

Abstract
Dehydroepiandrosterone (DHEA), the major precursor of androgens and estrogens, has several beneficial effects on the immune system, on memory function, and in modulating the effects of diabetes, obesity, and chemical carcinogenesis. Treatment of rats with DHEA influences expression of cytochrome P450 (P450) genes, including peroxisome proliferator-activated receptor α (PPARα)- and pregnane X receptor (PXR)-mediated induction of CYP4As and CYP3A23, and suppression of CYP2C11. DHEA treatment elevated the expression and activities of CYP3A4, CYP2C9, CYP2C19, and CYP2B6 in primary cultures of human hepatocytes. Induction of CYP3A4 in human hepatocytes was consistent with studies in rats, but induction of CYP2Cs was unexpected. The role of PXR in this response was studied in transient transfection assays. DHEA activated hPXR in a concentration-dependent manner. Because CYP2B6 induction by DHEA in human hepatocytes might involve either PXR or constitutive androstane receptor (CAR) activation, we performed experiments in primary hepatocytes from CAR knockout mice and observed that CAR was required for maximal induction of Cyp2b10 by DHEA. Furthermore, CAR-mediated Cyp2b10 induction by DHEA was inhibited by the inverse agonist of CAR, androstanol (5α-androstan-3α-ol). Further evidence for CAR activation was provided by cytoplasmic/nuclear transfer of CAR upon DHEA treatment. Elucidation of CAR activation and subsequent induction of CYP2B6 by DHEA presented an additional mechanism by which the sterol can modify the expression of P450s. The effect of DHEA on the activation of the xenosensors PPARα, PXR, and CAR, and the consequent potential for adverse drug/toxicant interactions should be considered in humans treated with this nutriceutical agent.
Dehydroepiandrosterone (DHEA), the major secretory product of the adrenal cortex, is the most abundant steroid in humans and has multifunctional properties: it is a precursor of sex steroid hormones and a peroxisome proliferator at pharmacological dosages (Wu et al., 1989). DHEA is derived from cholesterol via a series of steps catalyzed by cytochrome P450 (P450) enzymes (Miller, 2002). Physiological concentrations of DHEA and its sulfate derivative in human plasma are in the micromolar range and increase significantly in individuals consuming high amounts of DHEA (Legrain et al., 2000). It is secreted primarily as 3β-sulfate conjugate (DHEA/DHEA-sulfate ratio 1:250 or 1:500 in plasma), which is taken up by target tissues and hydrolyzed by sulfatases back to DHEA (Labrie et al., 1997; Webb et al., 2006). DHEA is further metabolized to androgens and estrogens in testis and ovary (Labrie et al., 2005), or to hydroxylated metabolites in the liver. DHEA is hydroxylated to 7α/β-hydroxy- or 16α-hydroxymetabolites by microsomal P450 enzymes (Miller et al., 2004; Chalbot and Morfin, 2005). Some of the hydroxylated metabolites are further oxidized to oxo-derivatives (Fitzpatrick et al., 2001; Miller et al., 2004).
DHEA has been suggested to display beneficial effects in humans including reduced serum cholesterol, lower incidence of cardiovascular disease, weight loss, and improved control of plasma glucose in patients with diabetes (Kawano et al., 2003; Villareal and Holloszy, 2004). DHEA also protects against chemically induced carcinogenesis (Lubet et al., 1998) and certain immune deficiencies (Henderson et al., 1992) in animal models. Furthermore, DHEA has been documented to be a neuroactive steroid displaying neuroprotective and memory-enchancing effects (Baulieu et al., 2000). In addition to these beneficial effects, chronic administration of DHEA at high doses to rodents stimulates several pathophysiological changes such as hepatomegaly and hepatocellular carcinoma (Rao et al., 1992).
DHEA administration to rats at supraphysiological doses has been reported to increase the expression of several genes including CYP4As, CYP3As, NADPH:cytochrome c oxidoreductase, catalase, or fatty acyl-CoA oxidase (Wu et al., 1989; Prough et al., 1994; Webb et al., 1996; Gu et al., 2003). Several drugs including clofibrate and nafenopin also induce CYP4A transcription by activation of peroxisome proliferator-activated receptor α (PPARα). Although induction of CYP4A expression by DHEA requires activation of PPARα, DHEA does not appear to be a ligand for PPARα (Prough et al., 1994; Peters et al., 1996). Receptor activation is possibly the result of an effect of phosphorylation status of PPARα (Webb et al., 2006). Furthermore, studies with primary cultures of rat hepatocytes revealed that several oxidative metabolites of DHEA produced in liver are also able to activate CYP4A message and protein levels (Webb et al., 1996). 7-Oxidized metabolites (7-hydroxy-DHEA and 7-oxo-DHEA) do not serve as ligands for PPARα, but they are presumed to activate PPARα in a way similar to that of the parent compound. Treatment of rats with pharmacological doses of DHEA results in alteration in expression of other genes responsive to peroxisome proliferators, including fatty acyl-CoA oxidase or malic enzyme. DHEA exerts regulatory effects not only on peroxisome proliferation-associated genes, but also on CYP3A23 gene in rats (Singleton et al., 1999). This observation was due to DHEA activation of the pregnane X receptor (PXR), a member of the nuclear receptor superfamily known to mediate induction of CYP3A expression (Jones et al., 2000; Ripp et al., 2002). Negative regulation of CYP2C11 expression by peroxisome proliferators has also been demonstrated in rats, although with DHEA, PPARα coexpression does not seem to be required for negative regulation (Ripp et al., 2003). The main goal of the present study was to examine the P450-inducing capacity of DHEA and of two of its metabolites (7α-hydroxy-DHEA and 7-oxo-DHEA) in primary culture of human hepatocytes. In addition to PXR action, we provided evidence for a role of constitutive androstane receptor (CAR) in the induction of certain P450 genes by DHEA.
Materials and Methods
Chemicals
5-Androsten-3β-ol-17-one (DHEA), 5-androsten-3β,7α-diol-17-one (7α-hydroxy-DHEA), and 5-androsten-3β-ol-7,17-dione (7-oxo-DHEA) were purchased from Steraloids, Inc. (Newport, RI). Dexamethasone, SR-12813 (tetra-ethyl 2-(3,5-di-tert-butyl-4-hydroxyphenyl)ethenyl-1,1-bisphosphonate), 5α-androstan-3β-ol (androstanol), and dimethyl sulfoxide (DMSO) were the products of Sigma Chemie GmbH (Deisenhofen, Germany). 3,3′,5,5′-Tetrachloro-1,4-bis(pyridyloxy)benzene (TCPOBOP) was purchased from Bayer AG (Leverkusen, Germany). 6-(4-Chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde-O-(3,4-dichlorobenzyl)oxime (CITCO) was purchased from BIOMOL Research Laboratories (Plymouth Meeting, PA). Chemicals for hepatocyte isolation, cell culture media, and supplements were purchased from Sigma Chemie GmbH and Merck (Darmstadt, Germany).
Isolation and Culture of Human Hepatocytes
Human livers were obtained from kidney transplant donors at the Transplantation and Surgical Clinic, Semmelweis University (Budapest, Hungary). Permission of the Hungarian Regional Committee of Science and Research Ethics was obtained to use human tissues. Clinical histories of the donors are shown in Table 1. Liver cells were isolated by the method of Bayliss and Skett (1996). Hepatocytes having viability better than 90% as determined by trypan blue exclusion were used in the experiments. The cells were plated at a density of 1.7 × 105 cells/cm2 in plastic dishes precoated with collagen in medium described by Ferrini et al. (1998). After overnight culture, the medium was replaced by serum-free medium. Forty-eight hours after serum deprivation, cells were cultured in the presence or absence of inducers for 48 h. Hepatocytes were treated with dexamethasone (1 and 10 μM), DHEA (50 μM), 7α-hydroxy-DHEA (50 μM), or 7-oxo-DHEA (50 μM).
TABLE 1.
Clinical histories of human donors
| Donor | Age | Sex | Race | Cause of Death | Medication |
|---|---|---|---|---|---|
| yr | |||||
| HH-075 | 63 | Male | Caucasian | Subarachnoidal hemorrhage | Noradrenaline |
| HH-078 | 47 | Female | Caucasian | Subarachnoidal hemorrhage | Dopamine, noradrenaline |
| HH-079 | 44 | Female | Caucasian | Subarachnoidal hemorrhage | Clindamycin, noradrenaline |
| HH-080 | 29 | Female | Caucasian | Subarachnoidal hemorrhage | Ceftriaxone, vancomycin |
| HH-082 | 44 | Male | Caucasian | Stroke | Noradrenaline |
| HH-083 | 32 | Female | Caucasian | Subdural hemorrhage | |
| HH-086 | 39 | Male | Caucasian | Cerebral contusion | Ceftriaxone, mannitol, noradrenaline |
Preparation and Culture of Primary Mouse Hepatocytes
The first breeding pair of CAR knockout C57BL/6J mice was a generous gift of D. D. Moore from the Department of Molecular and Cellular Biology, Baylor College of Medicine (Houston, Texas) (Wei et al., 2000). Further breeding of CAR knockout mice was carried out at the Biozentrum, University of Basel (Basel, Switzerland). Primary hepatocytes of control (CAR+/+) and CAR knockout (CAR−/−) mice were prepared by a two-step collagenase method (Bayliss and Skett, 1996). Liver cells were plated at a density of 3 × 105 cells/well on 12-well plates coated with collagen and maintained in medium described by Ferrini et al. (1998). Hepatocytes were exposed to TCPOBOP (10 μM), DHEA (25 μM), or vehicle (DMSO 0.1%) for 24 h.
P450 Enzyme Assays
Microsomal fraction from cultured human hepatocytes was prepared by differential centrifugation (van der Hoeven and Coon, 1974). Protein content of microsomes was determined by the method of Lowry et al. (1951), with bovine serum albumin as the standard. Published methods were followed to determine selective P450 enzyme activities: mephenytoin N-demethylation for CYP2B6 (Heyn et al., 1996), tolbutamide 4-hydroxylation for CYP2C9 (Miners and Birkett, 1996), mephenytoin 4′-hydroxylation for CYP2C19 (Srivastava et al., 1991), nifedipine oxidation (Guengerich et al., 1986), and midazolam 1′- and 4-hydroxylation (Kronbach et al., 1989) for CYP3A4/5. The incubation mixture contained NADPH-generating system (1 mM NADPH, 10 mM glucose 6-phosphate, 5 mM MgCl2, and 2 units/ml glucose-6-phosphate dehydrogenase), microsomes, and various selective substrates for P450 forms (mephenytoin, tolbutamide, midazolam, or nifedipine). The rates of enzyme activity were linearly dependent upon the amount of microsomal protein added for the 10- to 30-min incubation period. The metabolic extraction procedures and high-performance liquid chromatography analyses were performed according to the published methods. P450 enzyme assays were performed in triplicate and means ± standard deviations were calculated. For comparison among untreated and treated groups, statistical analysis of the results was carried out using a two-tailed t test with p < 0.05 as the criterion for significance. Due to high variation in basic P450 activities of human hepatocytes, the entire experiment was repeated in hepatocytes isolated from five to six donors to confirm the results.
RNA Extraction and Quantitative RT-PCR
Total RNA was isolated from human and mouse hepatocytes using TRIzol reagent (Invitrogen, Carlsbad, CA). Ten million liver cells were homogenized in 1 ml of TRIzol reagent, and total RNA was extracted according to the manufacturer's instructions. The RNA was precipitated using ethanol and stored at −80°C for further analyses. The primers used for RT-PCR analyses are shown in Table 2. RNA (3 μg) was reverse-transcribed into single-stranded cDNA using Transcriptor First Strand cDNA synthesis kit (Roche Diagnostics GmbH, Mannheim, Germany), and then real-time PCR with human cDNA was performed using FastStart TaqDNA polymerase (LightCycler TaqMan Master, Roche Diagnostics GmbH) and UPL (Universal Probe Library) probes for CYP2B6, CYP2C9, CYP2C19, and CYP3A4 (Roche Diagnostics GmbH). Mouse cDNA was analyzed with FAM-labeled Taq probe for Cyp2b10 (Microsynth GmbH, Balgach, Switzerland) using AmpliTaq DNA Polymerase (TaqMan Universal PCR Master Mix, No Amperase UNG, Applied Biosystems, Foster City, CA). The quantity of target RNA relative to that of housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was determined. P450 mRNA levels were quantified by RT-PCR measurements in the same human hepatocytes in which P450 activities were measured. Statistical analysis of the results obtained by RT-PCR was carried out in a manner similar to that of P450 activities.
TABLE 2.
Sequences of PCR primers and probes
| Primer | Sequence | Probe | Sequence |
|---|---|---|---|
| Human genes | |||
| CYP2B6 Forward | 5′-AAAGCGGAGTGTGGAGGA-3′ | CYP2B6 | FAM-5′-AGGAGGAG-3′-BHQ |
| Reverse | 5′-AAGGTGGGGTCCATGAGG-3′ | ||
| CYP2C9 Forward | 5′-GTGCACGAGGTCCAGAGATAC-3′ | CYP2C9 | FAM-5′-CTTCTCCC-3′-BHQ |
| Reverse | 5′-CAGGGAAATTAATATGGTTGTGC-3′ | ||
| CYP2C19 Forward | 5′-TGAAGGTGGAAATTTTAAGAAAAGTAA-3′ | CYP2C19 | FAM-5′-CAGCAGGA-3′-BHQ |
| Reverse | 5′-CCCTCTCCCACACAAATCC-3′ | ||
| CYP3A4 Forward | 5′-CATGGACTTTTTAAGAAGCTTGG-3′ | CYP3A4 | FAM-5′-CTCTGCCT-3′-BHQ |
| Reverse | 5′-TTCCATGTCAAACATACAAAAGC-3′ | ||
| GAPDH Forward | 5′-AGCCACATCGCTCAGACA-3′ | GAPDH | FAM-5′-TGGGGAAG-3′-BHQ |
| Reverse | 5′-GCCCAATACGACCAAATCC-3′ | ||
| Mouse genes | |||
| Cyp2b10 Forward | 5′-CAATGTTTAGTGGAGGAACTGCG-3′ | Cyp2b10 | FAM-5′-CCCAGGGAGCCCCCCTGGA-3′-TAMRA |
| Reverse | 5′-CACTGGAAGAGGAACGTGGG-3′ | ||
| GAPDH Forward | 5′-CCAGAACATCATCCCTGCATC-3′ | GAPDH | FAM-5′-CCGCCTGGAGAAACCTGCCAAGTATG-3′-TAMRA |
| Reverse | 5′-GGTCCTCAGTGTAGCCCAAGAT-3′ |
FAM, fluorescein labeling; BHQ, black hole quencher; TAMRA, 5-carboxytetramethylrhodamine-labeled probe.
Cell Culture and Transfection
HuH7 cell line was obtained from the European Collection of Cell Cultures (Salisbury, UK) and maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 1 mM glutamine, 10 mM sodium pyruvate, and 100 μg/ml penicillin and streptomycin. The pSG5, pSG5-hPXR, pGL3(CYP3A4/XREM[−7800/−7200]/−262/+11)LUC [containing the distal xenobiotic-responsive element module (XREM, −7800/−7200) linked to the proximal CYP3A4 promoter (−262/+11)], and pSV-β-galactosidase have been described elsewhere (Pascussi et al., 2001, 2003a). For reporter assays, 5 × 105 HuH7 cells were transiently transfected with 10 ng of expression plasmid (pSG5 or pSG5-hPXR) together with 100 ng of luciferase reporter constructs pGL3(CYP3A4/XREM[−7800/−7200]/−262/+11)LUC and 50 ng of pSV-β-galactosidase expression plasmid for transfection quality control using FeGENE6 transfection reagent (Roche Applied Science, Basel, Switzerland). Approximately 20 to 30% of the cells were transfected. After 16 h, the medium was changed, and the cells were treated with DHEA (1, 10, and 50 μM), SR-12813 (1 μM) or solvent (DMSO 0.1%) in Dulbecco's modified Eagle's medium containing 5% delipidated and charcoal-treated calf serum. After 24 h of incubation, luciferase and β-galactosidase activities were measured as described previously (Pascussi et al., 2003a). Values are expressed as the mean ± standard deviation for four experiments measured in triplicate and represent luciferase activity divided by the β-galactosidase activity of each extract. Reporter gene activity of the DMSO-treated cells served as a control and was set to unity.
Nuclear Translocation Assay
Mouse hepatocytes were cultured on glass coverslips coated with collagen. After 4 h of attachment, serum-free medium was added to the cells. Transfection was carried out in Opti-MEM I Reduced Serum Medium (Invitrogen) using Lipofectamine 2000 transfection reagent (Invitrogen). Cells were incubated with the pEGFP-c1-hCAR plasmid kindly provided by M. Negishi (Laboratory of Reproductive and Developmental Toxicology, National Institute of Environmental Health Sciences, Research Triangle Park, NC). Eighteen hours after transfection, hepatocytes were exposed to DHEA (50 μM) or CITCO (100 nM) for 4 h, then washed twice with phosphate-buffered saline solution and fixed in 4% (w/v) paraformaldehyde. Cells were stained with DAPI (4′-6-diamidino-2-phenylindole) and then washed again with phosphate-buffered saline. The coverslips were transferred to microscope slides and visualized in Moviol mounting medium. The intracellular localization of CAR-GFP fusion protein was determined by fluorescence microscopy using a Leica DM5000B microscope (Leica Microsystems, Wetzlar GmbH, Wetzlar, Germany) and AnalySIS Pro software (Soft Imaging System GmbH, Münster, Germany).
Statistical Analysis
For comparison among several groups, statistical analysis of the results obtained in experiments with human and mouse hepatocytes and in cell line transfection was carried out using a paired two-tailed t test with p < 0.05 as the criterion for significance (GraphPad InStat version 3.0, GraphPad Software, San Diego, CA).
Results
Previous work has clearly demonstrated the P450-inducing effect of DHEA in rats (Prough et al., 1994; Singleton et al., 1999; Gu et al., 2003). Work by Peters et al. (1996) and Ripp et al. (2002) has shown that DHEA acts on CYP4As and CYP3A23 at the transcriptional level by PPARα- and PXR-mediated events, respectively. Oxidative metabolites of DHEA have also been reported to activate PPARα and consequently to induce CYP4A in rat hepatocytes (Webb et al., 2006). The present study further characterizes the actions of DHEA and of some of its metabolites, 7α-hydroxy-DHEA and 7-oxo-DHEA (Miller et al., 2004; Chalbot and Morfin, 2005), to induce P450s in adult human hepatocytes. We also attempted to elucidate the role of nuclear receptors, PXR and CAR, in the induction of P450s by DHEA.
P450 Induction by DHEA in Human Hepatocytes
It was ascertained in preliminary experiments that 50 μM DHEA was required for maximal induction of P450 activities in cultured human hepatocytes (data not shown). In subsequent studies, this concentration of DHEA, 7α-hydroxy-DHEA, or 7-oxo-DHEA was used for treatment of hepatocytes. The synthetic glucocorticoid, dexamethasone, was used as a reference compound at relatively high concentrations (1 and 10 μM). At supramicromolar concentrations, dexamethasone binds to and activates PXR, producing induction of P450s such as CYP3A4 (Pascussi et al., 2001). P450 enzyme activities and mRNA levels were determined in primary human hepatocytes isolated from several donors (as indicated in Figs. 1 and 2) because of high individual variance in basic activities or P450 expression of the cells. To evaluate significant inducibility of human hepatocytes by DHEA and its metabolites, a paired t test was performed with p < 0.05 as the criterion for significance. Figures 1 and 2 present P450 activities and mRNA levels relative to untreated hepatocytes (0.1% DMSO-treated cells).
Fig. 1.

Induction of CYP3A4 activities (a) and mRNA levels (b) in primary human hepatocytes. Human liver cells were treated for 48 h with dexamethasone (DXM, 1 and 10 μM), DHEA (50 μM), and its metabolites: 7α-hydroxy-DHEA (OH-DHEA, 50 μM) and 7-oxo-DHEA (oxo-DHEA, 50 μM). CYP3A4 activities were determined in microsomes prepared from hepatocytes isolated from five donors. Levels of CYP3A4 mRNA in human hepatocytes (n = 5 donors) were quantified and normalized to GAPDH as described under Materials and Methods. Statistical analysis of the results obtained in untreated and treated cells indicated significant increase in both the activities and mRNA levels of CYP3A4 as a consequence of various treatments. Controls for each independent experiment were assigned values of 1 and results of treatments are expressed relative to the controls. Error bars represent standard deviations from the mean of five donors.
Fig. 2.
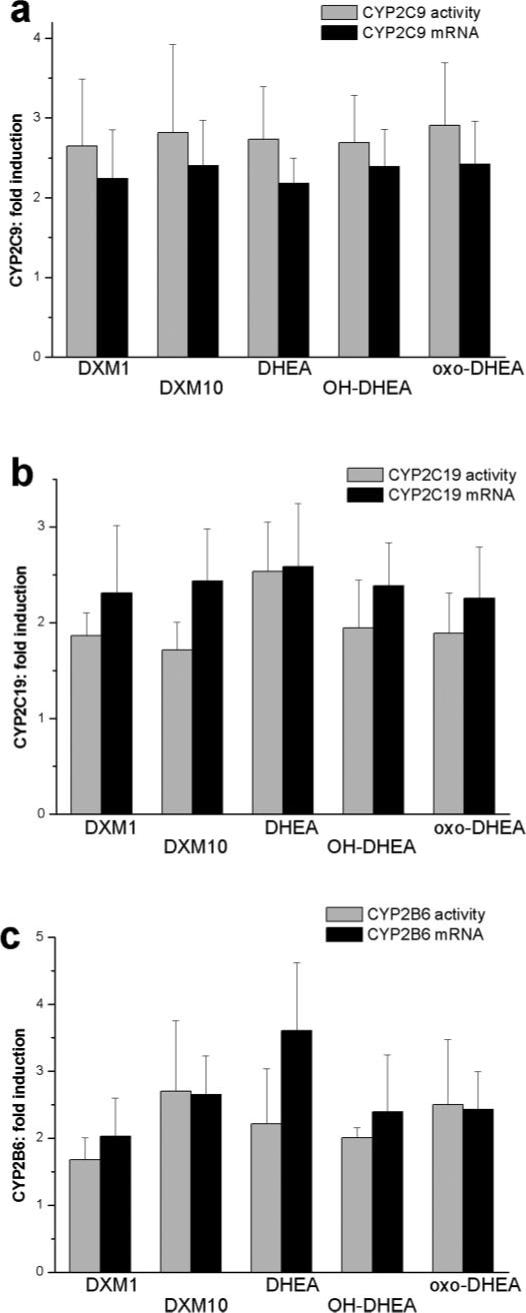
Induction of CYP2C9 (a), CYP2C19 (b) and CYP2B6 (c) activities and mRNA levels in primary human hepatocytes. Cells were treated for 48 h with dexamethasone (DXM, 1 and 10 μM), DHEA (50 μM) and its metabolites: 7α-hydroxy-DHEA (OH-DHEA, 50 μM) and 7-oxo-DHEA (oxo-DHEA, 50 μM). P450 activities were determined in microsomes prepared from hepatocytes isolated from six donors. Levels of CYP2C9, CYP2C19, and CYP2B6 mRNAs were quantified and normalized to GAPDH as described under Materials and Methods. Statistical analysis of the results obtained in untreated and treated cells indicated significant increase in both the activities and mRNA levels as a consequence of various treatments. Controls for each independent experiment were assigned values of 1 and results of treatments are expressed relative to the controls. Error bars represent standard deviations from the mean of six donors.
Nifedipine oxidation, and midazolam 1′- and 4-hydroxylation activities of CYP3A4 increased by 3- to 4-fold in dexamethasone-treated cells as expected from the work of others (Ledirac et al., 2000). Treatments with DHEA or its metabolites resulted in a more than 2-fold induction of CYP3A4 activities (Fig. 1a). The elevation in enzyme activities was nearly concomitant with the -fold changes in levels of CYP3A4 mRNA (Fig. 1b). Treatments of hepatocytes with DHEA or its two oxidative metabolites produced nearly identical increases in enzyme activities or mRNA levels of CYP3A4. These results suggested that DHEA and its metabolites caused transcriptional activation of CYP3A4 gene involving the action of nuclear receptors, most likely of PXR.
Tolbutamide hydroxylation activity and expression of CYP2C9 were also significantly induced by DHEA and two of its metabolites (Fig. 2a). The degree of the elevation of CYP2C9 activity and mRNA levels relative to the control cell population (2- to 3-fold) was identical with that of dexamethasone-treated cells. Similar results were observed for CYP2C19. DHEA treatment caused an appreciable 2- to 3-fold increase in mephenytoin 4-hydroxylation activity and mRNA levels of CYP2C19 (Fig. 2b). The two oxidative metabolites of DHEA also exerted the same CYP2C19-inducing effect as the parent compound. Furthermore, mephenytoin N-demethylation activity of CYP2B6 was potently induced by DHEA and its metabolites (Fig. 2c). Nearly identical increases in CYP2B6 activity were obtained in liver cells treated with DHEA, 7α-hydroxy-DHEA, and 7-oxo-DHEA. The 48-h treatment resulted in an approximately 2- or 2.5-fold increase similar to that of dexamethasone. Similar effects were observed at the level of CYP2B6 mRNA (Fig. 2c).
PXR Activation by DHEA
DHEA is known to induce expression of CYP4As through activation of PPARα. It is also an activator of PXR that mediates induction of CYP3A23 expression in rats (Ripp et al., 2002). It has been demonstrated that PXR is involved in regulation of the expression of several P450s (CYP2B6, CYP2C9, CYP2C19, and CYP3A4) in human (Pascussi et al., 2001, 2003b). Our results in human hepatocytes also suggested that DHEA transcriptionally activated P450 genes involving the action of PXR. To assess whether DHEA was able to activate human PXR, transient cotransfection assay was performed in the HuH7 cell line. The effect of DHEA on transcriptional activation of hPXR was investigated using reporter plasmid (pGL3(CYP3A4/XREM[−7800/−7200]/−262/+11)LUC). The reporter plasmid was cotransfected with hPXR. DHEA treatment resulted in a concentration-dependent increase in ligand-induced transactivation of reporter construct by hPXR (Fig. 3). However, the 3-fold induction observed at 50 μM DHEA was lower than the increase in luciferase activity caused by the well known PXR agonist SR-12813 (Jones et al., 2000). It should be noted that DHEA was able to slightly activate reporter plasmid without coexpression of hPXR to some extent (1.8-fold).
Fig. 3.

hPXR activation in response to DHEA and the PXR agonist SR-12813. HuH7 cells were transiently transfected with expression plasmid (pSG5 or pSG5-hPXR) together with luciferase reporter constructs pGL3(CYP3A4/XREM[−7800/−7200]/−262/+11)LUC and pSV-β-galactosidase expression plasmid for transfection quality control. The cells were treated for 24 h with DHEA (1, 10, and 50 μM), SR-12813 (1 μM), and normalized luciferase/β-galactosidase activities were determined as described under Materials and Methods; the activities are the mean ± S.D. (n = 4).
The Role of CAR in CYP2B Induction
The finding that DHEA was active in CYP2B6 induction in human hepatocytes raised the question of whether CAR activation may occur as a result of DHEA treatment or PXR activation by DHEA is responsible for CYP2B6 induction. Hepatocytes isolated from CAR knockout and wild-type mice were tested for their responsiveness to DHEA. RT-PCR analysis of RNA prepared from hepatocytes of wild-type mice revealed a substantial increase (approximately 8-fold) in mRNA of the mouse ortholog Cyp2b10 as a result of DHEA treatment compared with vehicle-treated controls (Fig. 4). Mouse hepatocytes from wild-type animals exhibited strong induction of Cyp2b10 (approximately 20-fold) after exposure to the mouse CAR activator TCPOBOP (10 μM). Lack of expression of CAR in hepatocytes from CAR-ablated mice resulted in abolition of Cyp2b10 induction by TCPOBOP. Although the induction of Cyp2b10 expression by DHEA was also reduced in primary hepatocytes of CAR knockout mice, it was not completely inhibited. Approximately 4-fold induction of Cyp2b10 by DHEA was still present in liver cells of CAR−/− mice (Fig. 4). These observations clearly established that CAR was involved in maximal induction of Cyp2b10 by DHEA, suggesting that both CAR and PXR may be required for the maximal inductive effect of DHEA on Cyp2b10 gene expression.
Fig. 4.
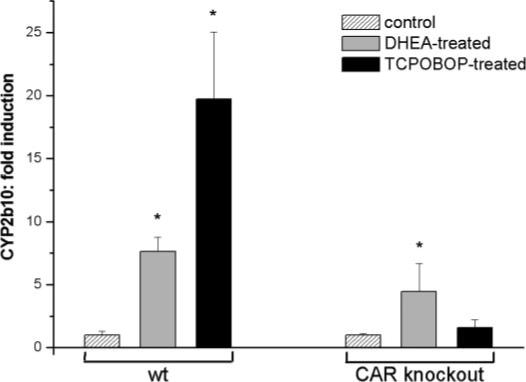
Cyp2b10 induction by DHEA and TCPOBOP in primary hepatocytes of wild-type and CAR knockout mice. Hepatocytes from wild-type (wt) and CAR knockout mice were treated with DHEA (25 μM) or TCPOBOP (10 μM). Levels of Cyp2b10 mRNA were quantified and normalized to GAPDH as described under Materials and Methods. The normalized levels of Cyp2b10 mRNA are expressed as the average ± S.D. *, significantly different from the controls, p < 0.05.
To evaluate the role of CAR in DHEA action, the effect of androstanol on Cyp2b10 induction was also investigated in mouse hepatocytes. Androstanol significantly reduced the induction of Cyp2b10 by the CAR ligand, TCPOBOP. Cyp2b10 mRNA levels of the cells treated with TCPOBOP decreased by approximately 50 to 60% in the presence of androstanol (Fig. 5). Androstanol was also observed to repress the induction of Cyp2b10 by DHEA. Androstanol treatment decreased Cyp2b10 mRNA levels by 40 to 50% in DHEA-induced hepatocytes. Thus, androstanol, the inverse agonist of CAR (Forman et al., 1998), reduced the expression of Cyp2b10 both in DHEA- and in TCPOBOP-induced cells.
Fig. 5.

The effect of androstanol on Cyp2b10 expression in DHEA- and TCPOBOP-induced mouse hepatocytes. Cyp2b10 of mouse hepatocytes was induced by DHEA (25 μM) or TCPOBOP (10 μM) in the presence or absence of the inverse agonist of CAR, androstanol. Levels of Cyp2b10 mRNA were quantified and normalized to GAPDH as described under Materials and Methods. The normalized levels of Cyp2b10 mRNA are expressed as the average ± S.D.
The capability of DHEA to trigger CAR translocation from cytoplasm to nucleus was also tested in mouse primary hepatocytes expressing human CAR. The subcellular localization of hCAR was determined by fluorescence microscopy in hepatocytes infected with plasmid coding for hCAR in fusion with enhanced green fluorescent protein (pEGFP-c1-hCAR). In control hepatocytes, hCAR-GFP was clearly located in the cytoplasm, whereas it was undetectable in the nuclei in the absence of stimulation (Fig. 6). After 4 h exposure to the selective human CAR agonist CITCO (Maglich et al., 2003), cytoplasmic/nuclear transfer of CAR was observed primarily in nuclei of the cells, as expected. In response to DHEA treatment, the pattern of CAR-GFP localization was similar to that seen after CITCO treatment (Fig. 6). The corresponding DAPI-stained and merged images provided further visual evidence indicating that DHEA promoted efficient CAR translocation into the nucleus of hepatocytes.
Fig. 6.

Subcellular localization of hCAR. Primary mouse hepatocytes were transfected with an expression vector containing GFP fused to human CAR. After transfection, cells were treated with 0.1% DMSO (control), CITCO (100 nM), or DHEA (50 μM). DAPI staining was used for visualization of nuclei as described under Materials and Methods.
Discussion
DHEA administration to rats affects the expression of several P450 genes. DHEA has been shown to increase the levels of CYP4As (Prough et al., 1994) and CYP3A23 (Singleton et al., 1999), and to suppress the expression of CYP2C11 (Ripp et al., 2003). Nuclear receptors such as PPARα and PXR appear to be involved in DHEA action in rats (Peters et al., 1996; Ripp et al., 2002). In addition, 7-oxidized metabolites of DHEA also induce CYP4A1 message and protein levels in rat hepatocytes contributing to the effect of the parent compound (Webb et al., 2006). In the present study, we therefore investigated the induction of P450s by DHEA and two of its oxidative metabolites in primary cultures of human hepatocytes and, in particular, the role of nuclear receptors PXR and CAR in DHEA action.
Human hepatocytes in culture as induction system were validated by testing the classical inducer compound, the steroid dexamethasone, on P450 gene expression. Concentrations of dexamethasone equal to or above 1 μM potently increased both the activities and mRNA levels of CYP3A4 and, to a lesser extent, of CYP2C9, CYP2C19, and CYP2B6. The present study then revealed that DHEA treatment also elevated the expression and activities of CYP3A4, CYP2C9, CYP2C19, and CYP2B6 similar to those of dexamethasone. In addition, the DHEA metabolite 7α-hydroxy-DHEA or 7-oxo-DHEA resulted in the same inducing action as did the parent compound. This means that biotransformation does not lead to inactivation of DHEA as P450 inducer.
The finding that DHEA potently induced CYP3A4 in human hepatocytes was consistent with previous studies in rats (Singleton et al., 1999). PXR activation has been proposed to mediate the increase in CYP3A expression (Ripp et al., 2002). But in contrast to studies in rats (Ripp et al., 2003), DHEA did not mediate suppression of CYP2Cs in human hepatocytes. Moreover, DHEA treatment led to up-regulation of both CYP2C9 and CYP2C19, suggesting the involvement of PXR. DHEA was able to activate hPXR in a concentration-dependent manner, although it appeared to be a weak PXR activator compared with the well known PXR agonist SR-12813.
DHEA-mediated transcription of CYP2B has not been unequivocally shown in previous studies. The current studies demonstrated an efficient CYP2B6 induction exerted by DHEA in primary cultures of human hepatocytes. The activation of human CAR by compounds such as phenobarbital and CITCO or mouse CAR by TCPOBOP has been well documented (Maglich et al., 2003). Experiments carried out in hepatocytes of CAR knockout mice demonstrated that ablated levels of CAR reduced, but did not abolish, response to DHEA. Partial inhibition of DHEA-mediated Cyp2b10 induction by the inverse agonist androstanol presented further evidence for the contribution of CAR to the enhancement of Cyp2b10 expression. The inactive nuclear receptor CAR, which is cytoplasmic in hepatocytes, undergoes rapid nuclear translocation upon treatment with CAR activators, phenobarbital, or CITCO. Although binding of CAR activators to the receptor may not occur, CAR translocation to the nucleus is the limiting step in the CAR activation process, followed by CAR/RXR heterodimer formation and binding of the proteins to the DNA of target genes (Waxman, 1999). The activation of CAR by DHEA was thus demonstrated in in vitro nuclear translocation assays. Using primary cultures of mouse hepatocytes expressing hCAR, the effect of DHEA on cytoplasmic/nuclear shuttling of CAR was compared with that of CITCO. In agreement with Maglich et al. (2003), the occurrence of translocation of hCAR from cytoplasm into the nucleus after CITCO treatment was well recognized. DHEA treatment also induced CAR translocation into the nucleus, presenting further evidence for CAR activation by DHEA. These findings suggested that CAR was required for maximal induction of Cyp2b10 by DHEA. However, in the absence of CAR, there still is an apparent receptor-mediated induction of P450 mRNA, protein, and activity that could be due to PXR (Ripp et al., 2003) or yet unidentified transcription factors.
In conclusion, we provide evidence for the induction of several P450s (CYP3A4, CYP2C9, CYP2C19, and CYP2B6) by DHEA and by two of its oxidative metabolites, 7α-hydroxy-DHEA and 7-oxo-DHEA, in human hepatocytes. Our results also provide convincing evidence for the activation of hPXR and hCAR by DHEA as prerequisite for the transcriptional activation of these P450 genes. In other studies, the activation of several nuclear receptors including PPARα, PXR, and estrogen receptor by DHEA and its metabolites has been demonstrated. CAR activation and subsequent induction of CYP2B6 by DHEA present an additional mechanism by which DHEA, a widely used nutriceutical, can modify the expression of P450s. Because the inductive response of the PXR and CAR system is associated with changes in the kinetics of numerous drugs and steroids and includes drug-drug interactions and adverse drug effects, the unrestricted use of DHEA as a panacea for various health problems should be carefully considered.
Acknowledgments
We are indebted to Maria Grenyi for skillful assistance in this study. We are also grateful to M. Negishi (National Institute of Environmental Health Sciences, Research Triangle Park, NC) and to D. D. Moore (Department of Molecular and Cellular Biology, Baylor College of Medicine, Houston, Texas) for providing pEGFP-c1-hCAR plasmid and CAR knockout mice, respectively.
This study was supported by János Bolyai Research Scholarship of the Hungarian Academy of Sciences (BO/00413/05) (K.M.), Slovenian-Hungarian Inter-governmental S&T Cooperation Programme (SLO-2/04) (K.K., D.R., K.M.), the European Community (LSHGCT-2005-512096, Steroltalk) (K.K., J.-M.P., D.R., U.A.M., K.M.), the National Institutes of Health (DK54774) (R.A.P.), Agence Nationale de la Recherche JCJC-05-47810 (J.-M.P.), and the Swiss National Science Foundation (V.T., U.A.M.).
ABBREVIATIONS
- DHEA
dehydroepiandrosterone or 5-androsten-3β-ol-17-one
- androstanol
5α-androstan-3α-ol
- CAR
constitutive androstane receptor
- CITCO
6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde-O-(3,4-dichlorobenzyl)oxime
- P450
cytochrome P450
- DAPI
4′-6-diamidino-2-phenylindole
- 7α-hydroxy-DHEA
5-androsten-3β,7α-diol-17-one
- 7-oxo-DHEA
5-androsten-3β-ol-7,17-dione
- DMSO
dimethyl sulfoxide
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GFP
green fluorescent protein
- PPARα
peroxisome proliferator-activated receptor α
- PXR
pregnane X receptor
- TCPOBOP
3,3′,5,5′-tetrachloro-1,4-bis(pyridyloxy)benzene
- SR-12813
tetra-ethyl 2-(3,5-di-tert-butyl-4-hydroxyphenyl)ethenyl-1,1-bisphosphonate
- RT-PCR
reverse transcriptase-polymerase chain reaction
- FAM
5-carboxyfluorescein
- XREM
xenobiotic-responsive element module
- h
human.
References
- Baulieu E-E, Thomas G, Legrain S, Lahlou N, Roger M, Debuire B, Faucounau V, Girard L, Hervy M-P, Latour F, Leaud M-C, Mokrane A, Pitti-Ferrandi H, Trivalle C, de Lacharriere O, Nouveau S, Rakoto-Arison B, Souberbielle J-C, Raison J, Le Bouc Y, Raynaud A, Girerd X, Forette F. Dehydroepiandrosterone (DHEA), DHEA sulphate, and aging: contribution of the DHEAge Study to a sociobiomedical issue. Proc Natl Acad Sci U S A. 2000;97:4279–4284. doi: 10.1073/pnas.97.8.4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayliss KM, Skett P. Isolation and culture of human hepatocytes. In: Jones GE, editor. Human Cell Culture Protocols. Humana Press; Totowa, NJ: 1996. pp. 369–390. [DOI] [PubMed] [Google Scholar]
- Chalbot S, Morfin R. Human liver S9 fractions: metabolism of dehydroepiandrosterone, epiandrosterone, and related 7-hydroxylated derivatives. Drug Metab Dispos. 2005;33:563–569. doi: 10.1124/dmd.104.003004. [DOI] [PubMed] [Google Scholar]
- Ferrini JB, Ourlin JC, Pichard L, Fabre G, Maurel P. Human hepatocyte culture. In: Phillips IR, Shephard EA, editors. Cytochrome P450 Protocols. Humana Press; Totowa, NJ: 1998. pp. 341–352. [Google Scholar]
- Fitzpatrick JL, Ripp SL, Smith NB, Pierce WM, Prough RA. Metabolism of DHEA by cytochrome P450 in rat and human liver microsomal fractions. Arch Biochem Biophys. 2001;389:278–287. doi: 10.1006/abbi.2001.2341. [DOI] [PubMed] [Google Scholar]
- Forman BM, Tzameli I, Choi HS, Chen J, Simha D, Seol W, Evans RM, Moore DD. Androstane metabolites bind to and deactivate the nuclear receptor CAR-beta. Nature. 1998;395:612–615. doi: 10.1038/26996. [DOI] [PubMed] [Google Scholar]
- Gu S, Ripp SL, Prough RA, Geoghegan TE. Dehydroepiandrosterone affects the expression of multiple genes in rat liver including 11β-hydroxysteroid dehydrogenase type 1: a cDNA array analysis. Mol Pharmacol. 2003;63:722–731. doi: 10.1124/mol.63.3.722. [DOI] [PubMed] [Google Scholar]
- Guengerich FP, Martin MV, Beaune PH, Kremers P, Wolff T, Waxman DJ. Characterization of rat and human liver microsomal cytochrome P-450 forms involved in nifedipine oxidation, a prototype for genetic polymorphism in oxidative drug metabolism. J Biol Chem. 1986;261:5051–5060. [PubMed] [Google Scholar]
- Henderson E, Yang JY, Swartz A. Dehydroepiandrosterone (DHEA and synthetic DHEA analogs are modest inhibitors of HIV-1 IIIB replication. AIDS Res Hum Retroviruses. 1992;8:625–631. doi: 10.1089/aid.1992.8.625. [DOI] [PubMed] [Google Scholar]
- Heyn H, White RB, Stevens JC. Catalytic role of cytochrome P4502B6 in the N-demethylation of S-mephenytoin. Drug Metab Dispos. 1996;24:948–954. [PubMed] [Google Scholar]
- Jones SA, Moore LB, Shenk JL, Wisely GB, Hamilton GA, McKee DD, Tomkinson NCO, LeCluyse EL, Lambert MH, Willson TM, Kliewer SA, Moore JT. The pregnane X receptor: a promiscuous xenobiotic receptor that has diverged during evolution. Mol Endocrinol. 2000;14:27–39. doi: 10.1210/mend.14.1.0409. [DOI] [PubMed] [Google Scholar]
- Kawano H, Yasue H, Kitagawa A, Hirai N, Yoshida T, Soejima H, Miyamoto S, Nakano M, Ogawa H. Dehydroepiandrosterone supplementation improves endothelial function and insulin sensitivity in men. J Clin Endocrinol Metab. 2003;88:3190–3195. doi: 10.1210/jc.2002-021603. [DOI] [PubMed] [Google Scholar]
- Kronbach T, Mathys D, Umeno M, Gonzalez FJ, Meyer U. Oxidation of midazolam and triazolam by human liver cytochrome P450IIIA4. Mol Pharmacol. 1989;36:89–96. [PubMed] [Google Scholar]
- Labrie F, Belanger A, Cusan L, Gomez J-L, Candas B. Marked decline in serum concentrations of adrenal C19 sex steroid precursors and conjugated androgen metabolites during aging. J Clin Endocrinol Metab. 1997;82:2396–2402. doi: 10.1210/jcem.82.8.4160. [DOI] [PubMed] [Google Scholar]
- Labrie F, Luu-The V, Belanger A, Lin S-X, Simard J, Pelletier G, Labrie C. Is dehydroepiandrosterone a hormone? J Endocrinol. 2005;187:169–196. doi: 10.1677/joe.1.06264. [DOI] [PubMed] [Google Scholar]
- Ledirac N, de Sousa G, Fontaine F, Agouridas C, Gugenheim J, Lorenzon G, Rahmani R. Effects of macrolide antibiotics on CYP3A expression in human and rat hepatocytes: interspecies differences in response to troleandomycin. Drug Metab Dispos. 2000;28:1391–1393. [PubMed] [Google Scholar]
- Legrain S, Massien C, Lahlou N, Roger M, Debuire B, Diquet B, Chatellier G, Azizi M, Faucounau V, Porchet H, Forette F, Baulieu E-E. Dehydroepiandrosterone replacement administration: pharmacokinetic and pharmacodynamic studies in healthy elderly subjects. J Clin Endocrinol Metab. 2000;85:3208–3217. doi: 10.1210/jcem.85.9.6805. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Lubet RA, Gordon GB, Prough RA, Lei X-D, You M, Wang Y, Grubbs CJ, Steele VE, Kelloff GJ, Thomas CF, Moon RD. Modulation of methylnitrosourea-induced breast cancer in Sprague Dawley rats by dehydroepiandrosterone: dose-dependent inhibition, effects of limited exposure, effects on peroxisomal enzymes, and lack of effects on levels of Ha-Ras mutations. Cancer Res. 1998;58:921–926. [PubMed] [Google Scholar]
- Maglich JD, Parks DJ, Moore LB, Collins JL, Goodwin B, Billin AN, Stoltz CA, Kliewer SA, Lambert MH, Willson TM, Moore JT. Identification of a novel human constitutive androstane receptor (CAR) agonist and its use in the identification of CAR target genes. J Biol Chem. 2003;278:17277–17283. doi: 10.1074/jbc.M300138200. [DOI] [PubMed] [Google Scholar]
- Miller KKM, Cai J, Ripp SL, Pierce WM, Rushmore TH, Prough RA. Stereo- and regioselectivity account for the diversity of dehydroepiandrosterone (DHEA) metabolites produced by liver microsomal cytochromes P450. Drug Metab Dispos. 2004;32:305–313. doi: 10.1124/dmd.32.3.305. [DOI] [PubMed] [Google Scholar]
- Miller WL. Androgen biosynthesis from cholesterol to DHEA. Mol Cell Endocrinol. 2002;198:7–14. doi: 10.1016/s0303-7207(02)00363-5. [DOI] [PubMed] [Google Scholar]
- Miners JO, Birkett DJ. Use of tolbutamide as a substrate probe for human hepatic cytochrome P450 2C9. Methods Enzymol. 1996;272:139–145. doi: 10.1016/s0076-6879(96)72017-7. [DOI] [PubMed] [Google Scholar]
- Pascussi J-M, Busson-Le Coniat M, Maurel P, Vilarem M-J. Transcriptional analysis of the orphan nuclear receptor constitutive androstane receptor (NR1I3) gene promoter: identification of a distal glucocorticoid response element. Mol Endocrinol. 2003a;17:42–55. doi: 10.1210/me.2002-0244. [DOI] [PubMed] [Google Scholar]
- Pascussi J-M, Drocourt L, Gerbal-Chaloin S, Fabre J-M, Maurel P, Vilarem M-J. Dual effect of dexamethasone on CYP3A4 gene expression in human hepatocytes. Eur J Biochem. 2001;268:6346–6357. doi: 10.1046/j.0014-2956.2001.02540.x. [DOI] [PubMed] [Google Scholar]
- Pascussi J-M, Gerbal-Chaloin S, Drocourt L, Maurel P, Vilarem MJ. The expression of CYP2B6, CYP2C9 and CYP3A4 genes: a tangle of networks of nuclear and steroid receptors. Biochim Biophys Acta. 2003b;1619:243–253. doi: 10.1016/s0304-4165(02)00483-x. [DOI] [PubMed] [Google Scholar]
- Peters JM, Zhou Y-C, Ram PA, Lee SST, Gonzalez FJ, Waxman DJ. Peroxisome proliferators-activated receptor α required for gene induction by dehydroepiandrosterone-3β-sulfate. Mol Pharmacol. 1996;50:67–74. [PubMed] [Google Scholar]
- Prough RA, Webb SJ, Wu H-G, Lapenson DP, Waxman DJ. Induction of microsomal and peroxisomal enzymes by dehydroepiandrosterone and its metabolite in rats. Cancer Res. 1994;54:2878–2886. [PubMed] [Google Scholar]
- Rao MS, Subbarao V, Yeldandi AV, Reddy JK. Hepatocarcinogenicity of dehydroepiandrosterone in the rat. Cancer Res. 1992;52:2977–2979. [PubMed] [Google Scholar]
- Ripp SL, Falkner KC, Pendleton ML, Tamasi V, Prough RA. Regulation of CYP2C11 by dehydroepiandrosterone and peroxisome proliferators: identification of the negative regulatory region of the gene. Mol Pharmacol. 2003;64:113–122. doi: 10.1124/mol.64.1.113. [DOI] [PubMed] [Google Scholar]
- Ripp SL, Fitzpatrick JL, Peters JM, Prough RA. Induction of CYP3A expression by dehydroepiandrosterone: involvement of the pregnane X receptor. Drug Metab Dispos. 2002;30:570–575. doi: 10.1124/dmd.30.5.570. [DOI] [PubMed] [Google Scholar]
- Singleton DW, Lei X-D, Webb SJ, Prough RA, Geoghegan TE. Cytochrome P-450 mRNAs are modulated by dehydroepiandrosterone, nafenopin and triiodothyronine. Drug Metab Dispos. 1999;27:193–200. [PubMed] [Google Scholar]
- Srivastava PK, Yun C-H, Beaune PH, Ged C, Guengerich FP. Separation of human liver microsomal tolbutamide hydroxylase and (S)-mephenytoin 4′-hydroxylase cytochrome P-450 enzymes. Mol Pharmacol. 1991;40:69–79. [PubMed] [Google Scholar]
- van der Hoeven TA, Coon MJ. Preparation and properties of partially purified cytochrome P-450 and reduced nicotinamide adenine dinucleotide phosphate-cytochrome P-450 reductase from rabbit liver microsomes. J Biol Chem. 1974;249:6302–6310. [PubMed] [Google Scholar]
- Villareal DT, Holloszy JO. Effect of DHEA on abdominal fat and insulin action in elderly women and men. J Am Med Assoc. 2004;292:2243–2248. doi: 10.1001/jama.292.18.2243. [DOI] [PubMed] [Google Scholar]
- Waxman DJ. P450 gene induction by structurally diverse xenochemicals: central role of nuclear receptors CAR, PXR, and PPAR. Arch Biochem Biophys. 1999;369:11–23. doi: 10.1006/abbi.1999.1351. [DOI] [PubMed] [Google Scholar]
- Webb SJ, Geoghegan TE, Prough RA, Miller KKM. The biological actions of dehydroepiandrosterone involves multiple receptors. Drug Metab Rev. 2006;38:89–116. doi: 10.1080/03602530600569877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb SJ, Xiao G-H, Geoghegan TE, Prough Regulation of CYP4A expression in rat by dehydroepiandrosterone and thyroid hormone. Mol Pharmacol. 1996;49:276–287. [PubMed] [Google Scholar]
- Wei P, Zhang J, Egan-Hafley M, Liang S, Moore DD. The nuclear receptor CAR mediates specific xenobiotic induction of drug metabolism. Nature. 2000;407:920–923. doi: 10.1038/35038112. [DOI] [PubMed] [Google Scholar]
- Wu H-Q, Masset-Brown J, Tweedie DJ, Milewich L, Frenkel RA, Martin-Wixtrom C, Estabrook RW, Prough RA. Induction of microsomal NADPH-cytochrome P-450 reductase and cytochrome P-450IVA1 (P-450LAω) by dehydroepiandrosterone in rats: a possible peroxisomal proliferators. Cancer Res. 1989;49:2337–2343. [PubMed] [Google Scholar]