

Abstract
Defects in soluble NSF attachment protein receptor (SNARE)-mediated granule exocytosis occur in islet beta cells, adipocytes, and/or skeletal muscle cells correlate with increased susceptibility to insulin resistance and diabetes. The serine/threonine kinase WNK1 (with no K (lysine)) has recently been implicated in exocytosis and is expressed in all three of these cell types. To search for WNK1 substrates related to exocytosis, we conducted a WNK1 two-hybrid screen, which yielded Munc18c. Munc18c is known to be a key regulator of accessibility of the target membrane (t-SNARE) protein syntaxin 4 to participate in SNARE core complex assembly, although a paucity of Munc18c-binding factors has precluded discovery of its precise functions. To validate WNK1 as a new Munc18c-interacting partner, the direct interaction between WNK1 and Munc18c was confirmed using in vitro binding analysis, and endogenous WNK1-Munc18c complexes were detected in the cytosolic and plasma membrane compartments of the islet beta cell line MIN6. This binding interaction is mediated through the N-terminal 172 residues of Munc18c and the kinase domain residues of WNK1 (residues 159–491). Expression of either of these two minimal interaction domains resulted in inhibition of glucose-stimulated insulin secretion, consistent with a functional importance for the endogenous WNK1-Munc18c complex in exocytosis. Interestingly, Munc18c failed to serve as a WNK1 substrate in kinase activity assays, suggesting that WNK1 functions in SNARE complex assembly outside its role as a kinase. Taken together, these data support a novel role for WNK1 and a new mechanism for the regulation of SNARE complex assembly by WNK1-Munc18c complexes.
WNK is an unusual novel member of the Ser/Thr kinase family, named WNK (with no K (lysine)), based upon its lack of an otherwise highly conserved lysine residue present within kinase core domains (1). Although three additional WNK family members have been identified in humans, named WNK2, WNK3, and WNK4 (2), WNK1 in particular has been linked to the inherited hypertension syndrome termed pseudohypoaldosteronism II (3). WNK1 heterozygous (+/−) knock-out mice are viable but display reduced blood pressure, consistent with the role of WNK1 in regulation of blood pressure in humans (4). WNK1 is a large ~230-kDa soluble protein that is widely expressed in diverse cell types and tissues and as such has been proposed to function outside this role in ion channel regulation (1, 5). WNK1 contains a kinase domain near its N terminus between residues 218 and 490 and a long C terminus with predicted coiled-coils (5). Analyses of protein kinase activities of various WNK1 truncation mutants also revealed the presence of an autoinhibitory domain located between residues 491 and 555 (6). Human WNK1 has been shown to become activated on Thr60 (7) (rodent residue is Thr58 (8)) in response to insulin-like growth factor and to insulin in adipocytes (9), although the function of WNK1 signaling in this endocrine cell type remains unknown.
In another endocrine cell type, insulin-secreting INS-1 beta cells, WNK1 has been implicated in vesicular trafficking via its ability to phosphorylate the calcium sensor protein synaptotagmin 2 (10, 11). Endogenous WNK1 and synaptotagmin 2 were shown to co-localize on a subset of secretory granules, and synaptotagmin 2 phosphorylation was enhanced by coexpression of WNK1 (10). Since synaptotagmin 2 is known to facilitate fusion of vesicles with the plasma membrane in neuronal cells (11), it is predicted that WNK1, by association, may be involved in this process as well.
The identification of synaptotagmin 2 as a WNK1 substrate was made by using the unique kinase domain of WNK1 as bait in a yeast two-hybrid screen (10). Using the same screening strategy, a second vesicular trafficking protein was also identified: Munc18c. Munc18c is a member of the SM (Sec1/Munc18) family of syntaxin-binding proteins and binds selectively to the syntaxin 4 isoform, whereas Munc18a and Munc18b isoforms bind to syntaxins 1–3 (12). Like synaptotagmin 2, Munc18c functions in insulin granule fusion, such that islets from Munc18c heterozygous (+/−) mice show impaired glucose-stimulated insulin secretion (13). However, distinct from syntaptotagmin 2 localization to the granule, Munc18c is a soluble ~67-kDa protein that exists in the cytosolic compartment and also at the plasma membrane, associated with membrane proteins syntaxin 4 and Doc2β (14). These differences in cellular locale of WNK1 substrates in islet beta cells suggest that WNK1 may function at multiple steps in vesicle trafficking and fusion. In addition, Munc18c functions in insulin-stimulated GLUT4 vesicle translocation and fusion in 3T3L1 adipocytes (15–17) and thus may be part of the mechanism by which WNK1 participates in insulin signaling. Although the precise role of Munc18c in vesicle/granule fusion has remained elusive, in part due to the lack of predictable domain structure and paucity of interacting partners, there is consensus that it functions as a scaffold to regulate syntaxin 4 conformation and availability for participation in SNARE core complex assembly (14, 18–20). Interestingly, Munc18c function and interaction with syntaxin 4 is altered by stimulus-induced tyrosine phosphorylation (21, 22) and protein kinase C-induced serine/threonine phosphorylation (23), supporting the idea that Munc18c associates with a kinase, such as WNK1.
In this study, we demonstrate that Munc18c is a WNK1-interacting protein and that endogenous Munc18c-WNK1 complexes are important for glucose-stimulated insulin secretion via a syntaxin 4-dependent mechanism. The complex association requires the N-terminal 172 residues of Munc18c and the kinase domain of WNK1. Paradoxically, although the kinase domain of WNK1 is essential for Munc18c association, activation by autophosphorylation of WNK1 is not required. These data provide new insight into the function of WNK1 in vesicle/granule exocytosis, outside its known function as a kinase, and reveal the identity of a novel Munc18c-interacting protein.
EXPERIMENTAL PROCEDURES
Materials
The rabbit polyclonal anti-WNK1 (Q256) and Munc18c antibodies were generated as described (1, 15). The monoclonal FLAG (M2) antibody was obtained from Sigma. Rabbit and mouse anti-GFP2 antibodies and pEGFP plasmids were acquired from Abcam (Cambridge, MA) and Clontech, respectively. The rabbit polyclonal anti-syntaxin 4 and mouse monoclonal anti-VAMP2 antibodies were purchased from Chemicon (Temecula, CA) and Synaptic Systems (Gottingen, Germany), respectively. The rabbit polyclonal anti-Myc antibody was purchased from Upstate Biotechnology, Inc. (Lake Placid, NY). The monoclonal anti-Myc (9E10) antibody and protein G-Plus-agarose were obtained from Santa Cruz Bio-technology, Inc. (Santa Cruz, CA). The MIN6 cells were a gift from Dr. John Hutton (University of Colorado Health Sciences Center). Goat anti-mouse and anti-rabbit horseradish peroxidase secondary antibodies and Transfectin lipid reagent were acquired from Bio-Rad. Enhanced chemiluminescence reagent was obtained from Amersham Biosciences (Pittsburgh, PA). The human C-peptide radioimmunoassay (RIA) kit was purchased from Linco Research Inc. (St. Charles, MO).
Plasmids
The pCMV5-Myc-WNK1 truncation mutants 1–491, 1–555, 1–220, and 159–555 were generated as previously described (1). An additional construct, pCMV5-Myc-WNK1-(1–491) K233M, was made by subcloning the K233M-mutagenized 1–491 fragment from pGEX-KS WNK1-(1–491) K233M (1) into the EcoRI and HindIII sites of the pCMV5 vector. The pMunc18c-GFP deletion constructs 1–172, 173–255, 256–332, and 333–592 and pcDNA3.1-FLAG-Munc18c were generated as previously described (14, 22). The pGEX4T-1-Munc18c construct was made by subcloning a PCR-generated full-length Munc18c fragment, using pcDNA3.1-Munc18c DNA as template, engineered with the 5′ SalI site and 3′ EcoRI site for insertion into the pGEX-4T–1 vector. Human proinsulin DNA was obtained as a gift from Dr. Chris Newgard (Duke University). All constructs were verified by DNA sequencing.
Cell Culture, Transient Transfection, and Secretion Assays
MIN6 beta cells were cultured as described previously (24). MIN6 beta cells at 50–60% confluence were transfected with 40 μ g of plasmid DNA/10-cm2 dish using Transfectin (Bio-Rad) to obtain ~50% transfection efficiency. After 48 h of incubation, cells were incubated for 2 h in modified Krebs-Ringer bicarbonate buffer (MKRBB; 5 mM KCl, 120 mM NaCl, 15 mM Hepes, pH 7.4, 24 mM NaHCO3, 1 mM MgCl2, 2 mM CaCl2, and 1 mg/ml bovine serum albumin) and stimulated with 20 mM glucose. Cells were subsequently lysed in Nonidet P-40 lysis buffer (25 mM Tris, pH 7.4, 1% Nonidet P-40, 10% glycerol, 50 mM sodium fluoride, 10 mM sodium pyrophosphate, 137 mM sodium chloride, 1 mM sodium vanadate, 1 mM phenylmethylsulfonyl fluoride, 10 μ g/ml aprotinin, 1 μ g/ml pepstatin, and 5 μ g/ml leupeptin), and lysates were cleared by microcentrifugation for 10 min at 4 °C for subsequent use in co-immunoprecipitation experiments. For measurement of human C-peptide release, MIN6 beta cells were transiently co-transfected with each plasmid plus human proinsulin cDNA, using transfectin with 2 μg of each DNA per 35-mm dish of cells at 50–60% confluence. Forty-eight hours later, cells were preincubated for 2 h in MKRBB buffer and stimulated with 20 mM glucose for 1 h, and human C-peptide was released into the buffer quantitated by RIA. CHO-K1 cells were cultured and electroporated as previously described (15). After 48 h of incubation, cleared detergent cell lysates were prepared for subsequent use in co-immunoprecipitation experiments.
Subcellular Fractionation
Subcellular fractions of beta cells were prepared as described previously (25). Briefly, MIN6 beta cells at 80–90% confluence were harvested into 1 ml of homogenization buffer (20 mM Tris-HCl, pH 7.4, 0.5 mM EDTA, 0.5 mM EGTA, 250 mM sucrose, 1 mM dithiothreitol, and 1 mM sodium orthovanadate containing protease inhibitors) and disrupted by 10 strokes through a 27-gauge needle. Homogenates were centrifuged at 900 × g for 10 min. Postnuclear supernatants were centrifuged at 5,500 × g for 15 min, and the subsequent supernatant was centrifuged at 25,000 × g for 20 min to obtain the secretory granule fraction in the pellet. The supernatant was further centrifuged at 100,000 × g for 1 h to obtain the cytosolic fraction. Plasma membrane (PM) fractions were obtained by mixing the postnuclear pellet with 1 ml of Buffer A (0.25 M sucrose, 1 mM MgCl2, and 10 mM Tris-HCl, pH 7.4) and 2 volumes of Buffer B (2 M sucrose, 1 mM MgCl2, and 10 mM Tris-HCl, pH 7.4). The mixture was overlaid with Buffer A and centrifuged at 113,000 × g for 1 h to obtain an interface containing the plasma membrane fraction. The interface was collected and diluted to 2 ml with homogenization buffer for centrifugation at 6,000 × g for 10 min, and the resulting pellet was collected as the plasma membrane fraction. All pellets were resuspended in 1% Nonidet P-40 lysis buffer to solubilize membrane proteins.
Co-immunoprecipitation and Immunoblotting
Antibodies were combined with cleared detergent lysate protein for 2 h at 4 °C followed by a second incubation with protein G-Plus-agarose for 2 h. Immunoprecipitates were subjected to 8.5 or 12% SDS-PAGE, followed by transfer to polyvinylidene difluoride membranes for immunoblotting. Chemiluminescence was detected using a Chemi-Doc gel documentation system (Bio-Rad).
Recombinant Proteins and Interaction Assays
Recombinant proteins GST-WNK1-(1–491) WT, GST-WNK1-(1–491) K233M, and GST-Munc18c were expressed in Escherichia coli and purified by glutathione-agarose affinity chromatography. Recombinant WNK1-(1–491) was obtained following thrombin cleavage (Novagen) of GST-WNK1-(1–491). The interaction of GST-Munc18c with thrombin-cleaved WNK1 was performed by incubating 2 μg of GST-Munc18c linked to Sepharose beads with 2 μg of recombinant WNK1 protein in Nonidet P-40 lysis buffer for 2 h at 4 °C. Following three washes with lysis buffer, proteins were eluted from the Sepharose beads and subjected to 10% SDS-PAGE followed by transfer to polyvinylidene difluoride membrane for immunoblotting.
In Vitro Kinase Assays
Kinase assays were performed using recombinant WNK1-(1– 491) and/or Munc18c proteins in 30 μl of 1× kinase buffer containing 20 mM Hepes (pH 7.6), 10 μM ATP, 10 mM MgCl2, 10 mM, 10 mM β-glycerophosphate, 1 mM dithiothreitol, 1 mM benzamidine, and 10 μCi of [γ-32P]ATP. GST-Munc18c and WNK1 were incubated for 15 and 30 min at 30 °C as described (10). Reactions were stopped by adding 7.5 μl of 5± SDS sample buffer followed by boiling for 2 min. Reactions (20 μl) were analyzed by SDS-PAGE and autoradiography.
Statistical Analysis
All data are expressed as mean ± S.E. Data were evaluated for statistical significance using Student’s t test.
RESULTS
WNK1 and Munc18c Associate in MIN6 Beta Cells
The kinase domain of WNK1 was used in the pLexA-bait strain (Clontech) to screen a library of pAD42-prey strains containing rat brain cDNA, as described previously (10, 26). Munc18c was detected in this screen. To confirm the WNK1-Munc18c interaction, a truncated form of WNK1-(1–491) containing the kinase domain (amino acids 218–490) fused to a C-terminal Myc tag (Fig. 1A) was co-electroporated with full-length Munc18c fused to EGFP (Munc18c-EGFP) or EGFP vector control into CHO-K1 cells for subsequent immunoprecipitation analyses. The CHO-K1 cells have high transfection efficiency (>70%) and contain undetectable levels of endogenous Munc18c protein. Anti-Myc (WNK1) immunoprecipitation resulted in specific co-immunoprecipitation of Munc18c-EGFP (Fig. 1B), confirming the yeast two-hybrid screen and indicating that the WNK1 truncation containing the N-terminal 491 residues was sufficient to confer association with Munc18c in mammalian cells.
FIGURE 1. The N terminus of WNK1 containing the kinase domain is sufficient to confer interaction with Munc18c.
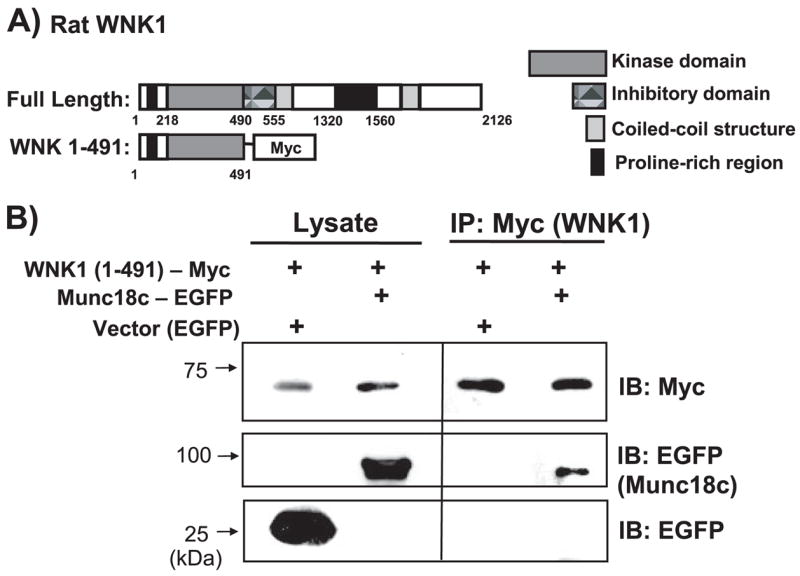
A, schematic diagram shows domains of full-length WNK1-(1–2126) and the truncated WNK1-(1– 491), which contains the kinase domain. B, CHO-K1 cells were electroporated with WNK1-(1– 491)-Myc and Munc18c-EGFP or EGFP-N3 vector DNA. Lysates were subsequently prepared and immunoprecipitated with anti-Myc antibody. Proteins were subjected to 10% SDS-PAGE and immunoblotted with anti-Myc and anti-EGFP antibodies. Data are representative of three independent experiments.
We next sought to determine if WNK1-Munc18c complexes formed in islet beta cells. WNK1 has previously been shown to localize to insulin granules and cytosolic fractions in INS-1 beta cells (10), whereas we and others have found Munc18c localized principally to plasma membrane and cytosolic compartments, with only trace amounts in the granule fraction of MIN6 beta cells (14, 27, 28). Given that both Munc18c and WNK1 reportedly exist in the cytosolic compartment, we investigated whether they associated in this locale. The MIN6 beta cell line was used for preparation of subcellular fractions and determination of WNK1 and Munc18c localization (Fig. 2A). Consistent with the locale of WNK1 in the INS-1 beta cells, WNK1 was found principally localized to the cytosolic (Cyt) fraction, with some presence in the storage granule (SG) and PM fractions. In the same fractions, Munc18c was also present, but its relative distribution in the cytosolic and PM fractions was reversed relative to that of WNK1. Immunodetection of the plasma membrane protein syntaxin 4 was used to validate integrity of sub-cellular fractionation in MIN6 beta cells (14).
FIGURE 2. WNK1 associates with Munc18c in MIN6 beta cells.
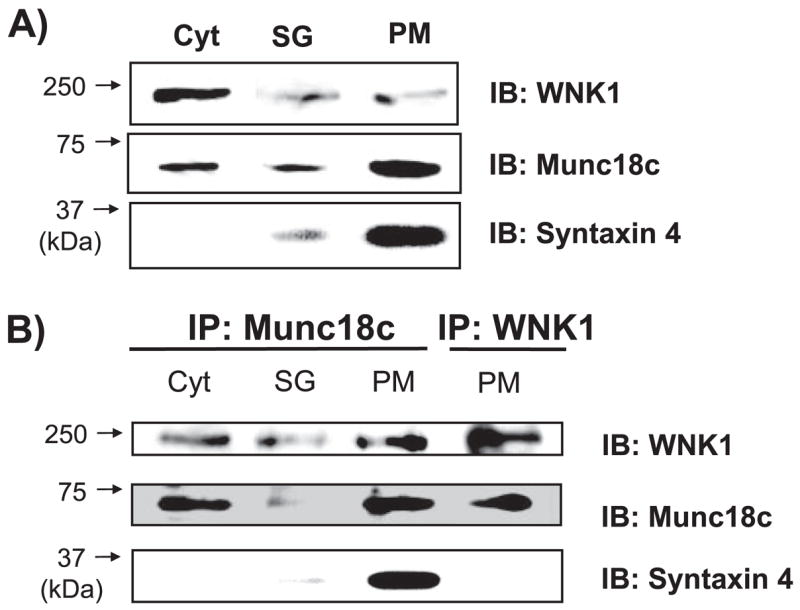
A, lysate prepared from cytosol (Cyt), storage granule (SG), and PM fractions isolated from MIN6 cells were resolved on 8.5% SDS-polyacrylamide gel (40 μg), transferred to polyvinylidene difluoride membrane, and immunoblotted with anti-WNK1, anti-Munc18c and anti-syntaxin 4 antibodies. B, cytosol fractions (2 mg) and granule (400 μg) were used for immunoprecipitation with anti-Munc18c antibody. One plasma membrane fraction preparation divided into two portions (200 μg each) was used for immunoprecipitation with anti-Munc18c or anti-WNK1 antibodies in parallel reactions. Proteins were resolved on 8.5% SDS-PAGE for subsequent immunoblotting with anti-WNK1, anti-Munc18c, and anti-syntaxin 4 antibodies. Data are representative of three independent experiments.
The fractions were subsequently divided for use in parallel immunoprecipitation reactions with Munc18c and WNK1 antibodies. Immunoprecipitation with anti-Munc18c antibody was capable of reproducibly co-immunoprecipitating WNK1 from the cytosolic and PM fractions (Fig. 2B, lanes 1–3). In two of the five cytosolic co-immunoprecipitation experiments, a glucose-induced decrease in Munc18c-WNK1 association was observed (data not shown). The lack of consistent detection of a decrease with glucose is reminiscent of that observed with Munc18c-syntaxin 4 complexes, which transiently associate/dissociate with changes in the phosphorylation state of Munc18c (22). In addition, reciprocal immunoprecipitation of WNK1 similarly resulted in co-precipitation of Munc18c from the PM fraction (Fig. 2B, lane 4). Anti-Munc18c co-immuno-precipitated syntaxin from the PM fraction as expected, although anti-WNK1 failed to precipitate syntaxin 4. These data suggest that Munc18c-WNK1 complexes existed primarily in the PM and cytosolic compartments and that those located at the PM excluded syntaxin 4.
Residues 1–172 of Munc18c Are Sufficient to Confer Binding to WNK1
To determine the minimal binding domain of Munc18c required for its interaction with WNK1, a series of Munc18-EGFP truncation constructs (Fig. 3A) was used as previously described (14, 22). Truncation constructs were co-elec-troporated with WNK1-(1–491)-Myc into CHO-K1 cells, and lysates were prepared for use in anti-Myc (WNK1) immunoprecipitation reactions. Although all four truncations of Munc18c were found to be expressed in cell lysates (Fig. 3B, lanes 1–4), only the truncation containing residues 1–172 of Munc18c was capable of co-precipitating with WNK1-Myc (Fig. 3B, lanes 5–8). This indicated that the N-terminal Munc18c-(1–172) residues are sufficient to confer interaction with WNK1.
FIGURE 3. The N terminus of Munc18c is sufficient for mediating the Munc18c-WNK1 interaction.
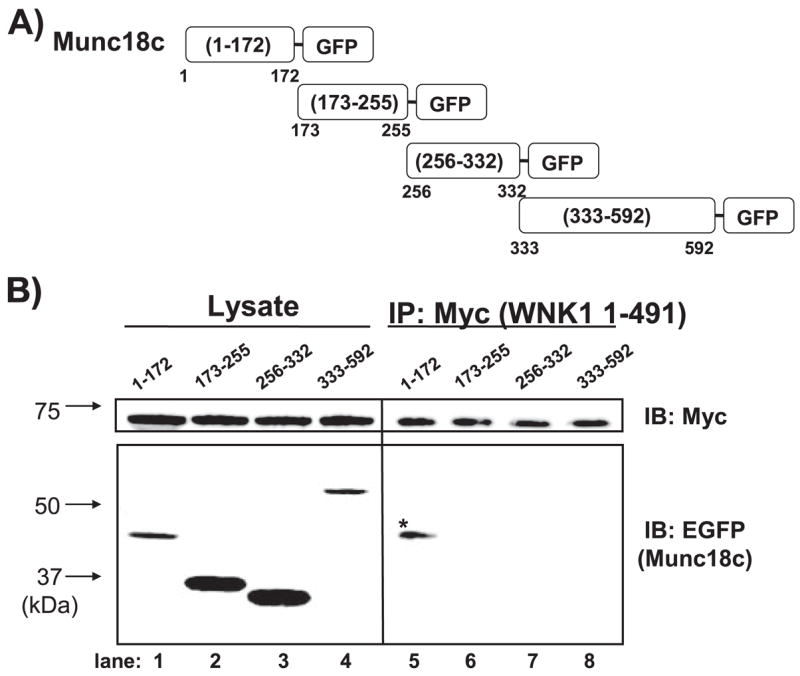
A, four fragments traversing the length of Munc18c were linked at the C terminus to EGFP. B, DNA constructs were co-electroporated into CHO-K1 cells with WNK1-(1– 491)-Myc, and 48 h later, detergent lysates were prepared for immunoprecipitation with anti-Myc antibody. Proteins were subjected to 12% SDS-PAGE and immunoblotted with anti-EGFP antibody to detect binding of Munc18c fragments. Equal precipitation of WNK1-Myc was confirmed by anti-Myc immunoblotting. Lysate proteins (50 μg/lane) show the expression of each Munc18c-EGFP fragment (left). Data are representative of at least three independent experiments.
Endogenous WNK1-Munc18c Complexes Are Essential for Syntaxin 4-mediated Insulin Granule Exocytosis
To determine whether WNK1-Munc18c complexes were functionally required for insulin secretion, we used the minimal binding domain of Munc18c (residues 1–172) to competitively inhibit formation of endogenous WNK1-Munc18c complexes. Co-immunoprecipitation assays revealed that 10-fold more Munc18c-(1–172) truncation protein was precipitated by WNK1-Myc compared with full-length Munc18c and that its overexpression reduced endogenous WNK1-Munc18c complex formation (n = 4; data not shown), suggesting its utility as a competitive inhibitor. Moreover, this small fragment of Munc18c fails to bind to syntaxin 4 (22) and thus should not perturb that association. MIN6 cells were co-transfected with pcDNA3.1-Munc18c-(1–172) or pcDNA3.1 vector control together with human proinsulin cDNA, and effects upon glucose-stimulated human C-peptide were assessed. Human C-peptide, cleaved from expressed human proinsulin, is synthesized and packaged in the MIN6 beta cells yet is immunologically distinct from endogenous mouse C-peptide and serves as a reporter of secretion from transfectable cells (29, 30). MIN6 cells transfected with vector control showed 141% of basal human C-peptide secretion in the response to glucose, whereas cells expressing Munc18c-(1–172) had significantly reduced glucose-stimulated secretion (Fig. 4A). Expression of Munc18c-(1–172) was without effect upon basal secretion (0.13 ± 0.02 ng/mg protein for Munc18c-1–172 versus 0.12 ± 0.01 ng/mg for control, n = 4). These data indicated that the endogenous WNK1-Munc18c complexes were functionally required for glucose-stimulated secretion but not necessarily for maintenance of basal insulin levels.
FIGURE 4. Munc18c-WNK1 complexes are essential for glucose-stimulated insulin granule exocytosis.
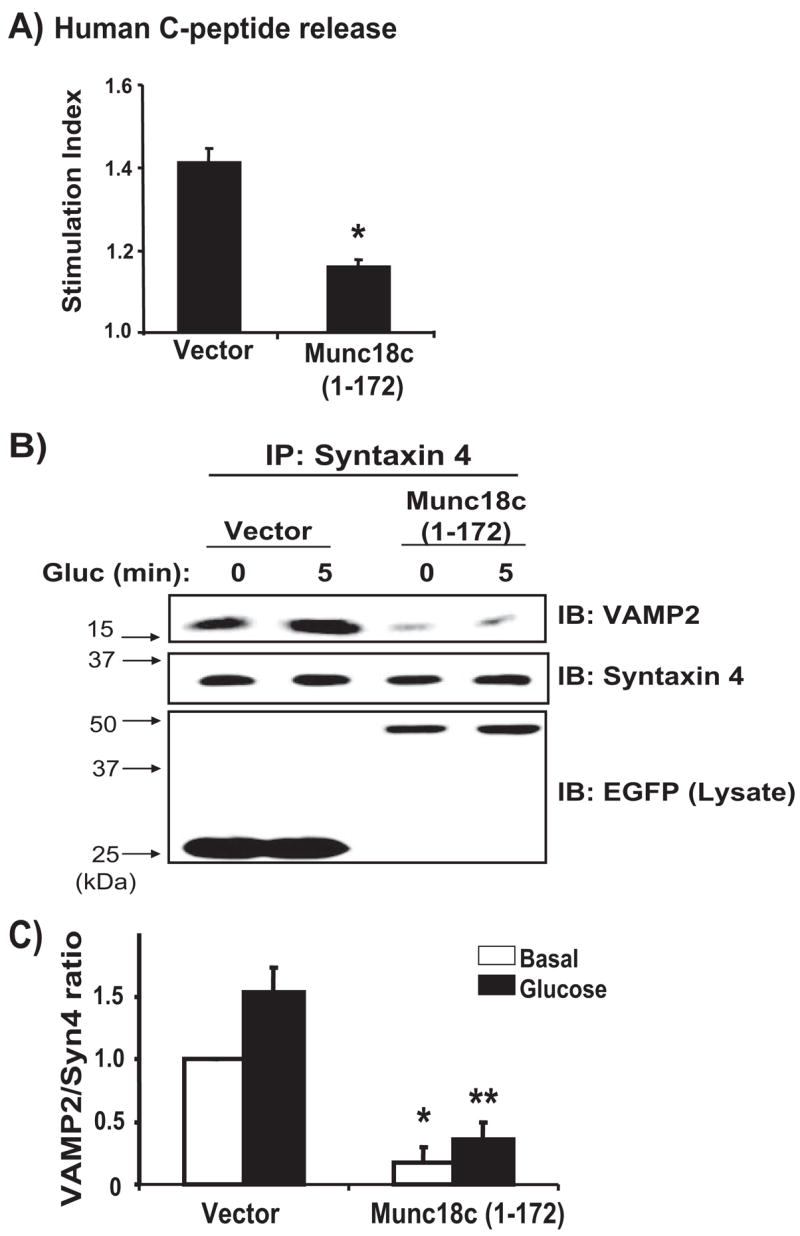
A, MIN6 cells were transiently transfected with either pcDNA3 vector or pcDNA3-Munc18c-(1–172) plus human proinsulin DNA, which served as a reporter of secretion specifically from transfectable cells. After 48 h of incubation, cells were preincubated in MKRBB for 2 h and left unstimulated (open bars) or stimulated with 20 mM glucose (black bars). Human C-peptide secreted into the media was measured by RIA and normalized for total cellular protein content. Data represent the stimulation indices of four independent experiments performed with multiple batches of DNA and are shown as the average ± S.E. *, p < 0.005 versus vector control. B, overexpression of Munc18c-(1–172) inhibits the association of endogenous VAMP2 with syntaxin 4. Detergent cell lysates prepared from MIN6 cells transiently transfected to express Munc18c-(1–172)-EGFP or vector control and left unstimulated or stimulated with 20 mM glucose for 5 min were used in immunoprecipitation reactions with anti-syntaxin 4 antibody. Immunoprecipitated proteins were resolved on 12% SDS-PAGE, and co-immunoprecipitated VAMP2 was detected by immunoblotting. C, optical density scanning quantitation of VAMP2/syntaxin. Data in each of three independent experiments were normalized to the level of association under basal conditions. *, p <0.001 versus vector basal conditions; **, p <0.001 versus vector stimulated.
To investigate the mechanism of this alteration in function, we assessed SNARE complex assembly (syntaxin-VAMP association) in MIN6 cell lysates from cells expressing Munc18c-(1–172)-EGFP or EGFP vector control. As we have shown previously (14, 22), glucose stimulation for 5 min increased the co-immunoprecipitation of VAMP2 granules with syntaxin 4 in lysates from vector-expressing cells (Fig. 4B, lanes 1 and 2). However, MIN6 cells overexpressing Munc18c-(1–172)-EGFP exhibited significantly less syntaxin 4-VAMP2 association, under both basal and glucose-stimulated conditions (Fig. 4B, lanes 3 and 4). Immunoblotting of lysate proteins confirmed equivalent expression levels of EGFP and Munc18c-(1–172)-EGFP proteins, indicating that the effect of the Munc18c-(1–172) protein was specific. Quantitation of the VAMP2/syntaxin 4 ratio from three independent experiments revealed the decreases in SNARE complexes to be on the order of 75% or ~3-fold fewer than those formed in cells expressing vector alone (Fig. 4C). This effect of the 1–172 region upon the basal VAMP2/syntaxin 4 ratio was unique, since we previously reported that expression of the Munc18c-(173–255) region impaired only the glucose-stimulated syntaxin 4-VAMP2 association (14). Since the abundance of SNARE complexes associated under basal conditions has been correlated with the abundance of insulin granules predocked at the PM for first phase insulin release (31), the data presented here suggest that endogenous WNK1-Munc18c complexes regulate syntaxin 4-mediated granule docking/fusion for both phases of glucose-stimulated insulin secretion. This would be consistent with our published data showing that syntaxin 4, a specific binding partner of Munc18c, is required in both first and second phase insulin release from islets (27).
The WNK1 Kinase Domain but Not Kinase Activity Is Required for Binding and Function with Munc18c
Our initial studies showed that the N-terminal 491 residues of WNK1, containing the kinase domain (residues 218–490) and an upstream proline-rich domain, located within the first 220 residues, were sufficient for interaction with Munc18c. Since WNK1 has previously been shown to bind proteins through the kinase and autoinhibitory domain (10) or through just the first 220 residues (8), we sought to further delineate the minimal binding domain of WNK1 needed for interaction with Munc18c using WNK1 truncations tagged with a C-terminal Myc epitope (Fig. 5A). The autoinhibitory domain spans residues 491–555 and inhibits the activation by autophosphorylation of WNK1 (6). Many protein kinases contain such a domain just beyond their catalytic domain as a means to suppress kinase activity until a stimulus displaces it from its inhibitory site (32, 33). CHO-K1 cells were co-electroporated with Munc18c-(1–172)-EGFP plus Myc-tagged forms of WNK1-(1–555), WNK1-(159–555), or WNK-(1–220), and lysates used for immunoprecipitation with anti-EGFP showed equivalent expression of each protein (Fig. 5B, lanes 1–3). Both WNK1-(1–555) and WNK1-(159–555) retained the ability to co-immunoprecipitate with Munc18c (Fig. 5B, lanes 4 and 5), indicating that the proline-rich domain is not required. Furthermore, WNK1-(1–220) failed to co-immunoprecipitate with Munc18c-EGFP (Fig. 5B, lane 6). Given the ability of the WNK1-(1–491) protein to bind, the minimal binding region of WNK1 required for Munc18c binding is contained within residues 220–491, the same residues that constitute the WNK1 kinase domain.
FIGURE 5. The kinase domain of WNK1 is required for Munc18c binding.
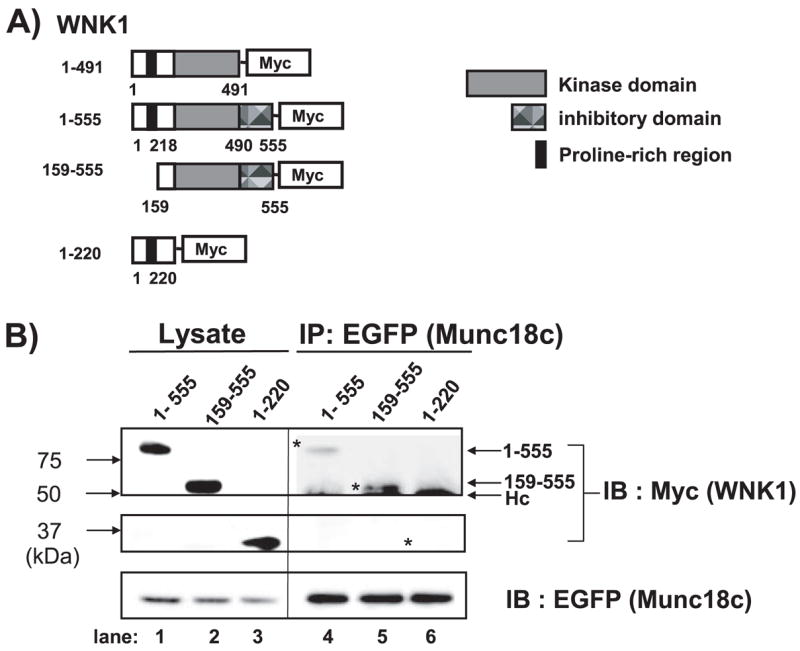
A, schematic diagram shows kinase domain-containing constructs WNK1-(1–491), WNK1-(1–555), and WNK1-(159 –555); WNK1-(1–220) lacks the kinase domain. B, CHO cells were electroporated with WNK1-(1–555)-Myc or WNK1-(159 –555) or WNK1-(1–220) and Munc18c-EGFP plasmids. Lysates were subsequently prepared and immunoprecipitated with anti-Myc antibody. Proteins were subjected to 10% SDS-PAGE and immunoblotted with anti-Myc and anti-EGFP antibodies. Hc, antibody heavy chain. Data are representative of three independent experiments.
Kinase substrates are known to bind in a direct manner and require particular amino acids in the kinase domain for interaction (34). Although the yeast two-hybrid data strongly suggested that Munc18c bound in a direct manner to WNK1, we used the GST pull-down approach with bacterially expressed and purified proteins for confirmation of this direct interaction. WT WNK1-(1–491) specifically bound to GST-Munc18c and not to the GST control (Fig. 6A, lanes 3 and 5). Interestingly, a kinase-dead form of WNK1-(1–491) bound in a similar and specific fashion to the GST-Munc18c (Fig. 6A, lanes 4 and 6). The kinase-dead WNK1 mutant contained a K233M mutation that preserved normal catalytic lysine arrangement but lacked autophosphorylation and kinase activity (6). Immunoblotting for GST showed equal loading of GST and GST-Munc18c proteins in all reactions and recognition of the WT and K233M forms of WNK1 protein input in the assay.
FIGURE 6. WNK1 kinase activity is not required for interaction with Munc18c.
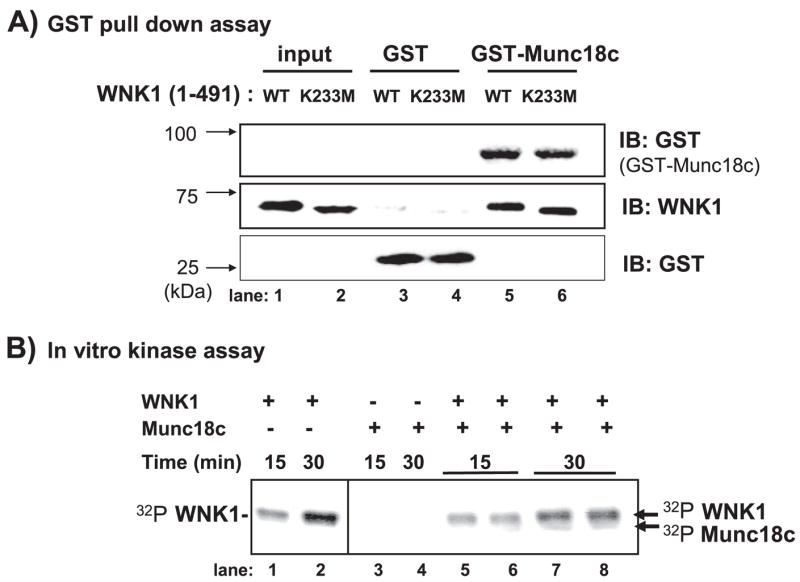
A, bacterially expressed GST-Munc18c or GST proteins were purified and linked to beads for in vitro binding studies with purified WNK1 WT and WNK1 K233M kinase-dead proteins. GST-precipitated proteins were subjected to 10% SDS-PAGE for subsequent immunoblotting with anti-WNK1 and anti-GST antibodies. B, in vitro kinase assays were performed with purified WNK1-(1– 491) and Munc18c proteins. Assays were terminated with Laemmli sample buffer and resolved by 10% SDS-PAGE. WNK1 autophosphorylation was detected at 15 min and increased at 30 min (lanes 1 and 2), yet no significant 32P incorporation in Munc18c (lanes 5– 8) was observed in five independent experiments.
The GST pull-down data indicated that Munc18c bound WNK1 in a direct manner but independent of the K233M mutation. To determine if Munc18c functioned as a WNK1 substrate, in vitro kinase reactions were performed as described previously (10, 26). In this assay, WNK1-(1–491) became auto-phosphorylated within 15 min, as detected by 32P incorporation and autoradiography, and was further increased after 30 min (Fig. 6B, lanes 1 and 2). However, the addition of Munc18c to the assay resulted in only trace levels of 32P incorporation at the position of Munc18c on the SDS-PAGE after 30 min (Fig. 6B, lanes 7 and 8). Taken together, these data suggested that the activation of WNK1 by autophosphorylation was not required for its interaction with Munc18c and that the WNK1-Munc18c complex may function in a capacity other than as just a kinase-substrate association reaction.
WNK1 Functions as a Munc18c Binding Partner Independent of Its Role as a Kinase
We next tested the ability of WNK1 to function as a Munc18c-binding protein within the cell. Although little is understood of the true physiological function of Munc18c to date, the identification of Munc18c binding partners can be made based upon their ability to “rescue” the inhibition of exocytosis that is caused by overexpression of Munc18c; non-Munc18c binding proteins or a non-PM-localized form of syntaxin 4 fails to rescue exocytosis (16, 22). As shown in Fig. 7A, Munc18c overexpression reduced glucose-stimulated human C-peptide release by ~75% (108% of basal versus 142% attained in vector-expressing cells, p < 0.001). However, the addition of WNK1 protein resulted in full restoration of glucose-stimulated human C-peptide release (Fig. 7A), consistent with its putative role as a Munc18c binding partner. Correspondingly, the addition of WNK1 to Munc18c-overexpressing cells also restored VAMP2-syntaxin 4 association in the co-immunoprecipitation assay (Fig. 7B). Compared with the normal levels of VAMP2 co-immunoprecipitated with syntaxin 4 in vector-transfected MIN6 cell lysates, levels of VAMP2 co-immunoprecipitated by syntaxin 4 from Munc18c-overexpressing cells were reduced substantially under both basal and glucose-stimulated conditions, as we have reported previously (14, 22). Quantitatively, the overexpression of Munc18c reduced the VAMP2/syntaxin 4 ratio by ~75% or greater under both basal and glucose-stimulated conditions and negated the glucose-stimulated increase in VAMP2/syntaxin 4 ratio normally observed (Fig. 7C). The addition of WNK1 restored VAMP2/syntaxin 4 association to levels observed in vector control cells. In all, these results suggested that WNK1, like syntaxin 4 and Doc2β, functions as a Munc18c-regulatory protein in SNARE complex assembly.
FIGURE 7. WNK1 functions as a Munc18c binding partner independent from its role as a kinase.
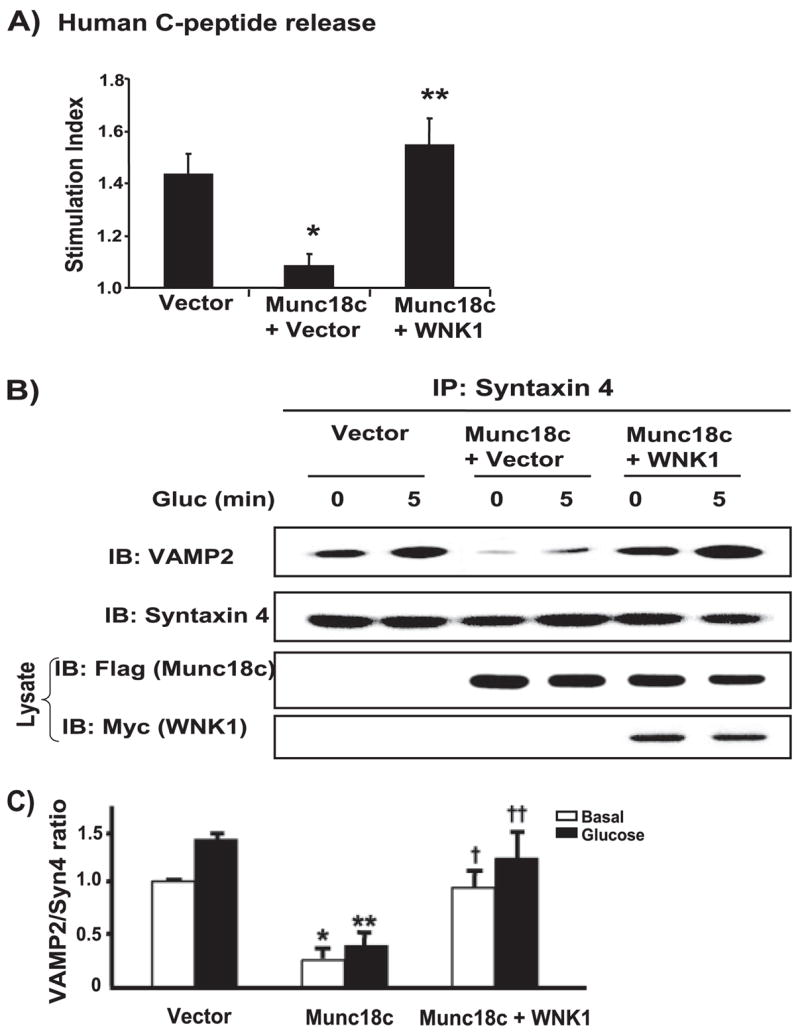
A, MIN6 cells were transfected with pcDNA3 vector control, pcDNA3-FLAG-Munc18c, or both pcDNA3-FLAG-Munc18c and pCMV5-WNK1-(1– 491) together plus human proinsulin DNA. After a 48-h incubation, cells were preincubated in MKRBB for 2 h, followed by stimulation with 20 mM glucose. Human C-peptide secreted into the media was measured by RIA and normalized for total cellular protein content. Data represent the stimulation indices of six independent experiments shown as the average ± S.E. *, p <0.001 versus vector control; **, p <0.001 versus Munc18c plus vector. B, co-overexpression of WNK1 with FLAG-Munc18c rescues VAMP2 association with syntaxin 4. Detergent cell lysates prepared from unstimulated (0) or glucose-stimulated (5 min) MIN6 cells transiently transfected with vector, Munc18c, or both Munc18c and WNK1 together were used for immunoprecipitation with anti-syntaxin 4 antibody. Immunoprecipitated proteins were resolved on 12% SDS-PAGE for immunodetection of VAMP2. Data are representative of four independent experiments. C, optical density scanning quantitation of VAMP2/syntaxin ratio in cells overexpressing Munc18c alone or Munc18c +WNK1-(1– 491). Data in each of four independent experiments were normalized to the level of association under basal conditions. *, p <0.01 versus vector transfected under basal conditions; **, p < 0.01 versus vector under glucose-stimulated conditions; †, p <0.01 versus Munc18c transfected under basal conditions; ††, p < 0.05 versus Munc18c transfected under glucose-stimulated conditions.
To investigate the relationship between WNK1 function as a Munc18c-regulatory protein and its known function as a kinase, we tested the ability of kinase-dead K233M WNK1 overexpression to relieve the inhibition upon glucose-stimulated insulin secretion caused by increased Munc18c expression in the human C-peptide secretion assay. Consistent with our data showing that K233M binds Munc18c as does WT WNK1, K233M WNK1 rescued glucose-stimulated secretion equally well as did WT WNK1 (Fig. 8A). In contrast, WNK1-(1–220) failed to rescue secretion when added to Munc18c-overexpressing cells, consistent with its failure to bind to Munc18c (Fig. 8A). In parallel, the addition of WNK1 K233M restored the levels of VAMP2 co-precipitated by syntaxin 4 under both unstimulated and stimulated conditions, matching those levels attained by the addition of WT WNK1 (Fig. 8B, lanes 3–6). As expected, WNK1-(1–220) was without effect in restoring VAMP2 levels (Fig. 8B, lanes 7 and 8) and demonstrated that the rescue effect was specific for Munc18c-binding proteins. Immunoblotting showed that FLAG-Munc18c expression was unaltered by expression of vector or WNK1 proteins and that the various WNK1 proteins were similarly expressed. Quantitation of the VAMP2/syntaxin 4 ratio confirmed that WNK1 addition restored the ability of glucose to increase the amount of VAMP2 associated with syntaxin 4 (Fig. 8C). Thus, these results suggest that WNK1 functions as a Munc18c-interacting protein in cells but that an intact kinase domain and ability to function as a kinase are not necessary for its role as a Munc18c-regulatory protein in insulin granule exocytosis.
FIGURE 8. WNK1 function in Munc18c-regulated insulin granule exocytosis occurs independently of its kinase activity.
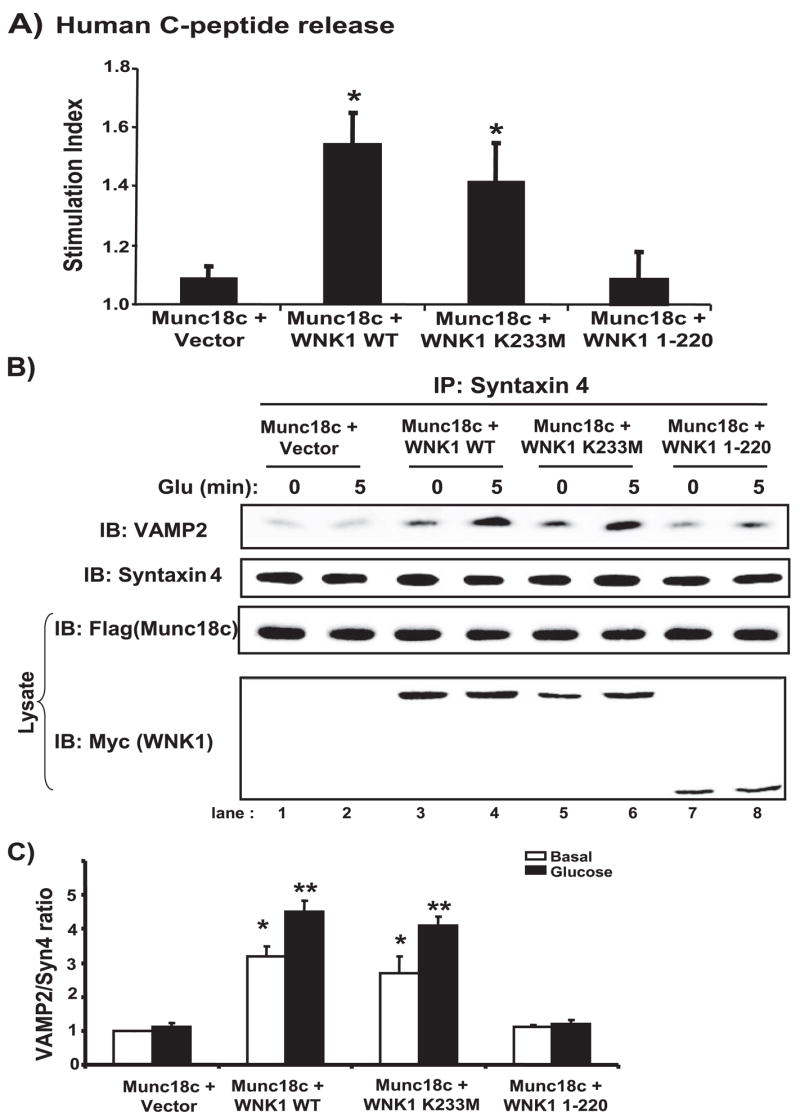
A, MIN6 cells were transfected with pcDNA3-FLAG-Munc18c or pcDNA3-FLAG-Munc18c plus pCMV5-WNK1-(1– 491) WT, pCMV5-WNK1-(1– 491, K233M kinase-dead form), or pCMV5-WNK1 (1–220) with reporter human proinsulin DNA. After a 48-h incubation, cells were preincubated in MKRBB for 2 h followed by stimulation with 20 mM glucose. Human C-peptide secreted into the media was measured by RIA and normalized for total cellular protein content. Data represent the stimulation indices of 3–5 independent experiments shown as the average ± S.E.; *, p < 0.01 versus Munc18c + vector. B, co-overexpression of WNK1 K233M with Munc18c rescues VAMP2 association with syntaxin 4, whereas WNK1-(1–220) fails to rescue. Detergent cell lysates prepared from unstimulated (0) or glucose-stimulated (5 min) MIN6 cells transiently transfected with Munc18c or Munc18c plus WNK1 WT, K233M, or WNK1-(1–220) were used for immunoprecipitation with anti-syntaxin 4 antibody. Immunoprecipitated proteins were resolved on 12% SDS-PAGE for immunodetection of VAMP2. Data are representative of three independent experiments. C, optical density scanning quantitation of VAMP2/syntaxin in cells overexpressing Munc18c minus/plus WNK1 WT, K233M, or 1–220 fragments. Data in each of three independent experiments were normalized to basal of Munc18c only = 1 and are shown as the average ± S.E. *, p < 0.005 versus basal of Munc18c + vector; **, p < 0.01 versus glucose-stimulated Munc18c + vector.
DISCUSSION
In this study, we have identified a new role for WNK1 in vesicle/granule exocytosis, as a novel binding partner for the syntaxin 4-regulatory protein Munc18c. Endogenous WNK1-Munc18c complexes were localized to PM and cytosolic compartments. WNK1 is the third binding partner identified for the Munc18c protein but only the first to bind to the cytosolic pool of Munc18c. Residues 1–172 of Munc18c and residues 159–491 containing only the kinase domain of WNK1 were sufficient to confer association, although mutation of the critical Lys233 residue in the kinase domain of WNK1 failed to impair its binding to Munc18c. Consistent with this, activation of WNK1 autophosphorylation was not required for its interaction with Munc18c, and virtually no phosphorylation of Munc18c was detected in WNK1 kinase assays. Competitive inhibition of endogenous WNK1-Munc18c complexes significantly reduced glucose-stimulated insulin release, correlated with a reduction in the abundance of VAMP2-granules associated with syntaxin 4. Last, WNK1 was found to function as a Munc18c binding protein in a “rescue assay,” with the K233M mutant function equal to that of the wild-type WNK1. Taken together, these data support a model whereby WNK1 binds Munc18c and functions in syntaxin 4-mediated insulin granule exocytosis in a manner independent of its role as a kinase.
The inability to detect phosphorylation of Munc18c by WNK1 was surprising, given that Munc18c has been reported to undergo Ser/Thr phosphorylation in endothelial cells (23) and adipocytes (35), combined with at least two algorithms predicting that the N terminus of Munc18c was rich in potential sites for this modification. Moreover, its neuronal counterpart Munc18-1 has been shown to undergo Ser/Thr phosphorylation, which then impacts its association with syntaxin 1A (36). Although it remains possible that Munc18c phosphorylation occurred at very low levels, which was obscured by the abundant phosphorylation of the similarly sized WNK1-(1–491) protein, immunoblotting consistently showed clear separation of these proteins on SDS-PAGE and therefore support the conclusion that Munc18c failed to function as a clear substrate for WNK1 in the in vitro kinase assays.
Notably, Munc18c is the first direct interacting protein of WNK1 that does not appear to function necessarily as a WNK1 substrate. WNK1 has been shown to bind directly to and phosphorylate the following substrates: synaptotagmin 2 (10), MEKK2 and MEKK3 (37), SGK1 (26), SPAK/OSR1 (38), WNK2 and WNK4 (39), Smad2 (40). A related family member and WNK1 substrate, WNK4, has been shown to bind to ROMK, and this interaction was independent of WNK4 kinase activity (41). However, ROMK bound to a region of WNK4 outside its kinase domain, unlike Munc18c, which binds to the kinase domain of WNK1. What is the purpose of the binding of a nonsubstrate protein through the specialized kinase domain? One possibility may be that WNK1 functions as a scaffold, recruiting Munc18c as a substrate for another kinase or phosphatase, as has been shown for other factors (42–44). In support of a role for WNK1 as a scaffold, WNK1 is known to contain 29 PXXP motifs, which could potentially attract other Src homology 3 domain-containing proteins. Further studies will be required to explore the role of WNK1 as a scaffold for Munc18c in fostering Munc18c post-translational modifications and/or cellular compartmentalization.
WNK1 is the third protein to be characterized as a Munc18c binding partner, the others being syntaxin 4 and Doc2β. However, unlike the other Munc18c-binding proteins, increased WNK1 expression alone failed to augment insulin secretion above normal levels, and small interfering RNA-mediated depletion of WNK1 failed to inhibit either insulin secretion (data not shown) or insulin-stimulated GLUT4 vesicle translocation in 3T3L1 adipocytes (9). One explanation for these differences may be that alteration of WNK1-Munc18c stoichiometry by overexpression or knockdown of WNK1 did not necessarily reflect the endogenous WNK1-Munc18c complex molar ratio. This idea is supported by studies showing that the stoichiometry of SNARE accessory proteins is particularly important for maintaining normal granule exocytosis (45, 46). Alternatively, small interfering RNA-mediated depletion of WNK1 may have preferentially eliminated a less essential pool of WNK1, since WNK1 exists in multiple cellular compartments. Further studies examining the distribution of WNK1 in control and small interfering RNA-depleted cells will provide insight as to whether there exist “essential” and “nonessential” pools of WNK1 in the cell.
WNK1 also differed from Doc2β function in Munc18c regulation in that disruption of the WNK1-Munc18c complex using the Munc18c-(1–172) competitive inhibitor caused a 75% reduction in VAMP2 association with syntaxin 4 under both basal and stimulated conditions (Fig. 4), whereas disruption of the endogenous Doc2β-Munc18c complex with the Munc18c-(173–255) competitive inhibitor abolished only the glucose-induced increase in VAMP2 association with syntaxin 4 and was entirely without effect upon basal level association (14). This difference may reflect the ability of the 1–172 competitive inhibitor to simultaneously disrupt Munc18c interactions with WNK1 and somewhat with Doc2β (data not shown), given that this region of Munc18c was able to support some binding to Doc2β, albeit the majority of Doc2β bound through the 173–255 region of Munc18c.
A key distinction between WNK1-Munc18c and Doc2β-Munc18c complexes is their cellular locale. Although both syntaxin 4 and Doc2β exclusively bind Munc18c in the PM compartment, WNK1 binds Munc18c in both the PM and cytosolic compartments. The Munc18 proteins are fairly equally distributed between the PM and cytosolic compartments of cells in which they have been studied to date (16, 47, 48), although their role in the cytosol has remained elusive due to lack of known cytosolic binding partners. Quantitatively, there is ~8-fold more protein in the cytosolic fraction than in the PM fraction, indicating that cytosolic WNK1-Munc18c complexes constitute the majority of these complexes in the cell. Thus, dissociation of these complexes using the Munc18c-(1–172) competitive inhibitor may have preferentially affected Munc18c in the cytosol and had less effect upon Munc18c at the PM. This notion is supported by our finding that in cells expressing the competitive inhibitor, there was not the expected increase in endogenous Munc18c co-immunoprecipitated by syntaxin 4 (data not shown), as was seen in analogous studies of Doc2β (14). Our finding that endogenous WNK1 and Munc18c present in the cytosol form complexes is the first clue as to the function of Munc18c in this cellular locale, although studies directed at dissociating just the cytosolic pool of WNK1-Munc18c complexes will be required to ascertain its particular function.
In summary, our finding that WNK1-Munc18c complexes are essential for VAMP2 association with syntaxin 4 may represent a new and key link between cardiovascular disease and insulin resistance. The pathogenesis of cardiovascular disease in insulin resistance is poorly understood. One commonality may lie in the use of syntaxin 4-based SNARE-mediated granule exocytosis by platelets, islet beta cells, adipocytes, and skeletal muscle cells to maintain normal hemostasis and glucose homeostasis. Thus, understanding how WNK1-Munc18c complexes function in exocytosis will advance our understanding of how glucose homeostasis is regulated and how it may be linked to cardiovascular function.
Acknowledgments
We are grateful to Dr. Chris Newgard and Dr. John Hutton for gifts of the human proinsulin cDNA and MIN6 beta cells, respectively. We thank Dr. Zhanxiang Wang for expertise and suggestions with this project.
Footnotes
The abbreviations used are: GFP, green fluorescent protein; EGFP, enhanced green fluorescence protein; IP, immunoprecipitation; IB, immunoblot; MKRBB, modified Krebs-Ringer bicarbonate buffer; PM, plasma membrane; SNARE, soluble NSF attachment protein receptor; GST, glutathione S-transferase; WT, wild type; RIA, radioimmunoassay; CHO, Chinese hamster ovary.
This work was supported by National Institutes of Health (NIH) Grant DK-067912 and American Diabetes Association Grant 1-03-CD-10) (to D. C. T.), American Heart Association Postdoctoral Fellowship 0720042Z (to E. O.) and NIH grant GM53032 (to M. H. C.).
References
- 1.Xu B, English JM, Wilsbacher JL, Stippec S, Goldsmith EJ, Cobb MH. J Biol Chem. 2000;275:16795–16801. doi: 10.1074/jbc.275.22.16795. [DOI] [PubMed] [Google Scholar]
- 2.Verissimo F, Jordan P. Oncogene. 2001;20:5562–5569. doi: 10.1038/sj.onc.1204726. [DOI] [PubMed] [Google Scholar]
- 3.Wilson FH, Disse-Nicodeme S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, Gunel M, Milford DV, Lipkin GW, Achard JM, Feely MP, Dussol B, Berland Y, Unwin RJ, Mayan H, Simon DB, Farfel Z, Jeunemaitre X, Lifton RP. Science. 2001;293:1107–1112. doi: 10.1126/science.1062844. [DOI] [PubMed] [Google Scholar]
- 4.Zambrowicz BP, Abuin A, Ramirez-Solis R, Richter LJ, Piggott J, BeltrandelRio H, Buxton EC, Edwards J, Finch RA, Friddle CJ, Gupta A, Hansen G, Hu Y, Huang W, Jaing C, Key BW, Jr, Kipp P, Kohlhauff B, Ma ZQ, Markesich D, Payne R, Potter DG, Qian N, Shaw J, Schrick J, Shi ZZ, Sparks MJ, Van Sligtenhorst I, Vogel P, Walke W, Xu N, Zhu Q, Person C, Sands AT. Proc Natl Acad Sci U S A. 2003;100:14109–14114. doi: 10.1073/pnas.2336103100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu BE, Lee BH, Min X, Lenertz L, Heise CJ, Stippec S, Goldsmith EJ, Cobb MH. Cell Res. 2005;15:6–10. doi: 10.1038/sj.cr.7290256. [DOI] [PubMed] [Google Scholar]
- 6.Xu BE, Min X, Stippec S, Lee BH, Goldsmith EJ, Cobb MH. J Biol Chem. 2002;277:48456–48462. doi: 10.1074/jbc.M207917200. [DOI] [PubMed] [Google Scholar]
- 7.Vitari AC, Deak M, Collins BJ, Morrice N, Prescott AR, Phelan A, Humphreys S, Alessi DR. Biochem J. 2004;378:257–268. doi: 10.1042/BJ20031692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu BE, Stippec S, Lazrak A, Huang CL, Cobb MH. J Biol Chem. 2005;280:34218–34223. doi: 10.1074/jbc.M505735200. [DOI] [PubMed] [Google Scholar]
- 9.Jiang ZY, Zhou QL, Holik J, Patel S, Leszyk J, Coleman K, Chouinard M, Czech MP. J Biol Chem. 2005;280:21622–21628. doi: 10.1074/jbc.M414464200. [DOI] [PubMed] [Google Scholar]
- 10.Lee BH, Min X, Heise CJ, Xu BE, Chen S, Shu H, Luby-Phelps K, Goldsmith EJ, Cobb MH. Mol Cell. 2004;15:741–751. doi: 10.1016/j.molcel.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 11.Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, Sudhof TC. Cell. 1994;79:717–727. doi: 10.1016/0092-8674(94)90556-8. [DOI] [PubMed] [Google Scholar]
- 12.Tellman JT, McIntosh S, James DE. J Biol Chem. 1995;270:5857–5863. doi: 10.1074/jbc.270.11.5857. [DOI] [PubMed] [Google Scholar]
- 13.Oh E, Spurlin BA, Pessin JE, Thurmond DC. Diabetes. 2005;54:638–647. doi: 10.2337/diabetes.54.3.638. [DOI] [PubMed] [Google Scholar]
- 14.Ke B, Oh E, Thurmond DC. J Biol Chem. 2007;282:21786–21797. doi: 10.1074/jbc.M701661200. [DOI] [PubMed] [Google Scholar]
- 15.Thurmond DC, Ceresa BP, Okada S, Elmendorf JS, Coker K, Pessin JE. J Biol Chem. 1998;273:33876–33883. doi: 10.1074/jbc.273.50.33876. [DOI] [PubMed] [Google Scholar]
- 16.Thurmond DC, Kanzaki M, Khan AH, Pessin JE. Mol Cell Biol. 2000;20:379–388. doi: 10.1128/mcb.20.1.379-388.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thurmond DC, Pessin JE. EMBO J. 2000;19:3565–3575. doi: 10.1093/emboj/19.14.3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu SH, Latham CF, Gee CL, James DE, Martin JL. Proc Natl Acad Sci U S A. 2007;104:8773–8778. doi: 10.1073/pnas.0701124104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.D’Andrea-Merrins M, Chang L, Lam AD, Ernst SA, Stuenkel EL. J Biol Chem. 2007;282:16553–16566. doi: 10.1074/jbc.M610818200. [DOI] [PubMed] [Google Scholar]
- 20.Rickman C, Medine CN, Bergmann A, Duncan RR. J Biol Chem. 2007;282:12097–12103. doi: 10.1074/jbc.M700227200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmelzle K, Kane S, Gridley S, Lienhard GE, White FM. Diabetes. 2006;55:2171–2179. doi: 10.2337/db06-0148. [DOI] [PubMed] [Google Scholar]
- 22.Oh E, Thurmond DC. J Biol Chem. 2006;281:17624–17634. doi: 10.1074/jbc.M601581200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fu J, Naren AP, Gao X, Ahmmed GU, Malik AB. J Biol Chem. 2005;280:3178–3184. doi: 10.1074/jbc.M410044200. [DOI] [PubMed] [Google Scholar]
- 24.Thurmond DC, Gonelle-Gispert C, Furukawa M, Halban PA, Pessin JE. Mol Endocrinol. 2003;17:732–742. doi: 10.1210/me.2002-0333. [DOI] [PubMed] [Google Scholar]
- 25.Nevins AK, Thurmond DC. J Biol Chem. 2005;280:1944–1952. doi: 10.1074/jbc.M409528200. [DOI] [PubMed] [Google Scholar]
- 26.Xu BE, Stippec S, Chu PY, Lazrak A, Li XJ, Lee BH, English JM, Ortega B, Huang CL, Cobb MH. Proc Natl Acad Sci U S A. 2005;102:10315–10320. doi: 10.1073/pnas.0504422102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spurlin BA, Thurmond DC. Mol Endocrinol. 2006;20:183–193. doi: 10.1210/me.2005-0157. [DOI] [PubMed] [Google Scholar]
- 28.Wheeler MB, Sheu L, Ghai M, Bouquillon A, Grondin G, Weller U, Beaudoin AR, Bennett MK, Trimble WS, Gaisano HY. Endocrinology. 1996;137:1340–1348. doi: 10.1210/endo.137.4.8625909. [DOI] [PubMed] [Google Scholar]
- 29.Kashima Y, Miki T, Shibasaki T, Ozaki N, Miyazaki M, Yano H, Seino S. J Biol Chem. 2001;276:46046–46053. doi: 10.1074/jbc.M108378200. [DOI] [PubMed] [Google Scholar]
- 30.Mulder H, Lu D, Finley Jt, An J, Cohen J, Antinozzi PA, McGarry JD, Newgard CB. J Biol Chem. 2001;276:6479–6484. doi: 10.1074/jbc.M010364200. [DOI] [PubMed] [Google Scholar]
- 31.Daniel S, Noda M, Straub SG, Sharp GW, Komatsu M, Schermerhorn T, Aizawa T. Diabetes. 1999;48:1686–1690. doi: 10.2337/diabetes.48.9.1686. [DOI] [PubMed] [Google Scholar]
- 32.Knighton DR, Pearson RB, Sowadski JM, Means AR, Ten Eyck LF, Taylor SS, Kemp BE. Science. 1992;258:130–135. doi: 10.1126/science.1439761. [DOI] [PubMed] [Google Scholar]
- 33.Knighton DR, Zheng JH, Ten Eyck LF, Ashford VA, Xuong NH, Taylor SS, Sowadski JM. Science. 1991;253:407–414. doi: 10.1126/science.1862342. [DOI] [PubMed] [Google Scholar]
- 34.Biondi RM, Nebreda AR. Biochem J. 2003;372:1–13. doi: 10.1042/BJ20021641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Macaulay SL, Grusovin J, Stoichevska V, Ryan JM, Castelli LA, Ward CW. FEBS Lett. 2002;528:154–160. doi: 10.1016/s0014-5793(02)03279-9. [DOI] [PubMed] [Google Scholar]
- 36.Fujita Y, Sasaki T, Fukui K, Kotani H, Kimura T, Hata Y, Sudhof TC, Scheller RH, Takai Y. J Biol Chem. 1996;271:7265–7268. doi: 10.1074/jbc.271.13.7265. [DOI] [PubMed] [Google Scholar]
- 37.Xu BE, Stippec S, Lenertz L, Lee BH, Zhang W, Lee YK, Cobb MH. J Biol Chem. 2004;279:7826–7831. doi: 10.1074/jbc.M313465200. [DOI] [PubMed] [Google Scholar]
- 38.Vitari AC, Deak M, Morrice NA, Alessi DR. Biochem J. 2005;391:17–24. doi: 10.1042/BJ20051180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Z, Yang CL, Ellison DH. Biochem Biophys Res Commun. 2004;317:939–944. doi: 10.1016/j.bbrc.2004.03.132. [DOI] [PubMed] [Google Scholar]
- 40.Lee BH, Chen W, Stippec S, Cobb MH. J Biol Chem. 2007;282:17985–17996. doi: 10.1074/jbc.M702664200. [DOI] [PubMed] [Google Scholar]
- 41.Kahle KT, Wilson FH, Leng Q, Lalioti MD, O’Connell AD, Dong K, Rapson AK, MacGregor GG, Giebisch G, Hebert SC, Lifton RP. Nat Genet. 2003;35:372–376. doi: 10.1038/ng1271. [DOI] [PubMed] [Google Scholar]
- 42.Bhattacharyya RP, Remenyi A, Good MC, Bashor CJ, Falick AM, Lim WA. Science. 2006;311:822–826. doi: 10.1126/science.1120941. [DOI] [PubMed] [Google Scholar]
- 43.Bhattacharyya RP, Remenyi A, Yeh BJ, Lim WA. Annu Rev Biochem. 2006;75:655–680. doi: 10.1146/annurev.biochem.75.103004.142710. [DOI] [PubMed] [Google Scholar]
- 44.Coghlan VM, Perrino BA, Howard M, Langeberg LK, Hicks JB, Gallatin WM, Scott JD. Science. 1995;267:108–111. doi: 10.1126/science.7528941. [DOI] [PubMed] [Google Scholar]
- 45.Nagamatsu S, Nakamichi Y, Yamamura C, Matsushima S, Watanabe T, Ozawa S, Furukawa H, Ishida H. Diabetes. 1999;48:2367–2373. doi: 10.2337/diabetes.48.12.2367. [DOI] [PubMed] [Google Scholar]
- 46.Chan CB, MacPhail RM, Sheu L, Wheeler MB, Gaisano HY. Diabetes. 1999;48:997–1005. doi: 10.2337/diabetes.48.5.997. [DOI] [PubMed] [Google Scholar]
- 47.Houng A, Polgar J, Reed GL. J Biol Chem. 2003;278:19627–19633. doi: 10.1074/jbc.M212465200. [DOI] [PubMed] [Google Scholar]
- 48.Perez-Branguli F, Muhaisen A, Blasi J. Mol Cell Neurosci. 2002;20:169–180. doi: 10.1006/mcne.2002.1122. [DOI] [PubMed] [Google Scholar]