

Abstract
In the CNS, steroid hormones play a major role in the maintenance of brain homeostasis and it’s response to injury. Since activated microglia are the pivotal immune cell involved in neurodegeneration, we investigated the possibility that microglia provide a discrete source for the metabolism of active steroid hormones. Using RT-PCR, our results showed that mouse microglia expressed mRNA for 17β-hydroxysteroid dehydrogenase type-1 and steroid 5α-reductase type-1, which are involved in the metabolism of androgens and estrogens. Microglia also expressed the peripheral benzodiazepine receptor and steroid acute regulatory protein; however, the enzymes required for de novo formation of progesterone and DHEA from cholesterol were not expressed. To test the function of these enzymes, primary microglia cultures were incubated with steroid precursors, DHEA and AD. Microglia preferentially produced delta-5 androgens (Adiol) from DHEA and 5α-reduced androgens from AD. Adiol behaved as an effective estrogen receptor agonist in neuronal cells. Activation of microglia with pro-inflammatory factors, LPS and INFγ did not affect the enzymatic properties of these proteins. However, PBR ligands reduced TNFα production signifying an immunomodulatory role for PBR. Collectively, our results suggest that microglia utilize steroid-converting enzymes and related proteins to influence inflammation and neurodegeneration within microenvironments of the brain.
Keywords: LPS, cytokines, neurosteroids, steroidogenesis, PBR, DHEA, Adiol, estrogen
1. Introduction
The brain is under the constant influence of steroid hormones. Among these, sex hormones are known to affect multiple CNS processes such as the differentiation and maturation of specific brain regions, learning and behavior, neurogenesis, neurotransmission, synaptogenesis, and neuroprotection [1–7]. Sex hormones are derived from the steroid precursors dehydroepiandrosterone (DHEA) and androstenedione (AD), which originate from the metabolism of cholesterol and its further conversion to pregnenolone [8, 9]. At one time sex steroids were thought to originate exclusively from the gonads and the adrenals; however, it is now accepted that local steroid synthesis occurs in a number of tissues [10], including the central nervous system [11]. It is further thought that steroids metabolized in the nervous system exert intracrine and paracrine effects on homeostasis, as suggested from studies in hippocampal neurotransmission [12–14], cerebellar development [15], or neuroprotection following neurotoxic challenge [16, 17].
Certain types of brain cells are capable of metabolizing steroids [18], yet the role of microglia in this process has not been studied. Microglia are the resident brain macrophages, and as such have important immunological and pathological functions [19]. Through their production of cytokines and superoxides, activated microglia may underlie pathological processes such as Alzheimer’s disease [20], Parkinson’s disease [21], and multiple sclerosis [22].
Steroid metabolism in the CNS has been implicated as an adaptive coping mechanism following brain damage [3]. Additionally, neurodegenerative conditions have been negatively correlated with brain steroid levels [23]. As androgens and estrogens can exert pro- or anti-inflammatory actions [24–26], the steroid profile in a tissue may determine the extent of inflammation [27]. While microglia are critical mediators of brain inflammation, it is unknown if they participate in the steroid-metabolizing actions of the brain.
In previous studies we have reported that the microglia cell line, BV2, exhibits steroid-converting capacity [28–30]. Given the neuroprotective properties of steroids and the critical role that microglia play during brain damage responses, and in the steady state [31, 32], we have characterized the expression of the main proteins and enzymes in the steroidogenic pathway by real-time PCR (RT-PCR) from ex vivo isolated microglia and primary cultured cells. The function of key enzymes involved in steroid metabolism was assessed by the metabolic conversion of the sex steroid precursors DHEA and AD and analysis of their metabolites by thin layer chromatography (TLC). Our results indicate that microglia express steroid-converting enzymes that contribute to the metabolism of steroids into active androgens and estrogens within the brain.
2. Materials and Methods
2.1. Animals
For these studies, the transgenic mouse line p7.2fms-EGFP (C57BL6/6 X CBA background) was used [33]. Enhanced green fluorescent protein (EGFP) expression is driven by the promoter and the regulatory elements of the c-fms gene that encodes the receptor for macrophage colony stimulating factor (CSF-1), resulting in EGFP expression in cells of the mononuclear phagocytic lineage, including microglia [33]. Animals were bred in Rockefeller University facilities under 12:12 ligh:dark cycle and free access to chow and water. All experimental procedures were approved by the Rockefeller University Animal Care and Use Committee. To induce inflammation, mice received a single injection with Salmonella typhimurium lipopoly-saccharides (LPS; 1–5 mg/kg, i.p.; Sigma, L2262; St. Louis, MO).
2.2. Ex vivo Microglia Isolation by fluorescence activated cell sorting (FACS)
Previously reported methods to obtain a single population of microglia by FACS were used [34]. In brief, adult mice (2–3 months of age) were anaesthetized with pentobarbital (750mg/kg) and rapidly decapitated. Brains were removed and placed on ice in Hank’s balanced salt solution (Gibco, Carlsbad, CA), and meninges, blood vessels and choroid plexus were carefully removed under a dissecting scope. Brain cell suspensions, obtained after incubation with type II-S collagenase (600U; Sigma) and DNAse (450U; Invitrogen, Carlsbad, CA) for 30min at 37°C in 15ml HBSS supplemented with 90 mM CaCl2, were homogenized by repetitive gentle pipetting with fire-polished Pasteur pipettes on ice, followed by filtering through a 40 mm cell strainer (BD, Franklin Lakes, NJ). Cells were washed by centrifugation and subject to Percoll gradient centrifugation as described previously [34]. Cells collected from the 30/70 interphase, were washed and re-suspended in 5% fetal calf serum (FCS)-PBS containing 100ng/ml propidium iodide (PI), before sorting in a FACS Vantage SE Flow Cytometer (BD), with smHighPurity precision. Post-sort analysis was performed to ensure the purity of the collection process.
2.3. Primary microglia (1 °MG) cultures and cell stimulation
Microglia cultures were prepared following standard protocols (23). Briefly, day 2-old mouse pup brains were dissected on ice, and the meninges were carefully removed. The forebrains were minced in 5% FCS-PBS, dissociated using fire polished Pasteur pipettes, and then passed through a 40mM nylon cell strainer (BD). Cells were washed once in buffer and seeded in culture media at a density of roughly two forebrains per 75mm flask. Culture media, 10%FCS DMEM (Gibco), was changed every 5 days and supplemented with 5ng/ml granulocyte-monocyte colony stimulating factor (GM-CSF; Sigma). After 2 weeks in culture at 37°C, 5 %CO2, cells were shaken at 125rpm for 5hrs at 37°C to harvest detached microglia. Microglia were counted and seeded in 6-well plates at a density of 1 million cells/well (for RNA-PCR) or in 24-well assay plates at a density of 0.25–0.3 million cells/well (for hormone metabolism-TLC, and cytokine assays). After plating, microglia were allowed to adhere for 1hr and then rinsed to remove non-adherent glial cells, fed and incubated as described above. The following day, cells were rinsed and incubated in DMEM for 24hr at 37°C with vehicle or the various treatments: LPS, interferon-γ (INFγ) (Sigma), dibutyril-cyclic adenosine monophosphate (db-cAMP) (Sigma), (see results). For PBR ligand stimulation, cultures were pre-treated for 10 minutes with Ro 5-4864 (Ro) and PK-11195 (PK) (Sigma), or corticosterone (Sigma) as positive control, before LPS+INFγ stimulation. INFγ was supplemented to LPS to obtain a robust nitric oxide (NO) response. LPS+INFγ conditioned media (LCM) was obtained by stimulating 1°MG cultures with 1%FCS DMEM plus 100ng/ml LPS+ 10ng/ml INFγ, collecting the supernatants 24hr later, and centrifuging at 2000rpm for 5min to clear any debris. LCM contains elevated levels of several inflammatory cytokines such as TNFα, IL-6 and NO, as well as IL1β, IL-12, MCP-1, MCP-5, and RANTES (Gottfried-Blackmore, unpublished results). This LCM was used to stimulate fresh cultures of 1°MG.
2.4. FACS Analysis of 1 °MG
After shaking for 5hr, microglia were collected and washed in FACS buffer (5% FCS PBS). Cells were then blocked for 15 min at 4°C with 5% mouse serum. Cells were then stained for 15 min at 4°C with phycoerythrin (PE) conjugated anti-CD11b (1:200) (BD), or its corresponding PE-conjugated isotype, anti-rat IgG2b (1:200) (BD). Finally, cells were washed 3X in FACS buffer and then analyzed using a BD FACSCalibur system (BD) under the FITC and PE channels. Data was analyzed using FlowJo software (Tree Star Inc., OR).
2.5. Real-time PCR
Adult microglia were sorted by FACS into RNA lysis buffer (Absolute RNA Microprep kit (Stratagene, La Jolla, CA)), frozen in dry ice, and kept at −80°C until processing. RNA was isolated using the Absolute RNA Microprep kit. RNA from 1°MG cultures was obtained by rinsing the cell cultures with PBS and then lysing cells in 350µl RNA lysis buffer (RNeasy Mini kit (Qiagen, Valencia, CA)). RNA was extracted using the RNeasy Mini kit protocol (Qiagen). Both extraction methods included a step with DNAse incubation (Qiagen). RNA was quantified with RiboGreen RNA Quantitation kit (Molecular Probes), following manufacturer instructions. 10ng of RNA were retrotranscribed with SuperScript II Reverse Transcriptase (Invitrogen) and 3µl of a 1:3 dilution of the cDNA were amplified by real-time PCR using SYBR Green master mix (AB) in a 7900HT SDS thermal cycler (AB). The steroidogenic protein transcripts quantified were peripheral benzodiazepine receptor (PBR), steroidogenic acute regulatory protein (StAR), cytochrome p450 side chain cleavage enzyme (p450scc), cytochrome p450 21-hydroxylase (p450c21), cytochrome p450 17-hydroxylase (p450c17), aryl sulfatase (Arsa), DHEA sulfotransferase (Sulft), 3β-hydroxysteroid dehydrogenase (isoforms 1,2,4) (3βHSD1,2,4), 17β-hydroxysteroid dehydrogenase type 1 (17βHSD1), cytochrome p450 aromatase (P450Arom), steroid 5α-reductase type 1 (5αR), 3α-hydroxysteroid dehydrogenase (3αHSD), and ribosomal protein L27A (L27A). Primers sequences were designed using Primer Express Software (ABI) and are indicated in Table 1; amplicons span at least one intron in order to avoid potential genomic DNA amplification. All primers were blasted on NCBI databases for target specificity and tested using appropriate positive control tissues such as ovary and adrenal glands. Each sample was run in triplicate and the average Ct (threshold cycle) was used to calculate the relative amount of product by the -ΔΔCt-method (AB), using the ribosomal L27A as a housekeeping gene. The ratio of enzyme Ct to L27A Ct values was calculated as a way of assessing the relative expression levels of each enzyme comparing 1°MG and ex vivo MG. In each experiment, both positive (1µg ovary mRNA/cDNA1:3) and negative (RT minus and water) controls were included to ensure that the PCR reaction was working properly.
Table 1. Oligonucleotide Primers designed for RT-PCR Amplification of Steroidogenic Enzymes.
Primers for RT-PCR used to determine the expression of steroidogenic enzymes. The table shows the name of the protein analyzed (name of gene in parenthesis), specific oligonucleotide sequence, and the exons spanned by each amplicon.
| Protein(Gene) | Sequence (5′-3′) | Bases | Exons | |
|---|---|---|---|---|
| StAR | Fwd | GAGCTCTCTGCTTGGTTCTCAA | 218-239 | 2–3 |
| (Star) | Rev | TTGAGTATGCCCAAGGCCTT | 325-316 | |
| PBR | Fwd | TGCAGAAACCCTCTTGGCATC | 173-193 | 2–3 |
| (Tspo) | Rev | TGAAACCTCCCAGCTCTTTCC | 286-266 | |
| p450scc | Fwd | CCTATTCCGCTTTTCCTTTGAGTCC | 636-660 | 3–4 |
| (Cyp11a1) | Rev | CGCTCCCCAAATATAACACTGCTG | 686-663 | |
| p450c21 | Fwd | TGCCCCATCGTGCAACTAGG | 1064-1083 | 8–9 |
| (Cyp21a1) | Rev | AGCCGGAGATGCTGCTAGCC | 1102-1083 | |
| p450c17 | Fwd | TCGGCCCCAGATGGTGACTC | 416-435 | 1–2 |
| (Cyp17a1) | Rev | TGGTCCGACAAGAGGCCTAGAG | 454-433 | |
| Sts | Fwd | CCACTACTGCAACGCCTACCT | Isles, et al. 2004 | |
| (Sts) | Rev | CGTGAAGTAGAAGGCCTTCCA | Hum. Mol. Gen. | |
| Arsa | Fwd | TTCACTGCAGATAACGGTCCTG | 2038-2059 | 5–6 |
| (Arsa-1) | Rev | AGGAGTAATGTGACCTGGCCA | 2178-2158 | |
| Sulft | Fwd | ACAGCTCTTTCCAAGCCATGA | 614-634 | 5–6 |
| (Sult2a2) | Rev | TCCCCAGTTGTGCCTTTTCT | 722-703 | |
| 3βHSD1 | Fwd | TCTGAAAGGTACCCAGAACCTATTGG | 433-458 | 3–4 |
| (Hsd3b1) | Rev | TTGCTTGAACACAGGCCTCCA | 476-456 | |
| 3βHSD2 | Fwd | AAAGGTACCCAGAACTTATTGGAGGC | 417-442 | 3–4 |
| (Hsd3b2) | Rev | GGCACACTGGCTTGGATACAGG | 463-442 | |
| 3βHSD4 | Fwd | GGTCGAAAACAGGAAGAGGAATTGTC | 118-144 | 1–2 |
| (Hsd3b4) | Rev | TGGTCTTTGTCTGCAGCTTGGAC | 163-140 | |
| 17βHSD1 | Fwd | AGTGTGGGAGGCTTGATGGGA | 458-478 | 3–4 |
| (Hsd17b1) | Rev | CACTTCGTGGAATGGCAGTCC | 496-479 | |
| Arom | Fwd | CTTTGGAGAACAATTCGCCCTTTC | 418-441 | 3–4 |
| (Cyp19a1) | Rev | GCCCGTCAGAGCTTTCATAAAGAA | 462-439 | |
| 5αR | Fwd | TGTTTCCTGACAGGCTTTGCCC | 436-457 | 2–3 |
| (Srd5a1) | Rev | CCATGCCCACTAACCACAGGG | 475-455 | |
| 3αHSD | Fwd | GCCATCGTGAAAAACAATGG | 692-711 | 6–7 |
| (Hsd3a1) | Rev | AATCAGCGCAGGAGTTCGA | 795-777 | |
| L27A | Fwd | TGTTGGAGGTGCCTGTGTTCT | 442-462 | 1–1 |
| (Rpl27a) | Rev | CATGGAGAGAAGGAAGGATGC | 542-522 | |
2.6. Incubation of tritiated hormones and steroid extraction
The day after seeding, cells were rinsed and incubated in 0.2ml DMEM containing [1,2,6,7-3H] DHEA (60 Ci/mmol) or [1,2,6,7-3H] AD (105 Ci/mmol) (Perkin Elmer Life Science, Shelton, CT) for 22–24hr. All incubations were conducted in a 5% CO2 atmosphere at 37°C. The reaction was stopped by vortexing the supernatant with acetone (0.2 ml) and ethyl acetate (0.5ml). A 0.2 ml portion of the organic phase extract was evaporated to dryness at room temperature, the residue was re-dissolved in methanol, and the yield of metabolites was determined after separation by thin layer chromatography (TLC).
2.7. TLC Identification of Metabolites
Products of tritiated steroid incubations were separated by TLC on silica gel containing a fluorescent indicator on pre-coated aluminum sheets (Fisher, Carlsbad, CA) using chloroform/ethyl-acetate/xylene (68/23/9 % by vol. for [3H] DHEA) and (62/21/17 % by vol. for [3H] AD). Non-radioactive steroids of known identity were added to the TLC sheets on lanes adjacent to the putative metabolites and were visualized and identified by their chromogenic properties after spraying with 5% (by vol.) sulphuric acid in methanol and heating on a hot plate. Unlabeled steroids E2 and E1were purchased from Sigma, and T, AD, 5αAD, DHEA, Adiol from Steraloids Inc. Purity of [3H]-DHEA and [3H]-AD (<98 %) was determined by TLC.
2.8. Cytokine and Nitric Oxide detection
After 24hr stimulation with PBR ligands and LPS+INFγ, 1°MG supernatants were collected, centrifuged (2,500rpm) for 5 minutes at 4°C in a tabletop microfuge, and then frozen at −20°C. For detection of TNFα and IL-6, ELISA was performed according to manufacturer’s instructions (eBioscience, SanDiego, CA). Nitric Oxide (NO) was determined indirectly though the Greiss Assay (Promega, Madison, WI).
2.9. Transfection and Luciferase Assay
EtC.1 cells were seeded in 24-well plates, 2×104 cells/well, in DMEM containing 10% charcoal stripped serum. 24hr later cells were transfected using Lipofectamine Plus (Invitrogen), following manufacturer’s instructions, with 0.4µg of plasmid DNA/well. The 3X ERE-Luciferase plasmid was a generous gift of Don McDonnell (Duke University Durham, NC), and the β-Galactosidase plasmid was from Promega (pSV-β-Gal control vector). 24hr after transfection, cells were incubated with various concentrations of E2 or Adiol for another 24hr. 100nM ICI pre-treatment was done for 30 minutes. Cell lysates were prepared and luciferase activity measured using Promega Luciferase Assay System according to manufacturer’s instructions (Promega). β-Gal activity was measured from cell lysates to normalize for transfection efficiency.
2.10. Statistical analysis
Statistical analysis was performed using StatView (SAS Institute Inc., Cary, NC). Experiments involving 2 groups were compared using a Student t-test. Experiments involving more than 2 groups were compared by Analysis of Variance (ANOVA), followed by posthoc analysis with Tukey Honestly Significant Difference (HSD) when variances were homogeneous (using Levene’s test); or with nonparametric Games-Howell test. Graphs show the mean ± the standard error of the mean (S.E.M.). P<0.05 was considered statistically significant. *, p<0.05, **, p<0.01 and ***, p<0.0001; (n.s.), non-significant.
3. Results
3.1. Purity of microglia assays
Exclusion of brain cells with high steroid-converting capacity, such as astrocytes [18], from our experiments with 1°MG cultures was verified by EGFP expression and by FACS analysis. Cultured 1°MG showed 99–98% purity by EGFP expression and staining with the macrophage-specific marker CD11b (Fig.1). Therefore, gene expression and enzymatic activity in our assays can be attributed mainly to microglia.
Fig. 1. Purity of in vitro cultures of primary microglia (1°MG) from cfms-EGFP mice.
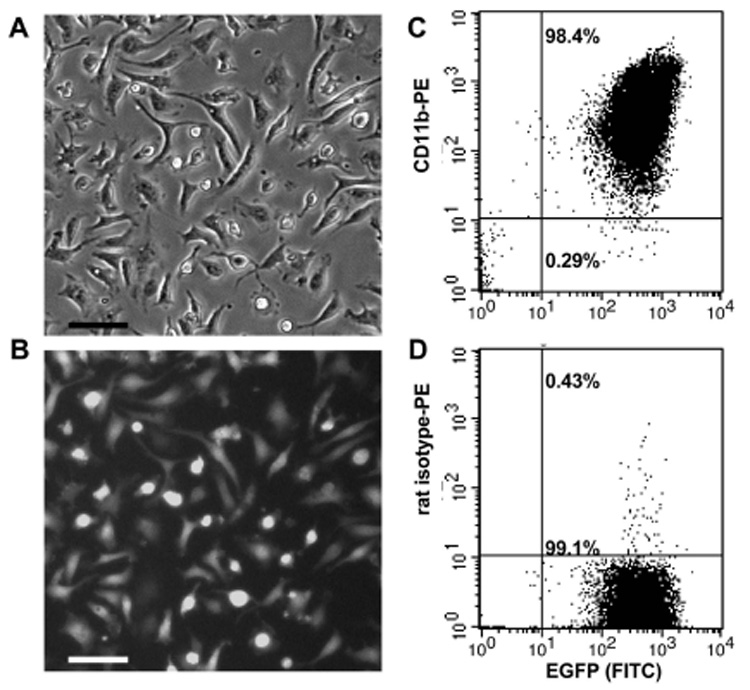
Purity of 1°MG cultures obtained from cfms-EGFP neonatal (day 2) mice was verified by fluorescence microscopy (A–B) for expression of EGFP, as well as by FACS analysis using EGFP expression and staining with the macrophage marker CD11b (C). Isotope control staining is shown in (D). Scale bar = 20µM.
3.2. Gene expression of steroid-converting enzymes in microglia
To investigate the expression of the main steroid-converting enzymes in mouse microglia, gene specific primers were designed for Real Time PCR (RT-PCR) (Table 1). Some of these enzymes have multiple isoforms (i.e. 3β-hydroxysteroid dehydrogenase has 7 isoforms in mouse). Selection of isoforms in this study was based on reported predominance (common vs. rare) and activity (dehydrogenase vs. oxo-reductase). RT-PCR analysis of RNA from adult, FACS-sorted, microglia (ex vivo MG) and from primary microglia cultures (1°MG) revealed positive expression of the peripheral benzodiazepine receptor (PBR), steroid acute regulatory protein (StAR), steroid (StS) and aryl (Arsa) sulfatase, 17β-hydroxysteroid dehydrogenase type 1 (17βHSD1), and 5α-reductase type 1 (5αR) (Table 2). However, cytochrome p450 side chain cleavage enzyme (p450scc), cytochrome p450 21-hydroxylase (p450c21), cytochrome p450 17-hydroxylase (p450c17), DHEA sulfotransferase (Sulft), 3βHSD type 1, 2, and 4, cytochrome p450 aromatase (P450Arom), and 3α-hydroxysteroid dehydrogenase (3αHSD) were not detected (Table 2). The ratio of specific gene Ct values to the Ct value of L27A, a housekeeping gene, was calculated as a way of assessing the relative expression levels of each gene within ex vivo MG and 1°MG (Table 2). The most abundantly expressed genes were the PBR (ratio of 0.80; 0.94) and Arsa (ratio of 0.85; 0.83), while the lowest expression was found for StAR (ratio of 0.68; 0.62) (Table 2). No differences were found between the expression levels of ex vivo MG and 1°MG (Student T-test; p=0.16–0.64), except for the PBR (p<0.0001) and StS (p<0.01), which were expressed at higher levels in 1°MG (Table.2). A summary of the expression of steroidogenic proteins in microglia is depicted in Figure 2.
Table 2. Relative comparison of steady-state microglia mRNA levels of proteins involved in steroid metabolism.
Expression levels of steroid converting proteins were evaluated by real-time PCR in microglia obtained from FACS-sorted cells from adult mouse brains (ex vivo MG), or from primary cultures (1°MG). Ovary tissue was used as a positive control. Values represent the mean (n=3) ± the SEM of the ratio of enzyme expression to the housekeeping ribosomal gene L27A. Cycle threshold values (Ct) are shown for L27A for reference.
| PBR | StAR | p450scc | p450c21 | p450c17 | StS | |
|---|---|---|---|---|---|---|
| ExVivo MG | 0.80 ± 0.03 | 0.68 ± 0.01 | ND | ND | ND | 0.67 ±0.02 |
| 1°MG | 0.94 ± 0.02 | 0.62 ± 0.01 | ND | ND | ND | 0.83 ± 0.00 |
| Ovary | 0.79 ± 0.02 | 0.79 ± 0.03 | 0.95 ± 0.0 | 0.55 ± 0.02 | 0.85 ± 0.0 | 0.66 ± 0.07 |
| Arsa | Sulft | 3βHSD1 | 3βHSD2 | 3βHSD4 | ||
| ExVivo MG | 0.85 ± 0.04 | ND | ND | ND | ND | |
| 1°MG | 0.83 ± 0.02 | ND | ND | ND | ND | |
| Ovary | 0.69 ± 0.02 | 0.54 ± 0.01 | 0.73 ± 0.03 | 0.69 ± 0.01 | 0.69 ± 0.02 | |
| 17βHSD | Arom | 5αR | 3αHSD | L27A(Ct) | ||
| ExVivo MG | 0.69 ± 0.01 | ND | 0.69 ± 0.01 | ND | 1.00 (21.5) | |
| 1°MG | 0.70 ± 0.02 | ND | 0.70 ± 0.01 | ND | 1.00 (23.9) | |
| Ovary | 0.77 ± 0.03 | 0.67 ± 0.04 | 0.64 ± 0.0 | 0.54 ± 0.01 | 1.00 (14.6) | |
Fig. 2. Expression summary of steroid converting proteins in microglia.
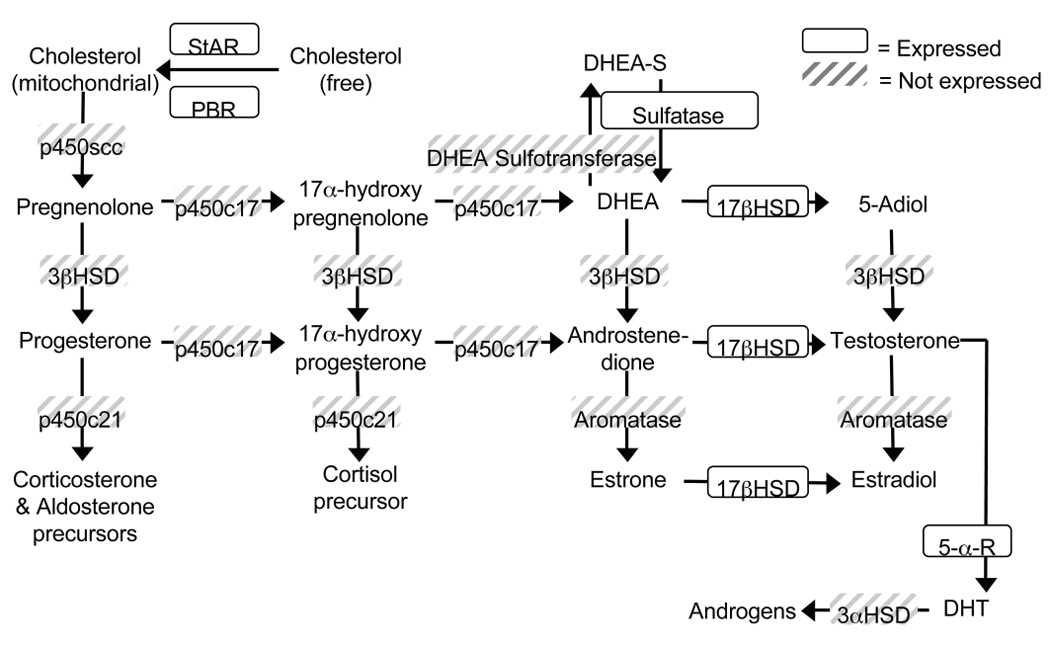
Diagram of the steroidogenic pathway showing the main steroidogenic enzymes and their steroid substrates. The boxed enzymes are those expressed in resting microglia, the shadowed ones were not expressed.
3.3. Inflammatory LPS stimulation modulates expression of steroid-converting enzymes in microglia
Intraperitoneal injections (i.p.) of LPS into mice leads to the activation of brain microglia, as indicated by changes in morphology and the expression of pro-inflammatory cytokines [34, 35]. Adult cfms-EGFP male mice were injected i.p. with saline or 1mg/kg LPS to evaluate the expression of the principal steroid-converting enzymes in activated microglia. Twenty-four hour in vivo stimulation with LPS led to an overall decrease in the expression of steroid-converting enzymes (Fig.3). StAR expression was decreased by 38.8±9.3% (p<0.05), 17βHSD1 by 71.4±0.8% (p<0.05), and 5αR by 66.6±1.8% (p<0.05) (Fig. 3). One notable exception was the PBR, whose expression was significantly increased 7.7±2.2-fold (p<0.05) (Fig.3). However, i.p. injection of LPS did not induce the expression of p450scc, p450c21, p450c17, Sulft, 3βHSD1,2,4, P450Arom, or 3αHSD in ex vivo microglia.
Fig. 3. In vivo LPS stimulation reduces steroidogenic enzyme expression in ex vivo microglia.
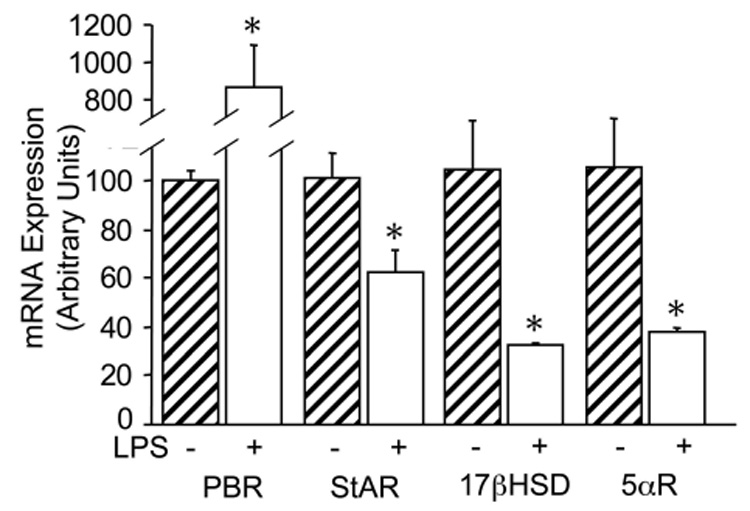
RT-PCR analysis of steroidogenic enzymes in FACS-sorted microglia from adult mice treated for 24hr with an IP injection of saline (hatched bars) or 1mg/kg LPS (open bars). Bars represent the % expression of each enzyme with respect to control samples and normalized to the housekeeping gene L27A. Values are the mean ± the SEM of three animals. *, P < 0.05 vs. control.
Since i.p. injection of LPS is known to induce high levels of circulating cytokines, we sought to assess whether the down regulation of steroid enzymes in microglia was due to a direct or indirect effect of LPS. 1°MG cultures were stimulated in vitro for 24hrs with different pro-inflammatory stimuli (LPS+INFγ, INFγ, and LPS-conditioned media (LCM), see methods) and cells were processed for RT-PCR analysis (Fig.4).
Fig. 4. LPS+INFγ increases the expression of steroidogenic enzymes in cultured 1°MG.
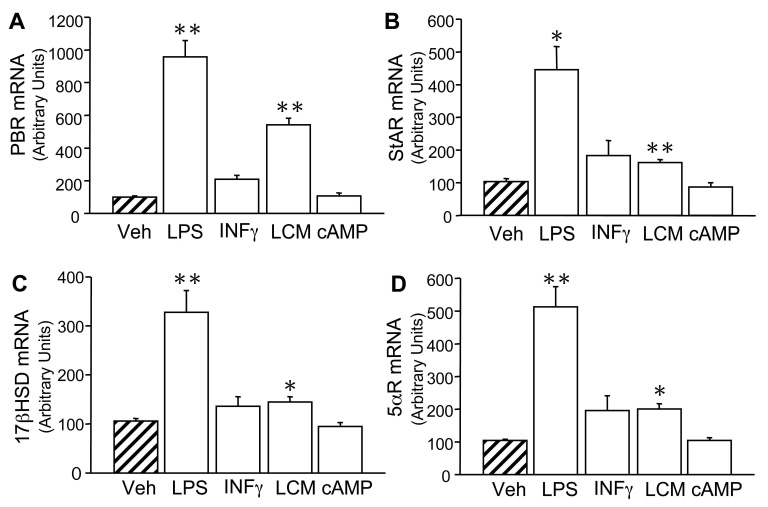
RT-PCR analysis of steroidogenic enzymes in cultured 1°MG. A–D) 1°MG cells were stimulated for 24hr with 100ng/ml LPS+ 10ng/ml INFγ (LPS), 10ng/ml INFγ (INFγ), LPS-conditioned media (LCM), 1mM db-cAMP (db-cAMP), or vehicle (Veh) (hatched bars). Bars represent the % expression of each enzyme with respect to vehicle and normalized to the housekeeping gene L27A. Values are the mean ± the SEM of 2–3 independent experiments done in triplicate. *, **, P < 0.05, P < 0.001 vs. Veh, respectively.
Stimulation with LPS+INFγ led to an increase in the expression of all steroid-converting enzymes expressed in 1°MG (Fig.4A–D). PBR expression was increased 9.6±1.0-fold (p<0.01) (Fig.4A), StAR increased 4.5±0.7-fold (p<0.05) (Fig.4B), 17βHSD1 increased 3.3±0.4-fold (p<0.01) (Fig.4C), and 5αR was increased 5.1±0.6-fold (p<0.01) (Fig.4D). INFγ stimulation alone had no significant effect (Fig.4) and LCM did not induce a down-regulation of steroid-converting enzymes. Instead, it mimicked the LPS+INFγ effect on 1°MG, but to a lesser extent (Fig.4). Finally, db-cAMP, a known inducer of steroidogenesis in various endocrine cells, was tested on 1°MG, yet it presented no effect on the expression of steroid-converting enzymes in 1°MG (Fig.4). None of the treatments in 1°MG induced the expression of p450scc, p450c21, p450c17, Sulft, 3βHSD1,2,4, P450Arom, or 3αHSD (data not shown).
3.4. Steroid-converting activity in microglia
The results from our RT-PCR analysis suggested that microglia express enzymes capable of metabolizing DHEA, androgens, and estrogens (Fig.2). The steroid-converting activity of microglia was evaluated by incubating 1°MG with the steroid intermediate dehydroepiandrosterone (DHEA) and measuring its metabolism. After 24hr incubation, 1°MG showed a 30±3.3% (p<0.0001) conversion of H3-DHEA, with Adiol being the only product detected of this conversion, accounting for 15.5±%1.3 (p<0.0001) of H3-radioactivity (Fig.5A). Gene expression experiments indicated that LPS+INFγ increased expression of steroid converting enzymes; however, microglia stimulation with LPS+INFγ had no effect on the conversion or profile of steroids produced from H3-DHEA (Fig.5B).
Fig. 5. Metabolism of H3-DHEA by 1°MG.
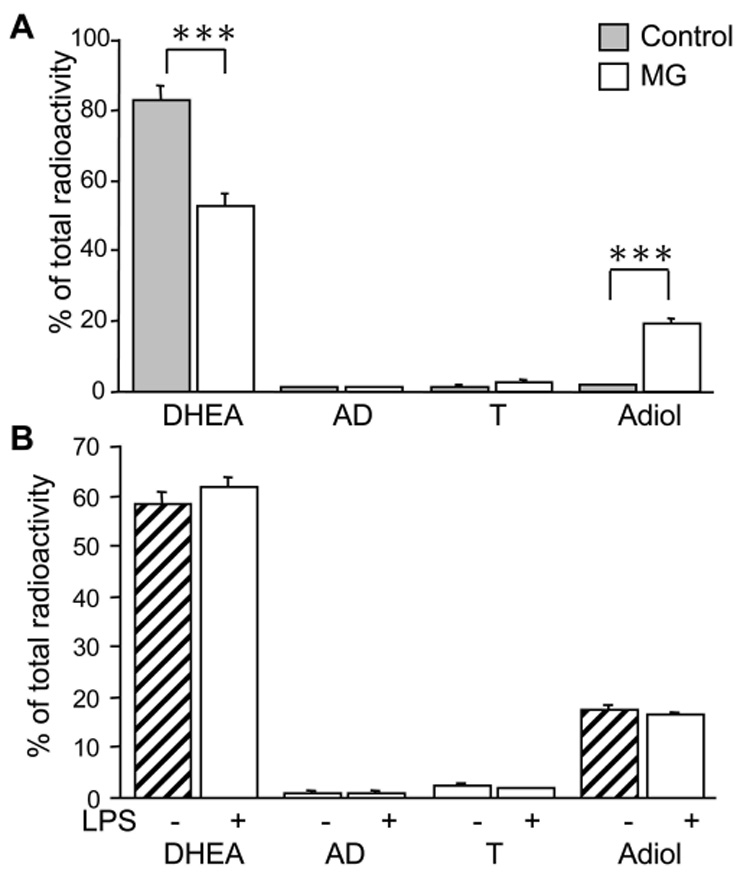
TLC analysis of H3-DHEA metabolism in 1°MG. Cells were incubated for 24hr with H3-DHEA and metabolites extracted from culture supernatants were resolved by TLC and counted on a scintillation counter. A) Resting microglia (MG, open bars) showed a significant conversion of DHEA compared to the no-cell controls (Control, grey bars), with the only product being Adiol. B) LPS (100ng/ml LPS+ 10ng/ml INFγ) stimulation of the cells (hatched bars) didn’t alter the metabolic activity or profile of steroids produced from DHEA in microglia. Bars represent the % of total H3 radioactivity collected from the TLC assay. Values are the mean ± the SEM of 2–3 independent experiments done in triplicate. ***, P < 0.0001 vs. MG.
Androstenedione (AD) is the main intermediary product from DHEA in the synthesis of androgens and estrogens. 1°MG incubated with H3-AD for 24hr showed a 36.7±1.7% (p<0.0001) conversion of this steroid (Fig.6A), with the main products of its conversion being T, 12.8±0.8% (p<0.0001); 5αAD, 3.8±0.8% (p<0.01); and DHT, 14.4±3.1% (p<0.01) (Fig.6A). Further, stimulation of the cells using LPS+INFγ did not affect the conversion or the profile of steroids produced from H3-AD (Fig.6B).
Fig. 6. 1°MG convert H3-AD into downstream steroids.
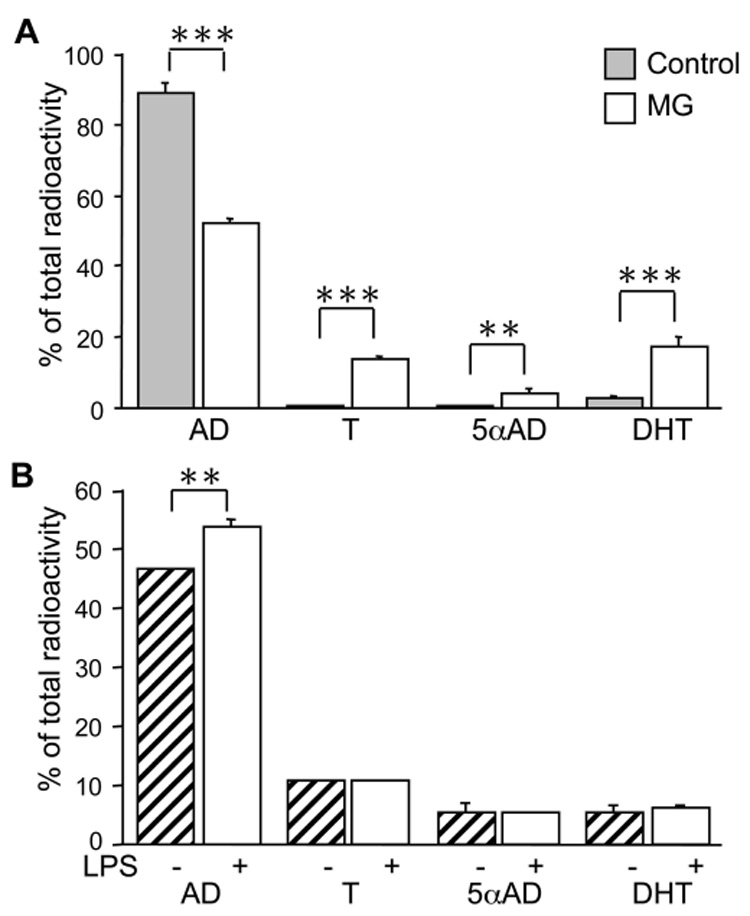
TLC analysis of H3-AD metabolism in 1°MG. Cells were incubated for 24hr with H3-AD and metabolites extracted from culture supernatants were resolved by TLC and counted on a scintillation counter. A) Resting microglia (MG, open bars) showed a significant conversion of AD compared to the no-cell controls (Control, grey bars). The main products of AD were testosterone (T), 5αAndrostanedione (5αAD), and dihydrotestosterone (DHT). B) LPS (100ng/ml LPS+ 10ng/ml INFγ) stimulation (hatched bars) slightly decreased the conversion of AD compared to vehicle treated cells (open bars), but not the production of downstream steroids. Bars represent the % of total H3 radioactivity collected from the TLC assay. Values are the mean ± the SEM of 2–3 independent experiments done in triplicate. **,***, P < 0.001, P < 0.0001 vs. MG, respectively.
3.5. PBR role in microglia
Of the genes studied, PBR was the most abundant transcript detected in microglia; additionally, it showed a robust induction by LPS+INFγ. The absence of cholesterol metabolizing enzymes, i.e. low levels of StAR and absence of p450scc and p450c17, suggest that PBR may play an alternate role in microglia. Indeed, the PBR has been associated with numerous biological functions such as cellular proliferation, porphyrin transport, heme biosynthesis, anion transport, apoptosis, and immunomodulation [rev. by [36]]. Stimulation of microglia with two selective PBR ligands, Ro and PK, led to a specific reduction of LPS-induced production of TNFα(16.2±10.8% decrease (p<0.05), and 40.9±8.9% (p<0.05) respectively), but had no effects on IL-6 and NO (Fig.7), except a 12.4±6.4% (p<0.05) increase by PK on NO (Fig.7). Despite the modulatory effects on these inflammatory products, PBR ligands had no effect the metabolism of DHEA or AD (data not shown).
Fig. 7. PBR ligands selectively modulate TNFα production in 1°MG.
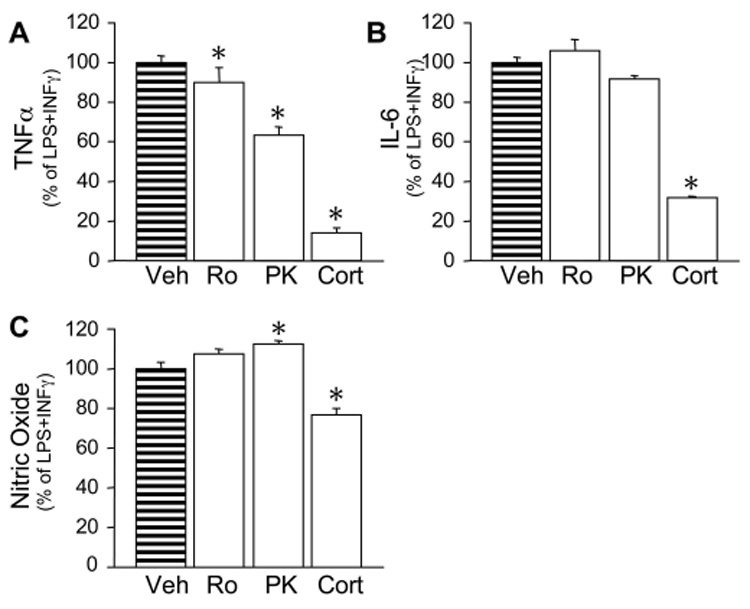
Cells were incubated for 24hr with LPS (100ng/ml LPS+ 10ng/ml INFγ) with a 10-minute pre-treatment of the PBR ligands Ro (10pM) and PK (10pM), or 1µM corticosterone as positive control. Culture supernatants were assayed for TNFα (A), IL-6 (B), and nitric oxide (NO) by ELISA and Greiss assay, respectively. Bars represent the % of total cytokines detected in the LPS controls. Mean cytokine levels measured for the LPS+INFγ controls were: TNFα (32ng/ml), IL-6 (70ng/ml) and NO (35mM). Un-stimulated cells had no detectable cytokine production. Bars represent the mean ± S.E.M. of 2–3 independent experiments done in triplicate. *, P < 0.05 vs. LPS+INFγ.
3.6. Adiol is an effective estrogen receptor agonist
The sole product of DHEA metabolism in 1°MG was Adiol. This steroid has been reported to have androgenic and estrogenic properties [37]. To determine whether Adiol could function as a specific estrogen receptor (ER) agonist, we utilized an ER-expressing neuronal cell line, EtC.1 [Gottfried-Blackmore, et al. submitted], transfected with a luciferase gene reporter coupled to 3 estrogen response elements (EREs). Incubation of these cells with Adiol induced the expression of the luciferase gene reporter at a dose of 1µM, but not at 10nM (Fig.8). Moreover, this induction was completely abrogated by pre-treatment of the cells with the specific ER-antagonist ICI-182,780 (100nM) (Fig.8). Microglia were not used for this assay because of their low expression of ERs and negligible responsiveness to E2 [Sierra et al. submitted].
Fig. 8. Adiol is an effective estrogen receptor agonist.
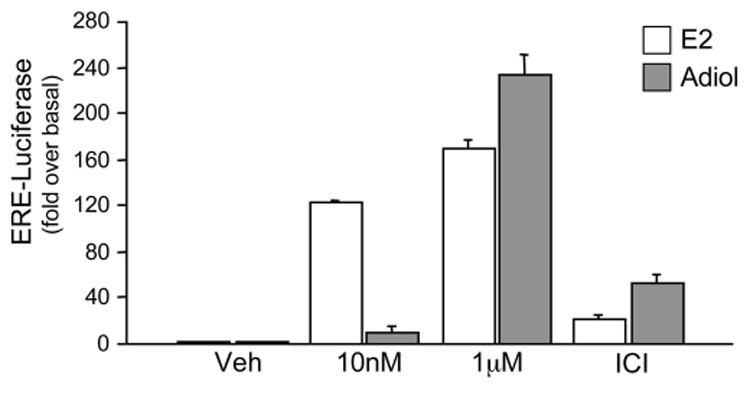
Estrogen receptor luciferase reporter assay reflected by luciferase expression from an ERE-Luciferase containing plasmid transfected into the neuronal cell line EtC.1. Bars represent luciferase activity determined by luminometry in cells treated with increasing doses of estrogen (E2) (open bars) or Adiol (grey bars). The third group of columns is from cells pre-treated 30 min with 100nM ICI 182,780 and then treated with the high doses of E2 and Adiol (ICI). Values are given in fold over basal ± the SEM, from one representative experiment done in triplicate.
4. Discussion
The involvement of sex hormones in neuroprotection is widely reported [38, 39]. Additionally, steroid-converting enzymes are up-regulated following neuronal damage, and blocking such enzymes increases neuronal death in a number of injury models [40]. These studies indicate the critical role of steroid metabolism in the brain’s responses to injury. Although the steroid-converting capacity of neurons, astrocytes and oligodendrocytes has been described to some extent, there are no reports addressing the contribution of microglia to brain steroidogenesis. In this study, microglia cells from adult and neonatal mice are reported to express steroid-converting enzymes and our data suggest microglia participate in the brain’s steroid-converting capacity.
In the brain it has been hypothesized that neurosteroidogenesis occurs involving the de novo synthesis from cholesterol of steroid hormone precursors, such as pregnenolone, progesterone and DHEA. At present, it remains controversial that neurosteroidogenesis can occur given conflicting data on expression of p450c17 in the adult brain [41, 42], required for synthesis of DHEA. Our results revealed that microglia do not express p450c17 nor p450scc, p450c21, or 3βHSD1-2, and therefore, do not have the capacity to synthesize steroids de novo from cholesterol.
Alternate roles for steroidogenic proteins
Despite the lack of p450scc and p450c17, microglia expressed low levels of StAR mRNA, which is the main mediator for cholesterol transfer to the mitochondria for the initiation of steroidogenesis [43, 44]. StAR expression in microglia may be playing a different role in these cells, such as cytosolic free sterol transfer, as has been suggested for other cholesterol binding proteins in macrophages [45]. Further studies will need to address this in microglia.
The PBR, like StAR, also participates in the transfer of cholesterol into the mitochondria [46]. In the brain, PBR expression is increased following nerve injury and can increase steroidogenesis locally [47]. In addition to its role in steroidogenesis [48], the PBR is reported to participate in other processes such as apoptosis, cancer, and immunomodulation [49–51]. This protein is widely expressed in monocytic cells [52], and has been identified as a marker of activated microglia [53]. Of the genes we studied, the most highly expressed in microglia was the PBR. In the CNS, PBR is mainly expressed in glial cells, and its expression levels increase following glial activation induced by inflammation or neuronal damage [rev. by [36]]. 1°MG and ex vivo MG challenged with inflammatory stimuli responded with an increased expression of PBR, consistent with previous reports. However, PBR stimulation with specific ligands, which increase steroidogenesis [47, 48], had no effect on DHEA or AD conversion in microglia. This may be due to the fact that these ligands affect the initial transfer of cholesterol into the mitochondria providing more substrate for p450scc, which is lacking in microglia. However, in our study we found that PBR ligands selectively decreased the production of TNFα in response to LPS+INFγ stimulation in 1°MG, in accordance with previous reports [54–56]. Interestingly, PBR ligands did not affect IL-6 and only slightly affected NO indicating that PRB ligands may preferentially interfere with signaling pathways involved in the expression of TNFα. Our data further support an immunomodulatory role of PBR in microglia, which may account for the neuroprotective effects reported for its ligands [57, 58].
Expression of steroid-converting enzymes and microglia LPS activation
Our results showed a significant reduction in all the steroid-converting enzymes expressed in ex vivo microglia after i.p. LPS stimulation. Systemic injection of LPS in vivo causes a rapid induction of circulating cytokines and inflammatory mediators [59], which can induce inflammatory genes in microglia [34, 35]. In vitro, LPS+INFγ caused an increase in steroid-converting enzyme expression, while single cytokine stimulation (INFγ), or stimulation with a combination of cytokines (LPS-conditioned media) showed only marginal effects. Our results would suggest that other in vivo factors may be causing the down-regulation of steroid-converting enzymes in microglia. Following the systemic rise of cytokines, activation of the Hypothalamic-Pituitary-Adrenal axis causes a 3–4 fold increase in circulating glucocorticoid levels (rev. by [60]). Increased glucocorticoid levels can block steroidogenesis in testicular Leydig cells [61, 62], and may have similar effects in brain microglia. We tested whether direct stimulation with corticosterone would decrease steroid-converting enzyme expression, however corticosterone failed to reduce StAR, 17βHSD1, or 5αR expression in vitro in both resting and LPS-activated microglia (data not shown). These results suggest other factors are involved in the in vivo reduction we observed. Another possibility for the discrepancy of the results is the developmental stage of the cells. Although primary microglia cultures are widely accepted to resemble adult cells, they may retain characteristics from developing cells. Such developmental differences could be pursued in future studies.
The LPS+INFγ activation of 1°MG caused increased enzyme expression in vitro, but did not affect the rate or metabolism of H3-DHEA and H3-AD conversion. The catalytic activity of several of these enzymes is determined by the redox potential set by the NADP+ to NADPH cofactor ratio [63]. Cofactor availability should be addressed in future studies comparing resting vs. activated microglia.
Macrophage secreted products, such as TNFα [64, 65], IL1β [66] and NO [67], can inhibit production of DHEA and androgens in the gonads via inhibition of p450c17 gene expression [68–70]. Our results indicate that 17βHSD1 and 5αR activity are not affected by inflammatory stimuli. This point is critical, considering that inflammatory conditions can dramatically reduce steroid metabolism in astrocytes co-cultured with microglia [41].
Steroid Metabolism in CNS Microglia
In this report, we show microglia expressed 17βHSD1 and 5αR, enzymes that are known to be involved in the metabolism of DHEA, androgens and estrogens. Conversion of H3-DHEA to AD did not occur in 1°MG consistent with absence of 3βHSD; however these cells converted H3-AD into T, DHT, and 5αAD, corroborating the activity of 17βHSD1 and 5αR in microglia. Similar findings have been reported in human alveolar macrophages where the principal metabolites of AD were 5αAD and T [71]. 5αR activity in microglia could be significant in amplifying the actions of androgens, as DHT is three times more efficient than T [72] and is considered a non-aromatizable androgen. Microglia had no detectable expression of aromatase, in contrast to differentiated monocytes/myeloid cells that show low levels of estrogen formation [73, 74].
In the CNS, DHEA has multiple effects reminiscent of sex hormones [75]. The absence of a specific DHEA receptor [76] has led investigators to suggest that DHEA is metabolized into active sex hormones that mediate these observed effects [28, 30, 74]. Neurons, astrocytes and oligodendrocytes can metabolize DHEA into sex hormones [18]. Recently we reported that the microglial cell line, BV2, is able to convert DHEA into Adiol and validated the identity of this product by high-performance liquid chromatography [30]. The current study corroborates these findings in 1°MG and shows the expression of 17βHSD1, required for this conversion, both in ex vivo MG and 1°MG.
DHEA levels are higher in brain than in circulation and have been negatively correlated with aging and neurodegeneration [77]. Moreover, conversion of DHEA in the brain, into metabolites like Adiol, may be reduced in patients with neurodegenerative disease [78]. Our data indicate that both resting and activated microglia can metabolize DHEA and specifically convert it to Adiol. This delta-5 steroid has reported androgenic and estrogenic properties in peripheral tissues [37, 79, 80] and in the male rat pituitary [81]. In this study we present evidence confirming that Adiol is an effective estrogen receptor agonist in ER-expressing granule neuronal cells (EtC.1). These results are significant given that microglia are situated among ER-expressing neurons and astrocytes [Sierra et al. submitted] suggesting that DHEA metabolism by microglia may be a source of active estrogens in the brain.
Microglia also expressed StS and Arsa. DHEA-sulfate, a predominant sulfated steroid in the brain with neuro-excitatory properties [82], cannot be utilized for sex steroid production in its sulfated form. The expression of StS and Arsa in microglia, consistent with that found in macrophages [83], suggests that microglia could modulate the levels of locally available DHEA [84] for androgen and estrogen conversion. Further studies will be required to elucidate these roles of StS and Arsa in microglia.
In conclusion, this report demonstrates for the first time that microglia express steroid-converting enzymes. Our data suggest that microglia depend on available steroid precursors for the conversion of androgens, rather than presenting de novo synthesis of sex hormones from cholesterol. Microglia differ from neurons, astrocytes and oligodendrocytes in the expression of steroid-converting enzymes, particularly those involved in the de novo formation of progesterone and DHEA from cholesterol. It is likely that some of these proteins may have alternate roles in microglia, such as StAR, or PBR, as demonstrated by the ability of PBR ligands to reduce TNFα production. Although inflammatory microglia are implicated in neural pathology, particularly by their accumulation at neurodegenerative loci, their steroid-converting activity may add to the adaptive neuroprotective mechanisms of the brain following damage. Therefore, understanding the impact of microglia steroid metabolism and the factors involved in its regulation will allow for better interventions and therapeutic use of steroid hormones.
Acknowledgements
We would like to thank Dr. M. Hardy and Dr. R. Ge (Population Council, Rockefeller University, NY) for help with enzyme inhibitors, for intellectually encouraging discussions, and revisions to this manuscript. We would also like to thank Dr. J. Idoyaga for assistance with flow cytometry analysis, and special thanks to Dr. D. Hume (University of Queensland, Australia) and Dr. J. Pollard (Albert Einstein College of Medicine, NY) for the kind gift of the 7.2fms-EGFP mice. Funding was obtained from NIA 5PO1AG16765-07.
Abbreviations
- DHEA
3β-hydroxy-5-androstene-17-one (dehydroepiandrosterone)
- AD
4-androstene-3,17-dione (androstenedione)
- 5αAD
5α-androstane-3,17-dione (5α-Adione)
- Adiol
5-androstene-3β,17β-diol (Δ5-Adiol)
- T
4-androsten-17β-ol-3-one (testosterone)
- DHT
5α-androstane-3-one-17β-ol (dihydrotestosterone)
- TLC
thin layer chromatography
- RT-PCR
real-time polymerase chain reaction
- PBR
peripheral benzodiazepine receptor
- StAR
steroidogenic acute regulatory protein
- P450scc
cytochrome p450 side chain cleavage enzyme
- P450c21
cytochrome p450 21-hydroxylase
- P450c17
cytochrome p450 17-hydroxylase
- Arsa
aryl sulfatase
- Sulft
DHEA sulfotransferase
- 3βHSD1,2,4
3β-hydroxysteroid dehydrogenase isoforms 1,2,4
- 17βHSD1
17β-hydroxysteroid dehydrogenase type 1
- p450Arom
cytochrome p450 aromatase
- 5αR
steroid 5α-reductase type 1
- 3αHSD
3α-hydroxysteroid dehydrogenase
- L27A
ribosomal protein L27A
- db-cAMP
dibutyril cyclic AMP
- LPS
lipopolysaccharide
- INFγ
interferon gamma
- NO
nitric oxide
- ERE
estrogen response element
- EGFP
enhanced green fluorescent protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bryant DN, et al. Multiple pathways transmit neuroprotective effects of gonadal steroids. Endocrine. 2006;29(2):199–207. doi: 10.1385/ENDO:29:2:199. [DOI] [PubMed] [Google Scholar]
- 2.Cooke BM, Woolley CS. Gonadal hormone modulation of dendrites in the mammalian CNS. J Neurobiol. 2005;64(1):34–46. doi: 10.1002/neu.20143. [DOI] [PubMed] [Google Scholar]
- 3.Garcia-Ovejero D, et al. Glia-neuron crosstalk in the neuroprotective mechanisms of sex steroid hormones. Brain Res Brain Res Rev. 2005;48(2):273–286. doi: 10.1016/j.brainresrev.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 4.Krause DN, Duckles SP, Pelligrino DA. Influence of sex steroid hormones on cerebrovascular function. J Appl Physiol. 2006;101(4):1252–1261. doi: 10.1152/japplphysiol.01095.2005. [DOI] [PubMed] [Google Scholar]
- 5.Scharfman HE, MacLusky NJ. The influence of gonadal hormones on neuronal excitability, seizures, and epilepsy in the female. Epilepsia. 2006;47(9):1423–1440. doi: 10.1111/j.1528-1167.2006.00672.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McEwen B. Estrogen actions throughout the brain. Recent Prog Horm Res. 2002;57:357–384. doi: 10.1210/rp.57.1.357. [DOI] [PubMed] [Google Scholar]
- 7.Galea LA, et al. Gonadal hormone modulation of hippocampal neurogenesis in the adult. Hippocampus. 2006;16(3):225–232. doi: 10.1002/hipo.20154. [DOI] [PubMed] [Google Scholar]
- 8.Endoh A, et al. The zona reticularis is the site of biosynthesis of dehydroepiandrosterone and dehydroepiandrosterone sulfate in the adult human adrenal cortex resulting from its low expression of 3 beta-hydroxysteroid dehydrogenase. J Clin Endocrinol Metab. 1996;81(10):3558–3565. doi: 10.1210/jcem.81.10.8855801. [DOI] [PubMed] [Google Scholar]
- 9.Mason JI, Bird IM, Rainey WE. Adrenal androgen biosynthesis with special attention to P450c17. Ann N Y Acad Sci. 1995;774:47–58. doi: 10.1111/j.1749-6632.1995.tb17371.x. [DOI] [PubMed] [Google Scholar]
- 10.Turgeon JL, et al. Complex actions of sex steroids in adipose tissue, the cardiovascular system, and brain: Insights from basic science and clinical studies. Endocr Rev. 2006;27(6):575–605. doi: 10.1210/er.2005-0020. [DOI] [PubMed] [Google Scholar]
- 11.Baulieu EE. Neurosteroids: of the nervous system, by the nervous system, for the nervous system. Recent Prog Horm Res. 1997;52:1–32. [PubMed] [Google Scholar]
- 12.Schumacher M, et al. Neurosteroids in the Hippocampus: Neuronal Plasticity and Memory. Stress. 1997;2(1):65–78. doi: 10.3109/10253899709014738. [DOI] [PubMed] [Google Scholar]
- 13.Paul SM, Purdy RH. Neuroactive steroids. Faseb J. 1992;6(6):2311–2322. [PubMed] [Google Scholar]
- 14.Majewska MD. Neurosteroids: endogenous bimodal modulators of the GABAA receptor. Mechanism of action and physiological significance. Prog Neurobiol. 1992;38(4):379–395. doi: 10.1016/0301-0082(92)90025-a. [DOI] [PubMed] [Google Scholar]
- 15.Tsutsui K. Biosynthesis and organizing action of neurosteroids in the developing Purkinje cell. Cerebellum. 2006;5(2):89–96. doi: 10.1080/14734220600697211. [DOI] [PubMed] [Google Scholar]
- 16.Schumacher M, et al. Local synthesis and dual actions of progesterone in the nervous system: neuroprotection and myelination. Growth Horm IGF Res. 2004;14 Suppl A:S18–S33. doi: 10.1016/j.ghir.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 17.Azcoitia I, et al. Brain aromatase is neuroprotective. J Neurobiol. 2001;47(4):318–329. doi: 10.1002/neu.1038. [DOI] [PubMed] [Google Scholar]
- 18.Zwain IH, Yen SS. Neurosteroidogenesis in astrocytes, oligodendrocytes, and neurons of cerebral cortex of rat brain. Endocrinology. 1999;140(8):3843–3852. doi: 10.1210/endo.140.8.6907. [DOI] [PubMed] [Google Scholar]
- 19.Aloisi F. Immune function of microglia. Glia. 2001;36(2):165–179. doi: 10.1002/glia.1106. [DOI] [PubMed] [Google Scholar]
- 20.Griffin WS. Inflammation and neurodegenerative diseases. Am J Clin Nutr. 2006;83(2):470S–474S. doi: 10.1093/ajcn/83.2.470S. [DOI] [PubMed] [Google Scholar]
- 21.Kim YS, Joh TH. Microglia, major player in the brain inflammation: their roles in the pathogenesis of Parkinson's disease. Exp Mol Med. 2006;38(4):333–347. doi: 10.1038/emm.2006.40. [DOI] [PubMed] [Google Scholar]
- 22.Raivich G, Banati R. Brain microglia and blood-derived macrophages: molecular profiles and functional roles in multiple sclerosis and animal models of autoimmune demyelinating disease. Brain Res Brain Res Rev. 2004;46(3):261–281. doi: 10.1016/j.brainresrev.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 23.Schumacher M, et al. Steroid hormones and neurosteroids in normal and pathological aging of the nervous system. Prog Neurobiol. 2003;71(1):3–29. doi: 10.1016/j.pneurobio.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 24.Straub RH, et al. Serum dehydroepiandrosterone (DHEA) and DHEA sulfate are negatively correlated with serum interleukin-6 (IL-6), and DHEA inhibits IL-6 secretion from mononuclear cells in man in vitro: possible link between endocrinosenescence and immunosenescence. J Clin Endocrinol Metab. 1998;83(6):2012–2017. doi: 10.1210/jcem.83.6.4876. [DOI] [PubMed] [Google Scholar]
- 25.Cutolo M, et al. Sex hormone modulation of cell growth and apoptosis of the human monocytic/macrophage cell line. Arthritis Res Ther. 2005;7(5):R1124–R1132. doi: 10.1186/ar1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jacobson JD, Ansari MA. Immunomodulatory actions of gonadal steroids may be mediated by gonadotropin-releasing hormone. Endocrinology. 2004;145(1):330–336. doi: 10.1210/en.2003-0510. [DOI] [PubMed] [Google Scholar]
- 27.Schmidt M, et al. Inflammation and sex hormone metabolism. Ann N Y Acad Sci. 2006;1069:236–246. doi: 10.1196/annals.1351.021. [DOI] [PubMed] [Google Scholar]
- 28.Jellinck PH, et al. Metabolism of dehydroepiandrosterone by rodent brain cell lines: relationship between 7-hydroxylation and aromatization. J Steroid Biochem Mol Biol. 2005;93(1):81–86. doi: 10.1016/j.jsbmb.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 29.Jellinck PH, et al. Dehydroepiandrosterone (DHEA) metabolism in the brain: identification by liquid chromatography/mass spectrometry of the delta-4-isomer of DHEA and related steroids formed from androstenedione by mouse BV2 microglia. J Steroid Biochem Mol Biol. 2006;98(1):41–47. doi: 10.1016/j.jsbmb.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 30.Jellinck PH, et al. Selective Conversion of Dehydroepiandrosterone (DHEA) by Microgila to 5-Androstenediol - A Steroid with Inherent Estrogen Properties. J Steroid Biochem. doi: 10.1016/j.jsbmb.2007.04.004. In press. [DOI] [PubMed] [Google Scholar]
- 31.Bessis A, et al. Microglial control of neuronal death and synaptic properties. Glia. 2007;55(3):233–238. doi: 10.1002/glia.20459. [DOI] [PubMed] [Google Scholar]
- 32.Raivich G. Like cops on the beat: the active role of resting microglia. Trends Neurosci. 2005;28(11):571–573. doi: 10.1016/j.tins.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 33.Sasmono RT, et al. A macrophage colony-stimulating factor receptor-green fluorescent protein transgene is expressed throughout the mononuclear phagocyte system of the mouse. Blood. 2003;101(3):1155–1163. doi: 10.1182/blood-2002-02-0569. [DOI] [PubMed] [Google Scholar]
- 34.Sierra A, et al. Microglia derived from aging mice exhibit an altered inflammatory profile. Glia. 2007;55(4):412–424. doi: 10.1002/glia.20468. [DOI] [PubMed] [Google Scholar]
- 35.Rivest S. Molecular insights on the cerebral innate immune system. Brain Behav Immun. 2003;17(1):13–19. doi: 10.1016/s0889-1591(02)00055-7. [DOI] [PubMed] [Google Scholar]
- 36.Casellas P, Galiegue S, Basile AS. Peripheral benzodiazepine receptors and mitochondrial function. Neurochem Int. 2002;40(6):475–486. doi: 10.1016/s0197-0186(01)00118-8. [DOI] [PubMed] [Google Scholar]
- 37.Poortman J, et al. Interaction of delta-5-androstene-3beta, 17beta-diol with estradiol and dihydrotestosterone receptors in human myometrial and mammary cancer tissue. J Clin Endocrinol Metab. 1975;40(3):373–379. doi: 10.1210/jcem-40-3-373. [DOI] [PubMed] [Google Scholar]
- 38.Stein DG, Hoffman SW. Estrogen and progesterone as neuroprotective agents in the treatment of acute brain injuries. Pediatr Rehabil. 2003;6(1):13–22. doi: 10.1080/1363849031000095279. [DOI] [PubMed] [Google Scholar]
- 39.Wise PM, et al. Estradiol is a neuroprotective factor in in vivo and in vitro models of brain injury. J Neurocytol. 2000;29(5–6):401–410. doi: 10.1023/a:1007169408561. [DOI] [PubMed] [Google Scholar]
- 40.Azcoitia I, et al. Brain steroidogenesis: emerging therapeutic strategies to prevent neurodegeneration. J Neural Transm. 2005;112(1):171–176. doi: 10.1007/s00702-004-0179-y. [DOI] [PubMed] [Google Scholar]
- 41.Zwain IH, Yen SS. Dehydroepiandrosterone: biosynthesis and metabolism in the brain. Endocrinology. 1999;140(2):880–887. doi: 10.1210/endo.140.2.6528. [DOI] [PubMed] [Google Scholar]
- 42.Baulieu EE, Robel P. Dehydroepiandrosterone (DHEA) and dehydroepiandrosterone sulfate (DHEAS) as neuroactive neurosteroids. Proc Natl Acad Sci U S A. 1998;95(8):4089–4091. doi: 10.1073/pnas.95.8.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sierra A. Neurosteroids: the StAR protein in the brain. J Neuroendocrinol. 2004;16(9):787–793. doi: 10.1111/j.1365-2826.2004.01226.x. [DOI] [PubMed] [Google Scholar]
- 44.Stocco DM. The role of the StAR protein in steroidogenesis: challenges for the future. J Endocrinol. 2000;164(3):247–253. doi: 10.1677/joe.0.1640247. [DOI] [PubMed] [Google Scholar]
- 45.Rodriguez-Agudo D, et al. Localization of StarD5 cholesterol binding protein. J Lipid Res. 2006;47(6):1168–1175. doi: 10.1194/jlr.M500447-JLR200. [DOI] [PubMed] [Google Scholar]
- 46.Hauet T, et al. PBR, StAR, and PKA: partners in cholesterol transport in steroidogenic cells. Endocr Res. 2002;28(4):395–401. doi: 10.1081/erc-120016814. [DOI] [PubMed] [Google Scholar]
- 47.Lacor P, et al. Regulation of the expression of peripheral benzodiazepine receptors and their endogenous ligands during rat sciatic nerve degeneration and regeneration: a role for PBR in neurosteroidogenesis. Brain Res. 1999;815(1):70–80. doi: 10.1016/s0006-8993(98)01105-6. [DOI] [PubMed] [Google Scholar]
- 48.Brown RC, Papadopoulos V. Role of the peripheral-type benzodiazepine receptor in adrenal and brain steroidogenesis. Int Rev Neurobiol. 2001;46:117–143. doi: 10.1016/s0074-7742(01)46061-2. [DOI] [PubMed] [Google Scholar]
- 49.Decaudin D. Peripheral benzodiazepine receptor and its clinical targeting. Anticancer Drugs. 2004;15(8):737–745. doi: 10.1097/00001813-200409000-00001. [DOI] [PubMed] [Google Scholar]
- 50.Galiegue S, Tinel N, Casellas P. The peripheral benzodiazepine receptor: a promising therapeutic drug target. Curr Med Chem. 2003;10(16):1563–1572. doi: 10.2174/0929867033457223. [DOI] [PubMed] [Google Scholar]
- 51.Papadopoulos V, et al. Peripheral-type benzodiazepine receptor in neurosteroid biosynthesis, neuropathology and neurological disorders. Neuroscience. 2006;138(3):749–756. doi: 10.1016/j.neuroscience.2005.05.063. [DOI] [PubMed] [Google Scholar]
- 52.Carayon P, et al. Involvement of peripheral benzodiazepine receptors in the protection of hematopoietic cells against oxygen radical damage. Blood. 1996;87(8):3170–3178. [PubMed] [Google Scholar]
- 53.Banati RB. Visualising microglial activation in vivo. Glia. 2002;40(2):206–217. doi: 10.1002/glia.10144. [DOI] [PubMed] [Google Scholar]
- 54.Taupin V, et al. Modulation of tumor necrosis factor-alpha, interleukin-1 beta, interleukin-6, interleukin-8, and granulocyte/macrophage colony-stimulating factor expression in human monocytes by an endogenous anxiogenic benzodiazepine ligand, triakontatetraneuropeptide: evidence for a role of prostaglandins. Mol Pharmacol. 1993;43(1):64–69. [PubMed] [Google Scholar]
- 55.Taupin V, et al. Increase in IL-6, IL-1 and TNF levels in rat brain following traumatic lesion. Influence of pre- and post-traumatic treatment with Ro5 4864, a peripheral-type (p site) benzodiazepine ligand. J Neuroimmunol. 1993;42(2):177–185. doi: 10.1016/0165-5728(93)90008-m. [DOI] [PubMed] [Google Scholar]
- 56.Choi HB, et al. Inhibition of lipopolysaccharide-induced cyclooxygenase-2, tumor necrosis factor-alpha and [Ca2+]i responses in human microglia by the peripheral benzodiazepine receptor ligand PK11195. J Neurochem. 2002;83(3):546–555. doi: 10.1046/j.1471-4159.2002.01122.x. [DOI] [PubMed] [Google Scholar]
- 57.Leonelli E, et al. Ro5-4864, a synthetic ligand of peripheral benzodiazepine receptor, reduces aging-associated myelin degeneration in the sciatic nerve of male rats. Mech Ageing Dev. 2005;126(11):1159–1163. doi: 10.1016/j.mad.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 58.Veiga S, Azcoitia I, Garcia-Segura LM. Ro5-4864, a peripheral benzodiazepine receptor ligand, reduces reactive gliosis and protects hippocampal hilar neurons from kainic acid excitotoxicity. J Neurosci Res. 2005;80(1):129–137. doi: 10.1002/jnr.20430. [DOI] [PubMed] [Google Scholar]
- 59.Chensue SW, et al. In vivo biologic and immunohistochemical analysis of interleukin-1 alpha, beta and tumor necrosis factor during experimental endotoxemia. Kinetics, Kupffer cell expression, and glucocorticoid effects. Am J Pathol. 1991;138(2):395–402. [PMC free article] [PubMed] [Google Scholar]
- 60.Besedovsky HO, del Rey A. Immune-neuro-endocrine interactions: facts and hypotheses. Endocr Rev. 1996;17(1):64–102. doi: 10.1210/edrv-17-1-64. [DOI] [PubMed] [Google Scholar]
- 61.Badrinarayanan R, et al. Corticosterone impairs the mRNA expression and activity of 3beta- and 17beta-hydroxysteroid dehydrogenases in adult rat Leydig cells. Biochem Cell Biol. 2006;84(5):745–754. doi: 10.1139/o06-074. [DOI] [PubMed] [Google Scholar]
- 62.Gao HB, et al. Suppression of endogenous corticosterone levels in vivo increases the steroidogenic capacity of purified rat Leydig cells in vitro. Endocrinology. 1996;137(5):1714–1718. doi: 10.1210/endo.137.5.8612506. [DOI] [PubMed] [Google Scholar]
- 63.Agarwal AK, Auchus RJ. Minireview: cellular redox state regulates hydroxysteroid ehydrogenase activity and intracellular hormone potency. Endocrinology. 2005;146(6):2531–2538. doi: 10.1210/en.2005-0061. [DOI] [PubMed] [Google Scholar]
- 64.Andreani CL, et al. Cytokine-mediated regulation of ovarian function. Tumor necrosis factor alpha inhibits gonadotropin-supported ovarian androgen biosynthesis. J Biol Chem. 1991;266(11):6761–6766. [PubMed] [Google Scholar]
- 65.Xiong Y, Hales DB. Differential effects of tumor necrosis factor-alpha and interleukin-1 on 3 beta-hydroxysteroid dehydrogenase/delta 5-->delta 4 isomerase expression in mouse Leydig cells. Endocrine. 1997;7(3):295–301. doi: 10.1007/BF02801322. [DOI] [PubMed] [Google Scholar]
- 66.Hurwitz A, et al. Cytokine-mediated regulation of ovarian function: interleukin-1 inhibits gonadotropin-induced androgen biosynthesis. Endocrinology. 1991;129(3):1250–1256. doi: 10.1210/endo-129-3-1250. [DOI] [PubMed] [Google Scholar]
- 67.Pomerantz DK, Pitelka V. Nitric oxide is a mediator of the inhibitory effect of activated macrophages on production of androgen by the Leydig cell of the mouse. Endocrinology. 1998;139(3):922–931. doi: 10.1210/endo.139.3.5773. [DOI] [PubMed] [Google Scholar]
- 68.Hales DB. Interleukin-1 inhibits Leydig cell steroidogenesis primarily by decreasing 17 alpha-hydroxylase/C17-20 lyase cytochrome P450 expression. Endocrinology. 1992;131(5):2165–2172. doi: 10.1210/endo.131.5.1425417. [DOI] [PubMed] [Google Scholar]
- 69.Li X, et al. Tumor necrosis factor-alpha inhibition of 17 alpha-hydroxylase/C17-20 lyase gene (Cyp17) expression. Endocrinology. 1995;136(8):3519–3526. doi: 10.1210/endo.136.8.7628389. [DOI] [PubMed] [Google Scholar]
- 70.Onami S, et al. Splenic macrophages can modify steroidogenesis of Leydig cells. Endocr J. 1996;43(5):477–485. doi: 10.1507/endocrj.43.477. [DOI] [PubMed] [Google Scholar]
- 71.Milewich L, Kaimal V, Toews GB. Androstenedione metabolism in human alveolar macrophages. J Clin Endocrinol Metab. 1983;56(5):920–924. doi: 10.1210/jcem-56-5-920. [DOI] [PubMed] [Google Scholar]
- 72.Kemppainen JA, et al. Distinguishing androgen receptor agonists and antagonists: distinct mechanisms of activation by medroxyprogesterone acetate and dihydrotestosterone. Mol Endocrinol. 1999;13(3):440–454. doi: 10.1210/mend.13.3.0255. [DOI] [PubMed] [Google Scholar]
- 73.Jakob F, et al. Expression and regulation of aromatase cytochrome P450 in THP 1 human myeloid leukaemia cells. Mol Cell Endocrinol. 1995;110(1–2):27–33. doi: 10.1016/0303-7207(95)03512-6. [DOI] [PubMed] [Google Scholar]
- 74.Schmidt M, et al. Conversion of dehydroepiandrosterone to downstream steroid hormones in macrophages. J Endocrinol. 2000;164(2):161–169. doi: 10.1677/joe.0.1640161. [DOI] [PubMed] [Google Scholar]
- 75.Majewska MD. Neuronal actions of dehydroepiandrosterone. Possible roles in brain development, aging, memory, and affect. Ann N Y Acad Sci. 1995;774:111–120. doi: 10.1111/j.1749-6632.1995.tb17375.x. [DOI] [PubMed] [Google Scholar]
- 76.Regelson W, Kalimi M. Dehydroepiandrosterone (DHEA)--the multifunctional steroid. II. Effects on the CNS, cell proliferation, metabolic and vascular, clinical and other effects. Mechanism of action? Ann N Y Acad Sci. 1994;719:564–575. doi: 10.1111/j.1749-6632.1994.tb56860.x. [DOI] [PubMed] [Google Scholar]
- 77.Weill-Engerer S, et al. Neurosteroid quantification in human brain regions: comparison between Alzheimer's and nondemented patients. J Clin Endocrinol Metab. 2002;87(11):5138–5143. doi: 10.1210/jc.2002-020878. [DOI] [PubMed] [Google Scholar]
- 78.Weill-Engerer S, et al. In vitro metabolism of dehydroepiandrosterone (DHEA) to 7alpha-hydroxy-DHEA and Delta5-androstene-3beta,17beta-diol in specific regions of the aging brain from Alzheimer's and non-demented patients. Brain Res. 2003;969(1–2):117–125. doi: 10.1016/s0006-8993(03)02288-1. [DOI] [PubMed] [Google Scholar]
- 79.Adams JB. Control of secretion and the function of C19-delta 5-steroids of the human adrenal gland. Mol Cell Endocrinol. 1985;41(1):1–17. doi: 10.1016/0303-7207(85)90138-8. [DOI] [PubMed] [Google Scholar]
- 80.Miyamoto H, et al. Delta5-androstenediol is a natural hormone with androgenic activity in human prostate cancer cells. Proc Natl Acad Sci U S A. 1998;95(19):11083–11088. doi: 10.1073/pnas.95.19.11083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Thieulant ML, et al. Binding and effects of 5 alpha-androstane-3 beta,17 beta-diol in the male rat pituitary. J Steroid Biochem. 1983;19(1A):241–246. doi: 10.1016/s0022-4731(83)80031-4. [DOI] [PubMed] [Google Scholar]
- 82.Baulieu EE, Robel P. Dehydroepiandrosterone and dehydroepiandrosterone sulfate as neuroactive neurosteroids. J Endocrinol. 1996;150 Suppl:S221–S239. [PubMed] [Google Scholar]
- 83.Weidler C, et al. Tumor necrosis factor inhibits conversion of dehydroepiandrosterone sulfate (DHEAS) to DHEA in rheumatoid arthritis synovial cells: a prerequisite for local androgen deficiency. Arthritis Rheum. 2005;52(6):1721–1729. doi: 10.1002/art.21112. [DOI] [PubMed] [Google Scholar]
- 84.Hennebold JD, Daynes RA. Regulation of macrophage dehydroepiandrosterone sulfate metabolism by inflammatory cytokines. Endocrinology. 1994;135(1):67–75. doi: 10.1210/endo.135.1.8013393. [DOI] [PubMed] [Google Scholar]