributes directly to cell death or represent an attempt to prevent it. In many studies, autophagic cell death has been defined by morphologic criteria and it has not been demonstrated that autophagy directly contributed to cell death. However, recent genetic studies disrupting the autophagic pathway have yielded increasing insight into the role of autophagy in cell survival and death in the heart. In this review, we discuss the emerging evidence that autophagy have a dual role in the heart where it can protect against or contribute to cell death depending on the stimulus.
Molecular Mechanism of Autophagy
Activation of the autophagsomal-lysosomal pathway starts with the formation of the autophagosome which involves a series of steps. The exact origin of the membrane is not clear but is thought to be from the endoplasmic reticulum in mammalian cells [19]. The induction step of autophagy involves isolation of a small isolation membrane (also called phagophore) to which necessary proteins will be recruited to form the mature autophagosome (Fig. 1). Electron microscopy studies suggests that isolation membranes appear to elongate as they curve to surround cytoplasmic material to be sequestered and then finally close to form autophagosomes with a diameter of 0.5–1.5 μm [15, 20]. This process is regulated by a system of evolutionarily conserved proteins (Atg proteins). Beclin 1 (Atg6) is part of a Phosphoinositide 3-kinase (PI3-K) complex and seems to play an important role during the initial steps of autophagosome formation by mediating the localization of other Atg proteins to the isolation membrane [21]. In addition, synthesis of the autophagosome requires the Atg5-Atg12 and LC3(Atg8)-phosphatidyl ethanolamine (PE) conjugation systems. The covalent conjugation between Atg12 and Atg5 is similar to that of ubiquitination where Atg12 is first activated by Atg7, an ubiquitin like E1 enzyme, and then transferred to Atg10 which functions like an E2 exzyme. Atg12 is then covalently conjugated to Atg5 at a lysine residue and Atg10 is released [22]. Finally, the Atg12-Atg5 complex interacts non-covalently with Atg16, and this complex then initiat es the elongation of the membrane by recruiting LC3-PE [23–25]. LC3 is synthesized as a larger precursor and is cleaved by the cysteine protease Atg4 to LC3-I which exposes a C-terminal glycine residue [26]. The processed LC3, like Atg12, is activated by Atg7 and then transferred to the E2-like protein Atg3 which catalyzes the covalent conjugation with PE [27, 28]. The recruitment of LC3-PE to the isolation membrane is dependent on Atg-5-Atg12 [23, 29]. The Atg5-Atg12-Atg16 complex dissociates from the mature autophagosome, as well as the LC3-PE located on the outer membrane of the autophagosome. Only the uncoated autophagosome can fuse with the lysosome [23, 26].
Figure 1.

Schematic model of autophagy. Induction of autophagy starts with the formation of the initial isolation membrane or phagophore and the subsequent recruitment of essential Atg proteins to start the elongation of the membrane. The membrane surrounds proteins or organelles in the cytosol to be sequestered and then fuses with the lysosome to form the autolysosome where the content is degraded.
Autophagy in the Heart
Autophagy is an important process in the heart and a defect in this process can be detrimental to the heart. For instance, conditional deletion of atg5 in the heart causes a disruption in autophagy and results in accumulation of abnormal organelles and rapid development of cardiac dysfunction [30]. Moreover, Danon’s cardiomyopathy is due to a deficiency in the lysosomal protein Lamp-2 which causes a disruption in the autophagosome-lysosome (Atg-Lys) pathway and accumulation of autophagosomes in the cardiac myocytes [18, 31]. Autophagy is dramatically enhanced in hearts of fasting mice, suggesting that autophagy is also an important survival response to starvation[32]. It is clear that a functional Atg-Lys pathway is essential for cellular homeostasis in the heart and that a defect in this pathway will result in adverse effects for the heart. In contrast, there are reports that upregulation of autophagy can be detrimental to the heart. Increased numbers of autophagosomes have been observed in cardiac tissues from patients with cardiovascular disorders such as dilated cardiomyopathy [33], aortic stenosis [34], and hibernating myocardium [35]. However, the functional significance of autophagy in those diseases is still not clear. Numerous autophagosomes are often seen in dying cells, but it is not clear whether autophagy directly contributed to cell death or was upregulated as an effort to prevent it.
Autophagy in Ischemia and Reperfusion
Increased autophagy has long been known to occur in hearts after ischemia and reperfusion. About 30 years ago, Sybers et al. observed an increase in autophagic vesicles after hypoxia with glucose deprivation combined with reoxygenation in buffer containing glucose in fetal mouse hearts in organ cultures [11]. In 1980, Decker and Widenthal reported that up to 40 min of ischemia and subsequent reperfusion caused upregulation of autophagy in Langendorff perfused rabbit hearts [8, 36]. They also noted that 60 min of ischemia resulted in large and likely dysfunctional lysosomes during reperfusion, suggesting that the prolonged ischemia impaired the autophagic-lysosomal pathway. They also reported that the increase in autophagy correlated with functional recovery and salvage of the myocardium after I/R, whereas extended ischemia, which seemed to impair the Atg-Lys pathway, correlated with irreversible damage and contractile dysfunction [8]. In addition, chronic ischemia was reported to induce autophagy and increase lysosomal activity in swine myocardium [37]. The same study found that areas of the heart with increased autophagy displayed fewer apoptotic cells, suggesting that induction of autophagy might have prevented apoptosis. The mTOR (mammalian target of rapamycin) has been identified as a negative regulator of autophagy in mammalian cells [38, 39]. Many studies have demonstrated that rapamycin is a potent stimulator of autophagy by inhibiting mTOR [39, 40], and rapamycin treatment has been shown to provide protection against I/R injury in Langendorff perfused rat hearts [41]. However, mTOR regulates multiple metabolic pathways in addition ot autophagy. Upregulation of autophagy has also been shown to be protective in vitro. Inhibition of autophagy in HL-1 myocytes caused increased cell death in response to simulated I/R (sI/R), whereas enhancement of the autophagic response was protective [4]. Similarly, Dosenko et al. reported that inhibiting autophagy in isolated cardiac myocytes during exposure to anoxia/reoxygenation caused an increase in cell death [42]. Moreover, Matsui et al. recently reported that glucose deprivation, a component of ischemia, resulted in upregulation of autophagy in isolated cardiac myocytes and that inhibition of autophagy enhanced glucose deprivation-mediated death [10]. These studies suggest that upregulation of autophagy during ischemia/reperfusion is cardioprotective and promotes survival of cells.
Some studies have reported that upregulation of autophagy promotes cell death during I/R. A study using RNAi against Beclin 1 or 3-methyladenine (3-MA) treatment to block autophagy found that this resulted in reduced cell death in isolated cardiac mocytes subjected to sI/R, suggesting that autophagy contributes to cell death [12]. Moreover, glucose deprivation of H9c2 cells, a cell line derived from rat cardiac myocytes, caused an increase in autophagosomes and inhibiting autophagy with 3-MA or LY294002 reduced cell death [43]. Although Matsui et al. found that autophagy was protective during ischemia, they observed that it switched to a detrimental role during reperfusion. They found that mice with heterozygous disruption of beclin 1 (beclin 1+/−), which exhibit reduced levels of autophagy during reperfusion, had decreased apoptosis and reduced infarct size compared to wild type mice [10]. Interestingly, it was recently reported that Beclin 1 contains a conserved pro-apoptotic BH3 domain [44, 45], suggesting that Beclin 1 might be an inducer of apoptosis. In support of this, reduced number of apoptotic cells is seen in beclin 1+/− hearts after I/R compared to wild type mice [10]. Clearly, more studies are needed to clarify the role of Beclin 1 in autophagy and apoptosis. These studies suggest that autophagy can have dual roles in the heart, and since autophagy is a degradation pathway, it is quite possible that constitutive and excessive autophagy could cause cell death by degrading too many essential proteins and organelles (Fig. 2). However, further studies are needed to elucidate under what conditions autophagy provides protection or cell death.
Figure 2.
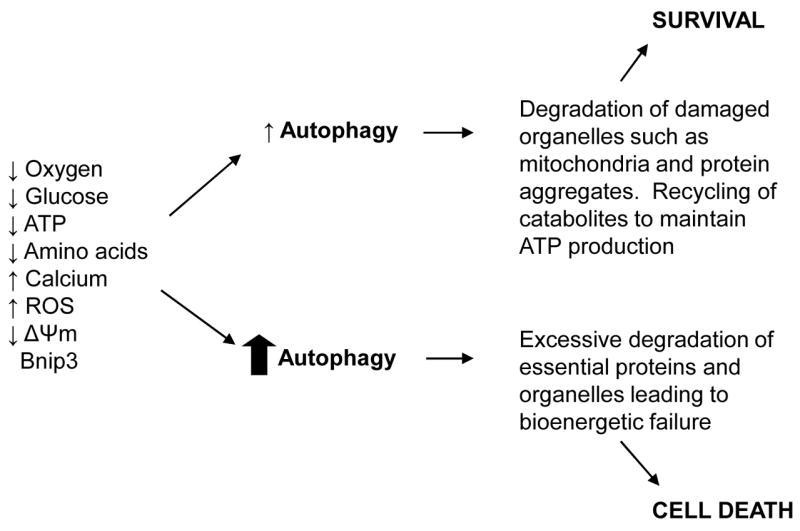
Exposure of cells to various stimuli leads to induction of autophagy which can be protective or detrimental to the cell. Low levels of autophagy can promote survival by degradation of intracellular contents to maintain ATP production and removal of damaged organelles, and protein aggregates. Excessive and long-term upregulation of autophagy eventually lead to destruction of essential proteins and organelles beyond a certain threshold leading to cell death.
Autophagy in Cardiac Hypertrophy and Heart Failure
β-adrenergic stimulation, which promotes apoptosis [46] and induces cardiac hypertrophy and heart failure [47], has been reported to inhibit autophagy [15]. Autophagy has also been shown to protect cells against β-adrenergic stimulation, where cardiac myocytes isolated from atg5 deficient mouse heart had increased sensitivity to isoproterenol stimulation compared to wild type cells [30]. Moreover, isoproterenol treatment for 7 days led to left ventricular dilation and cardiac dysfunction in autophagy deficient mice but not in wild type mice, suggesting that autophagy provides an important function in protecting cells against excessive β-adrenergic stimulation. Moreover, autophagy has been reported to be inhibited in the progression of cardiac hypertrophy [13, 15, 30]. For instance, Dammrich and Pfeifer observed that cardiac hypertrophy induced by aortic constriction led to inhibition of autophagy in cardiac myocytes undergoing hypertrophy and proposed that the inhibition of degradation was important in growing myocytes since autophagy-mediated protein degradation antagonizes cell growth [13]. Rapamycin, a potent activator of autophagy, was demonstrated to prevent cardiac hypertrophy induced by thyroid hormone treatment [48] or aortic banding [49]. McMullen et al. found that rapamycin treatment was even able to regress already established cardiac hypertrophy induced by pressure overload and improve cardiac function [50]. Although these studies did not directly investigate whether autophagy was upregulated, it is tempting to speculate that the induction of autophagy contributed to the protective effects of rapamycin in these models. It is possible that autophagy antagonizes cardiac hypertrophy by increasing protein degradation which would decrease cardiac mass. In addition, transverse aortic constriction (TAC)-induced hypertrophy reduced autophagy by week 1, and inhibition of autophagy by knockdown of atg7 using RNAi induced cardiac hypertrophy in isolated neonatal myocytes, and conditional deletion of atg5 in the heart resulted in increased cross-sectional area of the myocytes and heart-to-body weight ratio compared to wild type [30]. Interestingly, mice with conditional deletion of atg5 underwent the same degree of hypertrophy as wild type mice in response to TAC, suggesting that inhibition of basal autophagy is not required to yield an increase in cellular mass and that there might be alternative mechanisms present to compensate for the lack of autophagy in these mice.
In contrast, Zhu et al. found that increased autophagy in hypertrophied hearts played a role in the transition from hypertrophy to failure [16]. They reported that pressure-overload-induced hypertrophy caused upregulation of autophagy and that decreased autophagy in the beclin 1+/− mice correlated with decreased pathological remodeling, whereas enhancement of autophagy in Beclin 1 transgenic mice had diminished cardiac remodeling. The difference in the results is likely due to the differences in the mouse models used to study autophagy. The study by Nakai et al. used mice with cardiac specific deletion of atg5 which completely abrogates autophagy [30], whereas Zhu et al. used a mice heterozygous disruption of beclin 1 which results in reduced levels of autophagy in response to stress, and Beclin 1 transgenics which have enhanced levels of autophagy [16]. These studies are further complicated by the fact that Beclin 1 may also function as a pro-apoptotic BH3 molecule independent of its effect on autophagy [44, 45]. However, these studies suggest that the level of autophagy in the heart is an important factor in determining whether autophagy will be protective or detrimental.
Mechanisms of Cardioprotection
It is not clear exactly how autophagy provides protection against cell death, and several potential mechanisms have been proposed. Autophagy is used by cells to degrade organelles and it is possible that autophagy protects cells by removing damaged mitochondria which may be harmful to the cell. Damaged mitochondria can be dangerous to the cell by releasing proapoptotic factors such as cytochrome c which can activate apoptosis [51]. Therefore, removal of leaky mitochondria by autophagy may protect cells by preventing activation of apoptosis. Mitochondria sequestered inside autophagosomes are often seen in the myocardium after I/R. For instance, Sybers et al. noted numerous autophagosomes containing mitochondria in fetal hearts in organ culture after hypoxia/reoxygenation [11]. Decker and Wildenthal also observed that many autophagic vacuoles contained damaged mitochondria during reperfusion and proposed that autophagy is upregulated to remove damaged mitochondria [8]. In addition, we found many autophagosomes containing mitochondria in heart sections of Langendorff perfused rat hearts after I/R [52]. It has been reported that opening of the mitochondrial permeability transition pore (mPTP) triggered autophagy of damaged mitochondria in hepatocytes [3]. Since the mPTP opens when reperfusion is initiated after ischemia [53], it might serve as a signal to autophagosomes to sequester the mitochondria.
Another potential mechanism by which autophagy may promote survival is by maintaining energy homeostasis during ischemia (Fig. 2). Generation of ATP via oxidative phoshorylation is inhibited during ischemia and reduced ATP levels have been shown to induce autophagy [1, 10]. AMP kinase (AMPK) is involved in activating energy-generating pathways to maintain or restore ATP levels [54] and has been reported to upregulate autophagy [55–57]. AMPK is activated during ischemia and was recently shown to play a role in upregulaing autophagy during ischemia [10]. Degradation of proteins and organelles by the autophagic-lysosomal pathway generates free amino acids and fatty acids which can be used to maintain mitochondrial ATP production and protein synthesis and promote survival of cardiac cells. Matsui et al. found that glucose deprivation significantly reduced the cellular ATP content and upregulated autophagy in cardiac myocytes. In addition, treatment of cells with 3-MA to block autophagy during glucose deprivation exacerbated the reduction in cellular ATP and increased cell death, suggesting that autophagy promotes survival by maintaining ATP production during ischemia [10]. Another important metabolic benefit of autophagy is production of amino acids such as glutamate, which are necessary for regenerating NAD(P)H and glutathione synthesis, both essential for redox and cellular homeostasis.
Finally, it is possible that autophagy provides protection by degrading misfolded proteins that may be toxic to cells. Proteins are degraded by either the ubiquitin-proteasome system (UPS) or the autophagy-lysosome pathway. The UPS is responsible for the degradation of short-lived proteins [58], whereas autophagy regulates levels of long-lived proteins and organelles [7]. The UPS and autophagy are generally considered to be two separate degradation pathways, but recent studies have demonstrated that there may be cross-talk between the two pathways. Induction of autophagy has been reported to attenuate the toxicity induced by proteasome inhibition [59], suggesting that induction of autophagy is protective by removing protein aggregates. Many studies have found that inhibition of the UPS causes upregulation of autophagy, and that suppression of autophagy leads to accumulation of polyubiqutinated protein aggregates [60–64]. Also, rapamycin treatment to activate autophagy increased clearance of aggregate–prone proteins and reduced the appearance of protein aggregates in vitro and in vivo [39, 65, 66]. In the heart, cardiac specific deletion of Atg5 resulted in increased polyubiquitinated protein levels and proteasome activity [30]. These studies suggest that autophagy and the UPS are functionally coupled, but further studies are needed to determine the molecular players involved in the cross-talk between the two pathways.
Autophagy and Programmed Cell death
Recent studies involving manipulation of essential autophagy genes have provided some information about the role of autophagy in cell death. Autophagic cell death is often been observed when apoptosis is blocked and it appears that the same stimulus can induce both apoptotic and autophagic cell death. For instance, pro-apoptotic Bax and Bak are essential for apoptosis via the mitochondrial (intrinsic) cell death pathway and mouse embryonic fibroblasts lacking Bax and Bak (Bax/Bak dko MEFs) are resistant to cell death induced by stimuli that activate this pathway (i.e. staurosporine, etoposide, UV) [67]. Interestingly, Shimizu et al. found that etoposide treatment of Bax/Bak dko MEFs, which failed to trigger apoptotic cell death, induced extensive autophagy followed by delayed cell death [68]. Inhibition of autophagy using siRNA against Beclin-1 and Atg5 reduced etoposide-induced cell death, suggesting that these cells died an autophagic cell death. Another study found that lipopolysaccharide treatment induced caspase-independent cell death in macrophages that were inhibited by siRNA-mediated knockdown of Beclin-1 or pre-treatment with 3-MA or Wortmannin [69]. In addition, treatment with hydrogen peroxide or 2-methoxyestradiol (2-ME) to inhibit Superoxide Dismutase are both known to induce apoptosis in cells [70, 71], was recently shown to induce autophagic cell death in HEK293 and U87 cells [72]. This group found that inhibition of autophagy with 3-MA or downregulation of Beclin1, Atg5, or Atg7 using RNA interference significantly reduced hydrogen peroxide- or 2-ME mediated cell death. These studies suggest that inhibition of the apoptotic pathway can switch the cellular response from apoptosis to autophagic cell death. In autophagic cell death, it is likely that excessive autophagy destroys a large amount of proteins and organelles beyond a certain threshold, causing a bioenergetic catastrophe culminating in cell death (Fig. 2).
In addition, recent studies suggest that there is cross-talk between the apoptotic and autophagic pathways. For instance, Pyo et al. reported that Atg5 interacted with Fas-associated death domain (FADD) protein, an adaptor molecule involved in mediating receptor-mediated apoptosis, and mediated IFN-β-induced cell death in HeLa cells [73]. Interestingly, they found that overexpression of the Atg5 mutant, Atg5K130R, which cannot be conjugated to Atg12, was able to trigger cell death, but failed to induce autophagy. Another study found that Atg5 was cleaved by the protease calpain, which promoted the translocation of truncated Atg5 to the mitochondria where it interacted with Bcl-XL and triggered permeabilization of the outer mitochondrial membrane [6]. Similar to the study by Pyo et al., overexpression of truncated Atg5 induced apoptosis, but was unable to promote autophagy. These two studies suggest that Atg5 can directly cause cell death by activating the apoptotic pathway without the induction of autophagy. The role of autophagy in cell death is clearly complex and needs to be further investigated. It also complicates the interpretation of studies of Atg5, as knockdown may block apoptosis as well as autophagy.
Bcl-2 proteins and autophagy
The Bcl-2 family proteins are known to be important regulators of apoptosis in the heart, but there is increasing evidence that they can also regulate autophagy. The autophagy protein Beclin 1 was initially identified in a yeast two-hybrid system as a Bcl-2 interacting protein [74], and it was later reported that Bcl-2 binding to Beclin 1 disrupted autophagy. In addition, a mutant of Beclin 1 lacking the Bcl-2 binding domain was found to induce excessive autophagy and cell death when overexpressed in cells [5]. The same group found that transgenic mice overexpressing Bcl-2 in the heart had reduced levels of autophagy in the heart compared to wild type in response to starvation, suggesting that Bcl-2 functions as a negative regulator of autophagy by inhibiting Beclin 1 [5]. In contrast, in a study using the HL-1 cardiac myocyte cell line overexpression of wild type Bcl-2 did not have an effect on starvation-induced autophagic flux. Only Bcl-2 that was targeted specifically to the SR/ER reduced starvation-induced autophagy by reducing Ca2+ stores in the SR/ER [75]. Also, overexpression of the Beclin 1 mutant lacking the Bcl-2 binding domain suppressed starvation-induced autophagy suggesting that it was acting as a dominant negative protein. Shimizu et al. reported that overexpression of Bcl-2 or Bcl-XL increased autophagy in MEFs treated with etoposide [68]. These studies suggest that the interaction between Beclin-1 and Bcl-2/Bcl-XL provides a potential convergence point for apoptosis and autophagy. Thus, further studies are needed to clarify why Bcl-2 can act in opposing manner, but it is possible that the regulatory effects of Bcl-2 on autophagy differ depending on the cell type and/or stimulus.
In addition, there are reports that the pro-apoptotic BH3-only proteins can activate autophagy. The BH3-only protein Bad has been implicated in cardiac myocyte cell death and was found to be upregulated in hearts after I/R [76]. Recently, Maiuri et al. reported that knock down of Bad using siRNA reduced starvation-induced autophagy, whereas Bad overexpression induced autophagy [44]. Similarly, Bnip3, which contributes to I/R injury [52] and postinfarct remodeling [77], is a potent inducer of autophagy in cardiac cells [52]. It is not clear if Bad and Bnip3 directly activate autophagy or whether autophagy is upregulated as a consequence of their activation. Beclin 1 has recently been shown to contain a conserved BH3 domain [45], and this domain is essential for heterodimerization between the Bcl-2 proteins [78–80]. Pro-apoptotic BH3-only proteins such as Bad and Bid can activate apoptosis by displacing Bcl-2 and Bcl-XL from pro-apoptotic Bax and Bak allowing for their activation [81]. Interestingly, a pharmacological BH3 mimetic competitively inhibits the interaction between Beclin 1 and Bcl-2/Bcl-XL [44]. This suggests that BH3-only proteins can directly activate autophagy by displacing Bcl-2 from Beclin1. However, this need to be further investigated in the heart.
Conclusion
It is clear that autophagy can have dual roles in the heart. Although it is not known what factor determines whether autophagy will be protective or detrimental to the cell, it is likely that the level and duration of autophagy are important. For instance, low levels of autophagy during ischemia and early reperfusion might protect against cell death by providing the cell with free fatty acids and amino acids and removing damaged organelles, whereas high levels or long-term upregulation of autophagy during reperfusion can trigger cell death by excess degradation of essential proteins and organelles. Also, a complex interrelationship seems to exist between autophagy and the apoptotic cell death pathway, where regulators of apoptosis also function as regulators of autophagic activation. Since modulation of the autophagic pathway may represent a potential future therapeutic target to treat or prevent a variety of cardiovascular diseases, the relationship between the survival and death functions of autophagy in the heart needs to be further elucidated.
Acknowledgments
This manuscript was supported by funds from the California Tobacco-Related Disease Research Program of the University of California, New Investigator Award #14KT-0109, a Scientist Development Award from the AHA, and NIH grant HL087023 to ÅBG, and NIH grants HL071091, HL060590, HL085577, and AG025168 to RAG.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–48. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 2.Deretic V. Autophagy as an immune defense mechanism. Curr Opin Immunol. 2006;18:375–82. doi: 10.1016/j.coi.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 3.Elmore SP, Qian T, Grissom SF, Lemasters JJ. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J. 2001;15:2286–7. doi: 10.1096/fj.01-0206fje. [DOI] [PubMed] [Google Scholar]
- 4.Hamacher-Brady A, Brady NR, Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem. 2006;281:29776–87. doi: 10.1074/jbc.M603783200. [DOI] [PubMed] [Google Scholar]
- 5.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–39. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 6.Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8:1124–32. doi: 10.1038/ncb1482. [DOI] [PubMed] [Google Scholar]
- 7.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. DevCell. 2004;6:463–77. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 8.Decker RS, Wildenthal K. Lysosomal alterations in hypoxic and reoxygenated hearts. I. Ultrastructural and cytochemical changes. Am J Pathol. 1980;98:425–44. [PMC free article] [PubMed] [Google Scholar]
- 9.Hamacher-Brady A, Brady NR, Gottlieb RA, Gustafsson AB. Autophagy as a Protective Response to Bnip3-Mediated Apoptotic Signaling in the Heart. Autophagy. 2006:2. doi: 10.4161/auto.2947. [DOI] [PubMed] [Google Scholar]
- 10.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–22. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 11.Sybers HD, Ingwall J, DeLuca M. Autophagy in cardiac myocytes. Recent Adv Stud Cardiac Struct Metab. 1976;12:453–63. [PubMed] [Google Scholar]
- 12.Valentim L, Laurence KM, Townsend PA, Carroll CJ, Soond S, Scarabelli TM, et al. Urocortin inhibits Beclin1-mediated autophagic cell death in cardiac myocytes exposed to ischaemia/reperfusion injury. J Mol Cell Cardiol. 2006;40:846–52. doi: 10.1016/j.yjmcc.2006.03.428. [DOI] [PubMed] [Google Scholar]
- 13.Dammrich J, Pfeifer U. Cardiac hypertrophy in rats after supravalvular aortic constriction. II. Inhibition of cellular autophagy in hypertrophying cardiomyocytes. Virchows Arch B Cell Pathol Incl Mol Pathol. 1983;43:287–307. doi: 10.1007/BF02932962. [DOI] [PubMed] [Google Scholar]
- 14.Miyata S, Takemura G, Kawase Y, Li Y, Okada H, Maruyama R, et al. Autophagic cardiomyocyte death in cardiomyopathic hamsters and its prevention by granulocyte colony-stimulating factor. Am J Pathol. 2006;168:386–97. doi: 10.2353/ajpath.2006.050137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pfeifer U, Fohr J, Wilhelm W, Dammrich J. Short-term inhibition of cardiac cellular autophagy by isoproterenol. J Mol Cell Cardiol. 1987;19:1179–84. doi: 10.1016/s0022-2828(87)80528-x. [DOI] [PubMed] [Google Scholar]
- 16.Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, et al. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007;117:1782–93. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shimomura H, Terasaki F, Hayashi T, Kitaura Y, Isomura T, Suma H. Autophagic degeneration as a possible mechanism of myocardial cell death in dilated cardiomyopathy. JpnCircJ. 2001;65:965–8. doi: 10.1253/jcj.65.965. [DOI] [PubMed] [Google Scholar]
- 18.Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T, et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease) Nature. 2000;406:906–10. doi: 10.1038/35022604. [DOI] [PubMed] [Google Scholar]
- 19.Dunn WA., Jr Studies on the mechanisms of autophagy: formation of the autophagic vacuole. JCell Biol. 1990;110:1923–33. doi: 10.1083/jcb.110.6.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seglen PO, Bohley P. Autophagy and other vacuolar protein degradation mechanisms. Experientia. 1992;48:158–72. doi: 10.1007/BF01923509. [DOI] [PubMed] [Google Scholar]
- 21.Kihara A, Noda T, Ishihara N, Ohsumi Y. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol. 2001;152:519–30. doi: 10.1083/jcb.152.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shintani T, Mizushima N, Ogawa Y, Matsuura A, Noda T, Ohsumi Y. Apg10p, a novel protein-conjugating enzyme essential for autophagy in yeast. Embo J. 1999;18:5234–41. doi: 10.1093/emboj/18.19.5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001;152:657–68. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mizushima N, Noda T, Ohsumi Y. Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. Embo J. 1999;18:3888–96. doi: 10.1093/emboj/18.14.3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abeliovich H, Dunn WA, Jr, Kim J, Klionsky DJ. Dissection of autophagosome biogenesis into distinct nucleation and expansion steps. J Cell Biol. 2000;151:1025–34. doi: 10.1083/jcb.151.5.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kirisako T, Ichimura Y, Okada H, Kabeya Y, Mizushima N, Yoshimori T, et al. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J Cell Biol. 2000;151:263–76. doi: 10.1083/jcb.151.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schlumpberger M, Schaeffeler E, Straub M, Bredschneider M, Wolf DH, Thumm M. AUT1, a gene essential for autophagocytosis in the yeast Saccharomyces cerevisiae. J Bacteriol. 1997;179:1068–76. doi: 10.1128/jb.179.4.1068-1076.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim J, Huang WP, Klionsky DJ. Membrane recruitment of Aut7p in the autophagy and cytoplasm to vacuole targeting pathways requires Aut1p, Aut2p, and the autophagy conjugation complex. J Cell Biol. 2001;152:51–64. doi: 10.1083/jcb.152.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suzuki K, Kirisako T, Kamada Y, Mizushima N, Noda T, Ohsumi Y. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. Embo J. 2001;20:5971–81. doi: 10.1093/emboj/20.21.5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–24. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 31.Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lullmann-Rauch R, et al. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature. 2000;406:902–6. doi: 10.1038/35022595. [DOI] [PubMed] [Google Scholar]
- 32.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. MolBiolCell. 2004;15:1101–11. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kostin S, Pool L, Elsasser A, Hein S, Drexler HC, Arnon E, et al. Myocytes die by multiple mechanisms in failing human hearts. Circ Res. 2003;92:715–24. doi: 10.1161/01.RES.0000067471.95890.5C. [DOI] [PubMed] [Google Scholar]
- 34.Hein S, Arnon E, Kostin S, Schonburg M, Elsasser A, Polyakova V, et al. Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: structural deterioration and compensatory mechanisms. Circulation. 2003;107:984–91. doi: 10.1161/01.cir.0000051865.66123.b7. [DOI] [PubMed] [Google Scholar]
- 35.Elsasser A, Vogt AM, Nef H, Kostin S, Mollmann H, Skwara W, et al. Human hibernating myocardium is jeopardized by apoptotic and autophagic cell death. J Am Coll Cardiol. 2004;43:2191–9. doi: 10.1016/j.jacc.2004.02.053. [DOI] [PubMed] [Google Scholar]
- 36.Decker RS, Poole AR, Crie JS, Dingle JT, Wildenthal K. Lysosomal alterations in hypoxic and reoxygenated hearts. II. Immunohistochemical and biochemical changes in cathepsin D. AmJPathol. 1980;98:445–56. [PMC free article] [PubMed] [Google Scholar]
- 37.Yan L, Vatner DE, Kim SJ, Ge H, Masurekar M, Massover WH, et al. Autophagy in chronically ischemic myocardium. Proc Natl Acad Sci U S A. 2005;102:13807–12. doi: 10.1073/pnas.0506843102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mousavi SA, Brech A, Berg T, Kjeken R. Phosphoinositide 3-kinase regulates maturation of lysosomes in rat hepatocytes. Biochem J. 2003;372:861–9. doi: 10.1042/BJ20021136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–95. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 40.Beugnet A, Tee AR, Taylor PM, Proud CG. Regulation of targets of mTOR (mammalian target of rapamycin) signalling by intracellular amino acid availability. Biochem J. 2003;372:555–66. doi: 10.1042/BJ20021266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khan S, Salloum F, Das A, Xi L, Vetrovec GW, Kukreja RC. Rapamycin confers preconditioning-like protection against ischemia-reperfusion injury in isolated mouse heart and cardiomyocytes. J Mol Cell Cardiol. 2006;41:256–64. doi: 10.1016/j.yjmcc.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 42.Dosenko VE, Nagibin VS, Tumanovska LV, Moibenko AA. Protective effect of autophagy in anoxia-reoxygenation of isolated cardiomyocyte? Autophagy. 2006;2:305–6. doi: 10.4161/auto.2946. [DOI] [PubMed] [Google Scholar]
- 43.Aki T, Yamaguchi K, Fujimiya T, Mizukami Y. Phosphoinositide 3-kinase accelerates autophagic cell death during glucose deprivation in the rat cardiomyocyte-derived cell line H9c2. Oncogene. 2003;22:8529–35. doi: 10.1038/sj.onc.1207197. [DOI] [PubMed] [Google Scholar]
- 44.Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. Embo J. 2007;26:2527–39. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oberstein A, Jeffrey PD, Shi Y. Crystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J Biol Chem. 2007;282:13123–32. doi: 10.1074/jbc.M700492200. [DOI] [PubMed] [Google Scholar]
- 46.Shizukuda Y, Buttrick PM. Subtype specific roles of beta–adrenergic receptors in apoptosis of adult rat ventricular myocytes. J Mol Cell Cardiol. 2002;34:823–31. doi: 10.1006/jmcc.2002.2020. [DOI] [PubMed] [Google Scholar]
- 47.Xiang Y, Kobilka BK. Myocyte adrenoceptor signaling pathways. Science. 2003;300:1530–2. doi: 10.1126/science.1079206. [DOI] [PubMed] [Google Scholar]
- 48.Kuzman JA, O'Connell TD, Gerdes AM. Rapamycin prevents thyroid hormone-induced cardiac hypertrophy. Endocrinology. 2007;148:3477–84. doi: 10.1210/en.2007-0099. [DOI] [PubMed] [Google Scholar]
- 49.Ha T, Li Y, Gao X, McMullen JR, Shioi T, Izumo S, et al. Attenuation of cardiac hypertrophy by inhibiting both mTOR and NFkappaB activation in vivo. Free Radic Biol Med. 2005;39:1570–80. doi: 10.1016/j.freeradbiomed.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 50.McMullen JR, Sherwood MC, Tarnavski O, Zhang L, Dorfman AL, Shioi T, et al. Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation. 2004;109:3050–5. doi: 10.1161/01.CIR.0000130641.08705.45. [DOI] [PubMed] [Google Scholar]
- 51.Gustafsson AB, Gottlieb RA. Mechanisms of apoptosis in the heart. J Clin Immunol. 2003;23:447–59. doi: 10.1023/b:joci.0000010421.56035.60. [DOI] [PubMed] [Google Scholar]
- 52.Hamacher-Brady A, Brady NR, Logue SE, Sayen MR, Jinno M, Kirshenbaum LA, et al. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007;14:146–57. doi: 10.1038/sj.cdd.4401936. [DOI] [PubMed] [Google Scholar]
- 53.Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion--a target for cardioprotection. Cardiovasc Res. 2004;61:372–85. doi: 10.1016/S0008-6363(03)00533-9. [DOI] [PubMed] [Google Scholar]
- 54.Dolinsky VW, Dyck JR. Role of AMP-activated protein kinase in healthy and diseased hearts. Am J Physiol Heart Circ Physiol. 2006;291:H2557–69. doi: 10.1152/ajpheart.00329.2006. [DOI] [PubMed] [Google Scholar]
- 55.Meley D, Bauvy C, Houben-Weerts JH, Dubbelhuis PF, Helmond MT, Codogno P, et al. AMP-activated protein kinase and the regulation of autophagic proteolysis. J Biol Chem. 2006;281:34870–9. doi: 10.1074/jbc.M605488200. [DOI] [PubMed] [Google Scholar]
- 56.Samari HR, Seglen PO. Inhibition of hepatocytic autophagy by adenosine, aminoimidazole-4-carboxamide riboside, and N6-mercaptopurine riboside. Evidence for involvement of amp-activated protein kinase. J Biol Chem. 1998;273:23758–63. doi: 10.1074/jbc.273.37.23758. [DOI] [PubMed] [Google Scholar]
- 57.Wang Z, Wilson WA, Fujino MA, Roach PJ. Antagonistic controls of autophagy and glycogen accumulation by Snf1p, the yeast homolog of AMP-activated protein kinase, and the cyclin-dependent kinase Pho85p. Mol Cell Biol. 2001;21:5742–52. doi: 10.1128/MCB.21.17.5742-5752.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hochstrasser M. Ubiquitin-dependent protein degradation. Annu Rev Genet. 1996;30:405–39. doi: 10.1146/annurev.genet.30.1.405. [DOI] [PubMed] [Google Scholar]
- 59.Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–63. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- 60.Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005;280:40282–92. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- 61.Rideout HJ, Lang-Rollin I, Stefanis L. Involvement of macroautophagy in the dissolution of neuronal inclusions. Int J Biochem Cell Biol. 2004;36:2551–62. doi: 10.1016/j.biocel.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 62.Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, et al. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol. 2007;171:513–24. doi: 10.2353/ajpath.2007.070188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–9. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 64.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–4. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 65.Berger Z, Ravikumar B, Menzies FM, Oroz LG, Underwood BR, Pangalos MN, et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet. 2006;15:433–42. doi: 10.1093/hmg/ddi458. [DOI] [PubMed] [Google Scholar]
- 66.Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002;11:1107–17. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- 67.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–30. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6:1221–8. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- 69.Xu Y, Kim SO, Li Y, Han J. Autophagy contributes to caspase-independent macrophage cell death. J Biol Chem. 2006;281:19179–87. doi: 10.1074/jbc.M513377200. [DOI] [PubMed] [Google Scholar]
- 70.Li L, Heldin NE, Grawe J, Ulmsten U, Fu X. Induction of apoptosis or necrosis in human endometrial carcinoma cells by 2-methoxyestradiol. Anticancer Res. 2004;24:3983–90. [PubMed] [Google Scholar]
- 71.Takeda M, Shirato I, Kobayashi M, Endou H. Hydrogen peroxide induces necrosis, apoptosis, oncosis and apoptotic oncosis of mouse terminal proximal straight tubule cells. Nephron. 1999;81:234–8. doi: 10.1159/000045282. [DOI] [PubMed] [Google Scholar]
- 72.Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ. 2008;15:171–82. doi: 10.1038/sj.cdd.4402233. [DOI] [PubMed] [Google Scholar]
- 73.Pyo JO, Jang MH, Kwon YK, Lee HJ, Jun JI, Woo HN, et al. Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem. 2005;280:20722–9. doi: 10.1074/jbc.M413934200. [DOI] [PubMed] [Google Scholar]
- 74.Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, et al. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol. 1998;72:8586–96. doi: 10.1128/jvi.72.11.8586-8596.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brady NR, Hamacher-Brady A, Yuan H, Gottlieb RA. The autophagic response to nutrient deprivation in the hl-1 cardiac myocyte is modulated by Bcl-2 and sarco/endoplasmic reticulum calcium stores. Febs J. 2007;274:3184–97. doi: 10.1111/j.1742-4658.2007.05849.x. [DOI] [PubMed] [Google Scholar]
- 76.Murriel CL, Churchill E, Inagaki K, Szweda LI, Mochly-Rosen D. Protein kinase Cdelta activation induces apoptosis in response to cardiac ischemia and reperfusion damage: a mechanism involving BAD and the mitochondria. J Biol Chem. 2004;279:47985–91. doi: 10.1074/jbc.M405071200. [DOI] [PubMed] [Google Scholar]
- 77.Diwan A, Krenz M, Syed FM, Wansapura J, Ren X, Koesters AG, et al. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J Clin Invest. 2007;117:2825–33. doi: 10.1172/JCI32490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang K, Gross A, Waksman G, Korsmeyer SJ. Mutagenesis of the BH3 domain of BAX identifies residues critical for dimerization and killing. Mol Cell Biol. 1998;18:6083–9. doi: 10.1128/mcb.18.10.6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Boyd JM, Gallo GJ, Elangovan B, Houghton AB, Malstrom S, Avery BJ, et al. Bik, a novel death-inducing protein shares a distinct sequence motif with Bcl-2 family proteins and interacts with viral and cellular survival-promoting proteins. Oncogene. 1995;11:1921–8. [PubMed] [Google Scholar]
- 80.Chittenden T, Flemington C, Houghton AB, Ebb RG, Gallo GJ, Elangovan B, et al. A conserved domain in Bak, distinct from BH1 and BH2, mediates cell death and protein binding functions. EMBO J. 1995;14:5589–96. doi: 10.1002/j.1460-2075.1995.tb00246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gustafsson AB, Gottlieb RA. Bcl-2 Family Members and Apoptosis, Taken to Heart. Am J Physiol Cell Physiol. 2006 doi: 10.1152/ajpcell.00229.2006. [DOI] [PubMed] [Google Scholar]