

Abstract
The thymidine analogue 4-thiothymidine (S4TdR) is a photosensitizer for UVA radiation. The UV absorbance spectrum of S4TdR and its incorporation into DNA suggests that it might act synergistically with nonlethal doses of UVA to selectively kill hyperproliferative or cancerous skin cells. We show here that nontoxic concentrations of S4TdR combine with nonlethal doses of UVA to kill proliferating cultured skin cells. Established cell lines with a high fraction of proliferating cells were more sensitive than primary keratinocytes or fibroblasts to apoptosis induction by S4TdR/UVA. Although S4TdR plus UVA treatment induces stabilization of p53, cell death, as measured by apoptosis or clonal survival, occurs to a similar extent in both p53 wild-type and p53-null backgrounds. Furthermore, different types of human papilloma virus E6 proteins, which protect against UVB-induced apoptosis, have little effect on killing by S4TdR/UVA. S4TdR/UVA offers a possible therapeutic intervention strategy that seems to be applicable to human papilloma virus–associated skin lesions.
Introduction
Skin cancer is the most frequently diagnosed neoplastic condition. The frequency of nonmelanoma skin cancer (NMSC) in Caucasian populations has risen steadily in recent years and is predicted to increase further (1-3). Solar radiation is the major etiologic factor in the development of the basal cell carcinomas and squamous cell carcinomas (SCC) which comprise the majority of NMSCs. A significant fraction of incident solar UVA (wavelengths, 320–400 nm) and longer wavelength UVB (290–320 nm) penetrates the epidermal layer into the underlying dermis. These contain the keratinocytes and fibroblasts, respectively. UV radiation also causes epidermal thickening, which protects the underlying cells. This protective process can be compromised in hyperproliferative skin lesions.
DNA damage is firmly implicated in the initiation of NMSC. The DNA cyclobutane pyrimidine dimers and (6-4)pyrimidine-pyrimidone photoproducts that are generated when DNA absorbs UVB (4) are associated with mutagenesis and cancer. The p53 tumor suppressor gene is commonly mutated in SCC, and the most frequent p53 mutations are transitions at dipyrimidine sites that are considered signatures for UV-induced DNA damage. Individuals with xeroderma pigmentosum, in whom the nucleotide excision repair pathway is disabled by mutation, are unable to remove mutagenic DNA photoproducts from their skin cells and have a greatly (2,000-fold) increased rate of skin cancer in sun-exposed areas (5). Although UVA is the main component of incident sunlight, it is less directly damaging to DNA because the constituent purine and pyrimidine bases do not absorb significantly at wavelengths >320 nm. The genotoxic effects of UVA are a consequence of indirect photochemical reactions and it is less mutagenic and carcinogenic than UVB.
There is mounting evidence that human papilloma viruses (HPV) are a cofactor with UV in the development of NMSC (6). HPVs infect keratinocytes, alter the rate of epithelial proliferation and interfere with normal differentiation (7-9). As a result, the natural barrier formed by the upper layers of the epidermis is weakened. Although cutaneous HPV infection is usually a benign, self-limiting process, it may occasionally be associated with malignant conversion (10). The involvement of HPV in anogenital cancer is widely acknowledged (11). The E6 and E7 proteins encoded by anogenital HPV types 16 and 18 inactivate the p53 and Rb proteins that are critical for an appropriate cellular response to DNA damage. There is a noteworthy association between HPV and skin tumors in immunosuppressed renal transplant recipients and patients with the rare genetic disorder epidermodysplasia verruciformis (6, 10, 12, 13). Individuals with epidermodysplasia verruciformis develop SCC on sun-exposed parts of the body and these tumors are frequently host to HPV5 or HPV8 (14, 15). Because of this, HPVs, notably HPV5 and HPV8, have become a major focus of investigations in NMSC. The E6 and E7 proteins from cutaneous HPVs do not seem to compromise p53 or Rb function in the same way as the anogenital HPV E6 proteins. However, they do inhibit UVB-mediated, p53-dependent apoptosis (16). Certain HPV types selectively inhibit p53-dependent transcription of proapoptotic genes (17). Others may delay the repair of UVB-induced thymine dimers (18). Importantly, the E6 protein of cutaneous HPVs can target the mitochondrial Bcl-2 homologous antagonist/killer (Bak) protein—a key regulator of apoptosis—for proteolytic degradation (19). The consequent abrogation of UVB-induced apoptosis may contribute to NMSC development.
Surgical excision is the most common and successful treatment for localized primary skin lesions. Alternative approaches such as electrodessication, curettage and cryosurgery, or radiotherapy offer poor cosmetic outcome. Phototherapy is also a treatment option. Therapeutic UVB and psoralen plus UVA light are used to treat inflammatory dermatoses such as psoriasis. In psoralen plus UVA, a nontoxic photoreactive drug is activated by subsequent exposure to UVA to cause extensive DNA damage that eventually kills the tumor cells. Psoralen plus UVA has been widely used to treat psoriasis (20) as well as head and neck cancers. Although highly effective and well tolerated, it has serious limitations. In particular, it involves high cumulative doses of UVA and is associated with an increased risk of NMSC (21, 22). Significantly, the effectiveness of psoralen plus UVA is compromised in p53 mutant cells (23). This is particularly important because >90% of skin tumors contain p53 mutations (24, 25). In addition, if apoptotic pathways under the control of Bak are involved in the toxicity of antitumor treatments, infection by HPVs of the epidermodysplasia verruciformis type may compromise therapeutic effectiveness. For this reason, there is a need to develop alternative ways to kill hyperproliferative skin cells.
The nontoxic thiopyrimidine nucleoside, 4-thiothymidine (S4TdR), was previously shown to sensitize established human cell lines to low doses of UVA (∼365 nm; ref. 26). S4TdR/UVA toxicity depends absolutely on S4TdR salvage by thymidine kinase and incorporation into DNA. We have examined cell killing by the S4TdR/UVA combination in more detail. In particular, we address the possible suitability of S4TdR/UVA as an intervention strategy in epidermal disease, including NMSC and whether it might evade the antiapoptotic action of HPV in virally infected cells.
Materials and Methods
Cells and Cell Culture
Cells were all grown in 10% CO2 at 37°C and media supplemented with 10% FCS. HT1080 fibrosarcoma cells which express both the codon 72 arginine and proline (arg/pro) isoforms of wild-type p53 (27), normal, and psoriatic human skin fibroblasts were grown in DMEM. RTS3b, a spontaneously immortalized p53-null keratinocyte cell line (28) was grown in DMEM/Ham's F12 (3:1) plus growth factors and normal human keratinocytes in the same medium on a feeder layer of gamma ray–irradiated (60 Gy) Swiss 3T3 fibroblasts (29). The normal skin fibroblasts and keratinocytes were derived from a 46-year-old patient following breast surgery (skin type I). All experiments with keratinocytes and fibroblasts were done with cells at passages 3 to 5. Stable polyclonal HT1080 cell lines expressing E6 protein from either HPV5 or HPV18 in the bicistronic vector pIres (Clontech) were generated and expression of viral mRNA verified by RT-PCR as previously described (18). All studies involving patient material were carried out with prior local ethics committee approval and informed consent.
Chemicals and Drug Treatments
All chemicals, except where specified, were purchased from Sigma Chemical Co. S4TdR was synthesized as described previously (30) and dissolved in sterile deionized water. All treatments with S4TdR were carried out for 48 h in medium containing 10% FCS which had been dialyzed extensively through a 2-kDa exclusion membrane Spectra/Por (Spectrum Laboratories; Medicell International, Ltd.).
UV Radiation
UVA irradiation was delivered from a UVH 253 lamp (UV Light Technology, Ltd.) with a maximum output of 100 mW/cm2, as described previously (26). Emission wavelengths ranged from 320 to 400 nm with a maximum at 365 nm. UVB radiation was delivered from a broadband CL-1000 (Ultra-violet Products, Ltd.) equipped with five F8T5 bulbs giving a spectral peak at 312 nm. The output was 1.2 mW/cm2. Cells were irradiated at 60% to 70% confluency.
Determination of Cell Survival
Treated cells were plated at clonal density (20–40 cell/cm2, depending on the cell type) in fresh growth medium and allowed to attach for 4 to 8 h prior to irradiation. The colony-forming efficiency of untreated cells was between 5% and 12%. Keratinocytes were plated onto lethally gamma-irradiated Swiss 3T3 fibroblasts. After UVA irradiation, cells were maintained in drug-free normal growth medium for 10 to 14 days. Colonies were then fixed, stained with crystal violet, and scored. For each determination, at least three independent experiments were done in triplicate dishes for each experimental point.
Dimethylthiazol-2-yl-2-5-Diphenyltetrazolium Bromide Assays
Cells were seeded in 96-well plates in medium containing dialyzed serum and S4TdR (0, 10, 30, or 100 μmol/L) for 48 h, then UVA- or UVB-irradiated. After irradiation, normal growth medium was replaced and incubation continued for 72 h. Cells were then incubated for 4 h in dimethylthiazol-2-yl-2-5-diphenyltetrazolium bromide solution (0.5 mg/mL) prepared in serum-free medium. Medium was removed and DMSO was added to solubilize the precipitate. Optical densities were measured at 570 nm.
Western Blot Analysis
Total cell lysates were resolved on SDS polyacrylamide gels and transferred onto polyvinylidene difluoride membranes (Amersham). Membranes were probed with primary antibodies against: p53 (DO-1, dilution 1:1,000; Cancer Research UK), Bak (Ab-2, dilution 1:250 for Western blots; Ab-1, dilution 1:50 for flow cytometry), Bax (2D2, dilution 1:1,000), Mdm2 (IF2, dilution 1:1,000), and α-tubulin (DM1A, dilution 1:4,000) from Calbiochem, E6AP (BL447, dilution 1:1,000) from Bethyl Laboratories, Inc., phospho-Ser15 (dilution 1:1,000), phospho-Ser392 (dilution 1:1,000) p53, p21 (DCS60, dilution 1:2,500), and cleaved caspase 3 (Asp175, dilution 1:1,000) from Cell Signaling Technology, Inc. Secondary antibodies were horseradish peroxidase–conjugated rabbit anti-mouse or goat anti-rabbit (used at dilution of 1:2,000; Dako UK, Ltd.). Reactive proteins were visualized by chemiluminescence with ECL plus (Amersham plc).
Flow Cytometry
Annexin V + propidium iodide (PI) staining. After UV treatment, adherent and nonadherent cells were pooled and treated with Alexa647-conjugated Annexin V (BD Biosciences) in 10 mmol/L of HEPES-NaOH (pH 7.4), 140 mmol/L of NaCl, and 2.5 mmol/L of CaCl2 for 15 min at room temperature in the dark. Following transfer to ice, PI (at a concentration of 50 μg/mL in PBS) was added for 5 min and cells were analyzed by flow cytometry. For analysis of S phase population by bromodeoxyuridine labeling, S4TdR-treated cells were pulse-labeled with bromodeoxyuridine (10 μmol/L) for 1 h at 37°C, harvested, fixed with 70% ethanol, and processed for analysis by flow cytometry. Flow cytometric detection of the open Bak isoform was done as described elsewhere (31). For each sample, 10,000 cells were analyzed and data were captured on a FACSCalibur (BD Biosciences) and analysis was done using CellQuest software (Becton Dickinson).
Microscopy
Cells were visualized on a Leica microscope (Leica Microsystems UK, Ltd.), using bright-field setting and a 200× magnification.
Statistical Analysis
The results are represented as the mean ± SD. Significant differences (P < 0.05) are indicated and were determined by Student's t test, either paired or unpaired, after one-way ANOVA (*, P < 0.05; **, P < 0.01).
Results
Cell Killing by S4TdR/UVA
Effect of Growth in S4TdR on Cell Cycle
S4TdR/UVA was previously shown to be cytotoxic towards SV40-transformed human fibroblast and lymphoma cell lines (26). To examine the effects of S4TdR/UVA in cells more relevant to human skin disorders, we compared the responses of HT1080, fibrosarcoma cell line (27), and RTS3b—a spontaneously immortalized keratinocyte cell line (28). These cell lines were compared with primary normal adult keratinocytes (normal human keratinocytes, NHK) and their patient-matched dermal fibroblasts (normal human fibroblasts, NHF), and to cultures of primary dermal psoriatic fibroblasts.
S4TdR/UVA toxicity depends on S4TdR incorporation into DNA. To maximize incorporation, cells were grown in medium containing S4TdR supplemented with FCS depleted of endogenous TdR by dialysis. Using flow cytometry, we first examined whether this had a significant effect on cell proliferation or on cycle distribution, as measured after pulse-labeling with bromodeoxyuridine. Neither HT1080 nor RTS3b cells were affected by growth in dialyzed serum, 100 μmol/L of S4TdR, or both. Irrespective of the growth conditions, approximately half of the cells (54–57%) were in S phase (Supplementary Table S1).4
The cell cycle distributions of NHK and NHF from normal adult skin differed substantially from those of the two established cell lines. As expected, both NHK and NHF contained a substantially lower fraction of S phase cells (12–18%) and a proportional increase in G0/G1 cells (62–79%). In NHK, neither of these variables were affected by growth in dialyzed serum or S4TdR. In both NHF and psoriatic human fibroblast, growth in dialyzed serum caused a small (<2-fold) decrease in the fraction of S phase cells. No detectable change in proliferative fraction occurred in the presence of S4TdR, however (Supplementary Table S1).4
Sensitization to UVA Killing
S4TdR sensitized both HT1080 and RTS3b cells to low doses of UVA. Cells grown for 48 h in a range of S4TdR concentrations were irradiated with 5 or 10 kJ/m2 of UVA and sensitivity determined by dimethylthiazol-2-yl-2-5-diphenyltetrazolium bromide assay. For both cell lines, survival was significantly reduced by 10 μmol/L of S4TdR combined with 5 kJ/m2 of UVA (Fig. 1). The same UVA dose was more toxic to both HT1080 and RTS3b cells that had been grown in 30 or 100 μmol/L of S4TdR. Neither UVA (5 or 10 kJ/m2) nor S4TdR alone (10, 30, 100 μmol/L) detectably affected survival. In control experiments, both cell lines were sensitive to UVB (0.15 kJ/m2) independently of growth in S4TdR. This is consistent with its lack of significant absorbance in the UVB region (26). Over the range of S4TdR and UVA doses examined, HT1080 cells were slightly more sensitive than RTS3b. This was also reflected in their similar slightly greater sensitivity to UVB.
Figure 1.
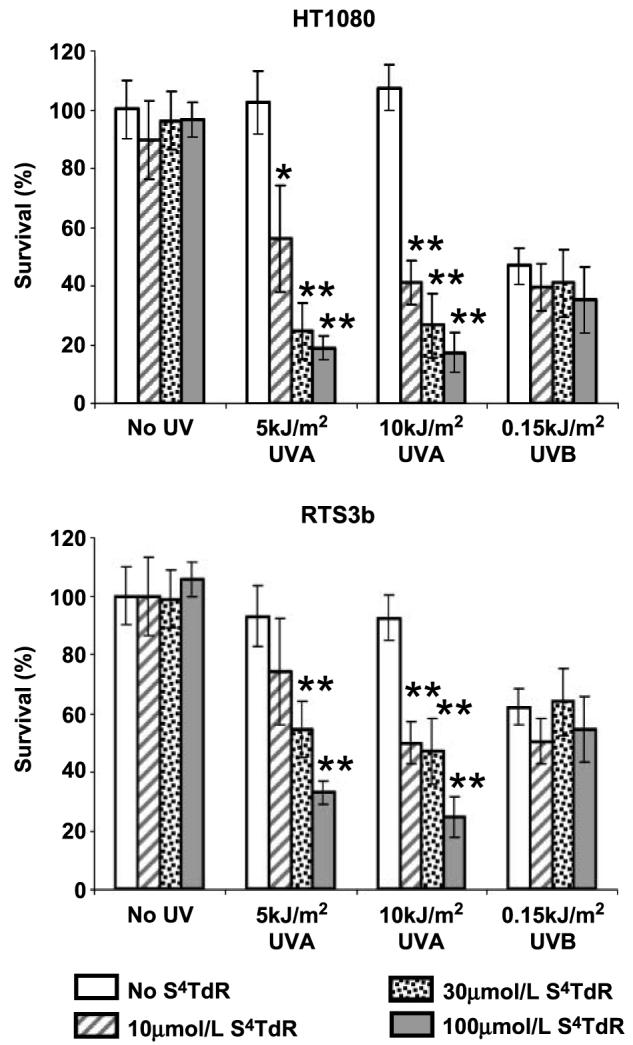
Survival of human fibrosarcoma and keratinocyte lines following S4TdR/UVA treatment. HT1080 and RTS3b cells were treated with 0, 10, 30, or 100 μmol/L of S4TdR for 48 h as described in Materials and Methods, and UVA- or UVB-irradiated at the doses indicated. Cells were grown further for 72 h in fresh medium and survival was measured by dimethylthiazol-2-yl-2-5-diphenyltetrazolium bromide assay. Columns, means; bars, SD (n = 4–6). UVA treatments: *, P < 0.05; **, P < 0.01, significant differences from the corresponding drug treatment in unirradiated control. Survival associated with UVB treatments were not significantly different among themselves, but were significantly different (P < 0.05) from the corresponding drug treatment in unirradiated controls.
Without the photosensitizer, neither the established cell lines nor primary keratinocytes (NHK) or fibroblasts (NHF) were significantly affected by UVA alone at doses <50 kJ/m2. Above this dose, survival, measured by clonal assay, declined. HT1080 and RTS3b were equally sensitive and their survival was reduced to ∼10% at 200 kJ/m2 (Fig. 2A). NHK were significantly more resistant to UVA than their immortal RTS3b counterparts and NHF. Approximately 40% of NHK survived a dose of 200 kJ/m2 (Fig. 2A). NHF were the most sensitive and only ∼1% survived a dose of 200 kJ/m2. This observation is consistent with the generally decreased susceptibility of NHK to UV irradiation reported by other laboratories (32).
Figure 2.
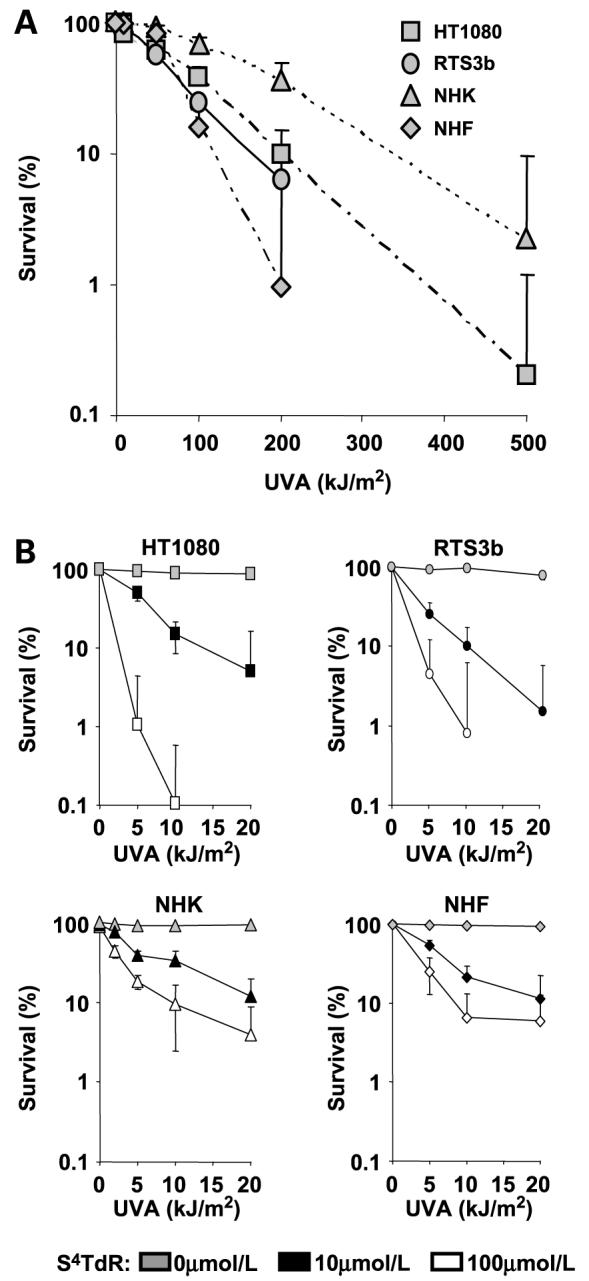
Sensitivity of human skin cells to UVA alone and to S4TdR/UVA assessed by clonal survival assay. A, HT1080, RTS3b, NHK, and NHF cells were plated at single colony density, and irradiated with UVA alone (0, 10, 50, 100, 200, and 500 kJ/m2). B, cells were treated with 0, 10, and 100 μmol/L of S4TdR (gray, black, and white symbols, respectively) as indicated in Materials and Methods, then plated at single colony density, and UVA-irradiated at 0, 5, 10, and 20 kJ/m2. Colonies were stained and counted after 10 to 14 days. All curves were determined from at least three independent experiments, each done in triplicate. Top, HT1080 cells were compared with RTS3b. Bottom, normal human keratinocytes (NHK) were compared with their patient-matched fibroblasts (NHF). Points, means; bars, SD (n = 4–6).
The ability of S4TdR to sensitize HT1080 and RTS3b cells to UVA was confirmed by clonal survival. A combination of 100 μmol/L of S4TdR and 10 kJ/m2 of UVA reduced the survival of both cell types by >99% (Fig. 2B, top). S4TdR (≤300 μmol/L) or UVA (≤20 kJ/m2) alone had no effect. The sensitivity of HT1080 and RTS3b cells to S4TdR/UVA treatment mirrors that previously reported for SV40-transformed MRC5VA human fibroblasts and Raji lymphoma cells (26), indicating that the synergy between the drug and UVA is a general phenomenon.
The S4TdR/UVA sensitivity of NHK and NHF was measured in the same way. At low S4TdR concentrations (30 μmol/L; ref. 10), their UVA sensitivity was similar to that of the two established cell lines (Fig. 2B, bottom). Higher UVA doses and higher S4TdR concentrations had significantly less effects on the primary keratinocytes and fibroblasts suggesting that incorporation of S4TdR into the DNA may be limiting. This is consistent with the measurably lower S phase fraction in these primary cells. There was no significant difference in the survival responses of NHK, NHF, or psoriatic human fibroblast to S4TdR/UVA (data not shown).
Morphologic Changes Following S4TdR/UVA
Microscopic inspection indicated that as early as 24 h after irradiation with 10 kJ/m2 of UVA, HT1080 cells grown in 100 μmol/L of S4TdR exhibited the membrane blebbing and cell fragmentation that are typical of apoptosis (Fig. 3A). Cells treated with UVA or S4TdR alone seemed normal. Induction of cell death was examined by flow cytometry 24 h after treatment. Representative Annexin V versus PI scatter plots are shown in Fig. 3B. Twenty-four hours after a combination of 10 kJ/m2 of UVA and 100 μmol/L of S4TdR, ∼40% of HT1080 cells were undergoing apoptosis (Annexin V–positive), consistent with the morphologic changes observed microscopically. A kinetics experiment (Supplementary Fig. S1)4 documented the sequential conversion of Annexin V–positive/PI-negative cells to Annexin V–positive/PI-positive cells, recorded at 16 and 24 h. Events in S4TdR/UVA-treated RTS3b cells were essentially the same (Fig. 3B). Neither UVA nor S4TdR alone induced significant changes in the pattern of staining. These findings confirm that S4TdR plus low-dose UVA treatment causes cell death to similar extents in HT1080 and RTS3b, and further indicates that death occurs by apoptosis.
Figure 3.

S4TdR/UVA treatment induces cell death by apoptosis. HT1080 and RTS3b cells were treated with S4TdR or UVA or combinations as indicated. A, 24 h posttreatment, bright-field images were captured (magnification, ×200). Cellular damage (arrows): shrinkage, rounding up and fractionating into several bodies indicative of cell death by apoptosis are visible only when S4TdR and UVA are used in combination. B, cells treated as described in A, were processed for flow cytometry analysis by double staining with Alexa647-conjugated Annexin V and PI, as described in Materials and Methods. Apoptotic cells were considered as the sum of Annexin V-positive/PI-negative (early apoptotic) and Annexin V-positive/PI-positive (late apoptotic) cells. C, extracts from HT1080 cells treated as in A, were assayed by Western blot and probed with antibodies against cleaved forms of caspase-3 and against total Bak (top). HT1080 cells treated as in A, were processed for the detection of the active form of Bak, as referred to in Materials and Methods (bottom). Isotype, control sample incubated with an isotype-matched irrelevant antibody. Cells were judged positive for the detection of the active form of Bak by setting a marker on isotype control. Columns, means from Bak-associated fluorescence; bars, SD (n = 4). The “S4TdR/UVA” treatment value was significantly different (P < 0.05) from the values for “no UV”, “S4TdR”, and “UVA” treatments.
The induction of apoptotic HT1080 cells inferred from microscopic observation and Annexin V/PI staining was confirmed by analysis of key apoptosis proteins. S4TdR/UVA treatment induced cleavage of caspase 3, the executioner caspase (Fig. 3C, top). In addition, flow cytometry analysis indicated that ∼60% of S4TdR/UVA-treated HT1080 cells were positive for an active form of Bak generated by a conformational change that follows the induction of apoptosis (Fig. 3C, bottom; Supplementary Fig. S2;4 refs. 33, 34). Less than 5% of cells treated with S4TdR or UVA alone were Bak isoform–positive. S4TdR/UVA treatment did not induce a significant change in the steady-state level of Bak protein, however (Fig. 3C, top). This indicates that events following S4TdR/UVA treatment differ from those following UVB radiation (19).
p53 DNA Damage Response
The p53 response was examined in HT1080 cells. UVA doses <50 kJ/m2 did not induce p53 stabilization (data not shown). At doses in excess of 100 kJ/m2, p53 stabilization was rapid and was detectable as early as 2 h postirradiation. p53 levels remained high for at least 24 h after irradiation. Extremely high UVA doses consistently abrogated the response, and after 500 kJ/m2, no p53 was detectable by Western blotting until at least 16 h postirradiation. But this dose was very toxic, as shown previously in Fig. 2A. The levels of α-tubulin, included as a loading control, were also severely reduced by this dose, indicating that reduced p53 expression may be a consequence of an inhibition of transcription/translation or general cell killing and proteolysis. Cells recovered from this inhibition, partial recovery was apparent at 16 h after irradiation, and by 24 h, both p53 and α-tubulin levels had increased significantly (Fig. 4A).
Figure 4.
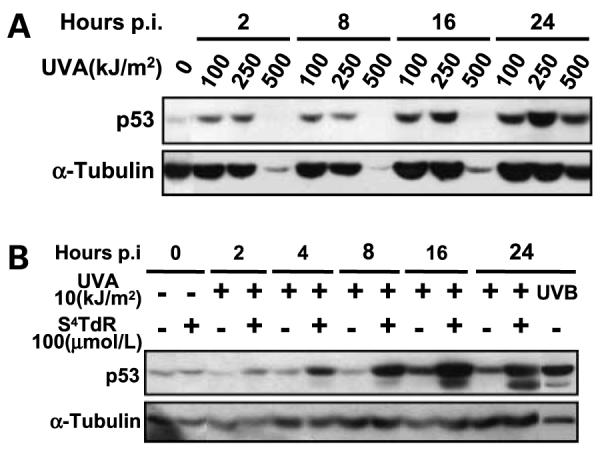
The p53 response to UVA and S4TdR/UVA treatment in HT1080 cells. Cells were treated as indicated on the figures and detailed in Materials and Methods. A, irradiation with UVA alone. B, treatment with 100 μmol/L of S4TdR or 10 kJ/m2 of UVA alone or in combination, or with 0.15 kJ/m2 of UVB, and then assayed by Western blot and probed for activation of p53. Blotting for the cytoskeleton protein α-tubulin was used as a loading control.
In contrast, very low doses of UVA (10 kJ/m2) induced p53 stabilization in cells pretreated with S4TdR, consistent with the formation of photochemical DNA damage. Induction was rapid and increased p53 protein levels were detectable 2 to 4 h after radiation (Fig. 4B). The response seemed to be maximal at 16 to 24 h.
HPV E6, the p53 Response, and Cell Death
E6 proteins of diverse HPV types protect HT1080 cells against UVB-induced apoptosis (16). To investigate whether HPV E6 influences the induction of p53 response and cell death by S4TdR/UVA, we constructed HT1080 cells stably expressing E6 from high-risk HPV5 (an epidermodysplasia verruciformis–associated HPV) or HPV18 (an anogenital type).
Neither S4TdR/UVA (Fig. 5A) nor UVB treatment (data not shown) induced detectable p53 stabilization in HT1080 cells expressing HPV18 E6. As expected, this compromised downstream events and the induction of both cyclin-dependent kinase inhibitor p21CIP1 and E3 ubiquitin ligase Mdm2 was also abrogated in HPV18 E6–expressing cells. In contrast, p53 stabilization seemed to be normal in HT1080 cells expressing HPV5 E6, which were comparable to the vector-only control cells in this regard. p53 stabilization was accompanied by Ser392 and Ser15 phosphorylation—both of which are associated with DNA damage and are required for p53 functions (35, 36). p21 and Mdm2 induction in HPV5 E6–expressing cells was also comparable to vector-only controls. Consistent with their lack of cytotoxicity at these doses, neither S4TdR nor UVA alone had any effect on the induction or stabilization of these proteins (data not shown).
Figure 5.
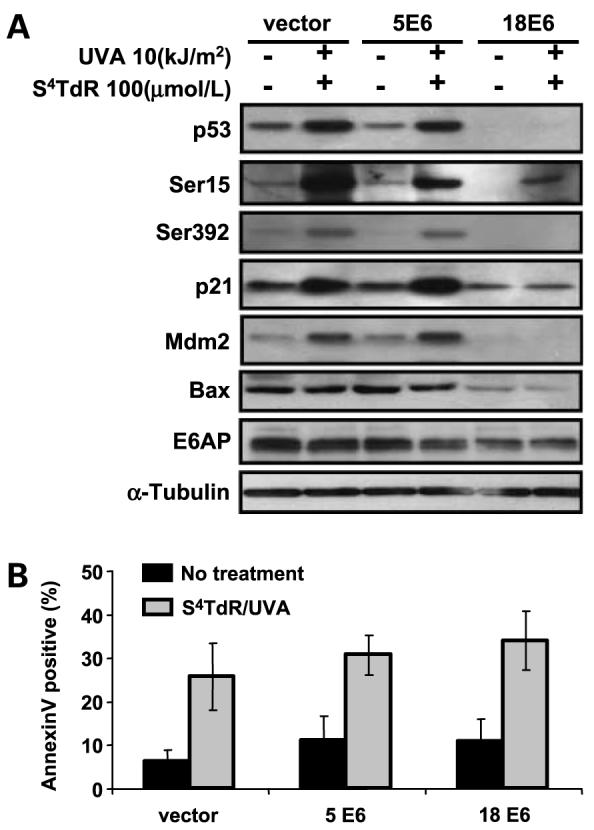
The p53 response in HT1080 sublines expressing HPV5 E6 or HPV18 E6. A, HT1080 sublines were treated without or with S4TdR/UVA as detailed in Materials and Methods. At 24 h posttreatment, total cell protein extracts were resolved on SDS-PAGE gel and probed for the proteins of interest. B, a fraction of the cells treated under A were also analyzed for apoptosis by Annexin V/PI staining followed by flow cytometry. Columns, mean percentages of Annexin V–positive cells are represented for each subline; bars, SD (n = 4). S4TdR/UVA treatment gave a significant increase of Annexin V (P < 0.05) over unirradiated controls within each cell line. Identical conditions compared across the lines were not significantly different.
Expression of HPV18 E6, but not HPV5 E6 reduced the basal level of the proapoptotic Bax protein (Fig. 5A). S4TdR/UVA had no detectable effect on Bax levels which remained unaltered 24 h after treatment of control, HPV5 E6, or HPV18 E6 cells. Levels of the ubiquitin ligase E6AP, which is responsible for the degradation of p53 in cells expressing E6 from anogenital type HPVs (37), were also not significantly altered 24 h after S4TdR/UVA treatment. Notwithstanding their effects on gene expression, neither HPV5 E6 nor HPV18 E6 detectably altered the resistance of HT1080 cells to S4TdR/UVA. This was apparent in measurements of apoptosis by Annexin V/PI staining (Fig. 5B) and was confirmed by measurements of viability by dimethylthiazol-2-yl-2-5-diphenyltetrazolium bromide assay at 72 h posttreatment (data not shown) and clonal survival (Fig. 6). Because both the HPV5 and HPV18 E6 proteins protect HT1080 cells against killing by UVB, these observations provide additional evidence that S4TdR/UVA and UVB activate different cell death pathways.
Figure 6.
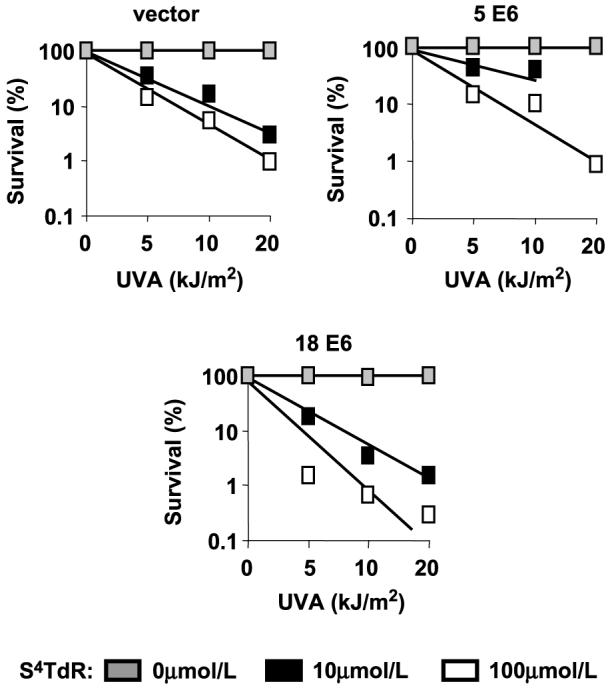
Survival of HT1080 sublines expressing E6 from HPV5 or HPV18 to S4TdR/UVA treatment, measured by colony-forming assay. Cells were treated with 0, or 10, 100 μmol/L of S4TdR as detailed in Materials and Methods, then plated at single colony density, and UVA-irradiated at 0, 5, 10, and 20 kJ/m2. Surviving colonies were counted after 10 to 14 days. All curves were determined from at least three independent experiments, each done in triplicate. There were no significant differences in cell-killing at the UVA doses used, when comparing the HPV lines to the vector control line.
Discussion
The synergistic cytotoxicity of S4TdR and UVA was previously shown to require incorporation of the thiopyrimidine into DNA via the thymidine salvage pathway (26). S4TdR-treated cells are killed by extremely low UVA doses—well below those required to cause death or mutation if given without the sensitizer. This suggested that it might be worth evaluating S4TdR/UVA as a potential treatment strategy for tumors or hyperproliferative disorders that are accessible to therapeutic radiation. The present investigation into the effects of S4TdR/UVA on a series of established and primary cells derived from skin represents a first step.
Overall, despite differences after UVA treatment alone, the response of these various cell types to combined S4TdR/UVA is quite uniform. The sensitivity of the established HT1080 and RTS3b cell lines did not differ significantly from that previously reported for the established SV40-transformed human fibroblast cell line MRC5VA. A marked resistance of primary keratinocytes and fibroblasts—particularly at higher S4TdR concentrations—is consistent with their reduced proliferative fraction and use of a UVA chromophore that depends on incorporation into DNA for its biological effect. Differences in response to UVA alone were more marked and we confirmed the relative resistance of NHK to UVA (32). Their increased ability to withstand high UVA doses is not paralleled by S4TdR/UVA resistance. This suggests that the death pathways engaged after UVA and S4TdR/UVA are likely to be different.
Cellular utilization of exogenous thymidine analogues is influenced by both the intracellular pool of thymidine nucleotides produced by de novo synthesis and the extracellular concentration of competitor TdR. The calf serum used for cell culture may contain high concentrations of TdR, so to obtain consistent results with S4TdR, serum was dialyzed before use. Human serum also contains TdR. At ≤0.1 μmol/L (38), the human plasma TdR concentration is orders of magnitude lower than that of calf serum, however. This suggests that, if reasonable serum levels of S4TdR could be achieved in human subjects, good cellular uptake and incorporation of the analogue are likely. The reported serum TdR concentration in mice is significantly higher, ∼1 μmol/L (39). The possibility that this higher concentration of competitor nucleoside might attenuate the effects of S4TdR merits consideration if mouse models or human xenografts are used to evaluate S4TdR/UVA as a therapeutic option.
The aim of anticancer treatments is to destroy the tumor cells with minimal effects on healthy surrounding tissue. In the case of S4TdR/UVA, the requirement for active thymidine salvage and DNA replication as well as precise targeting of the UVA would both favor the selective destruction of tumor tissue. Our findings that the two established transformed skin-related cell lines were more susceptible to S4TdR/UVA than primary keratinocytes and fibroblasts with a lower proliferative fraction is in line with this. It is noteworthy, in this regard, that SCCs are generally characterized by a high proliferative fraction and exhibit strong staining with proliferation-associated markers such as Ki-67 (40), and often show invasive properties. Interestingly, HPV-positive SCCs also have a high proliferative potential, but show a significantly lower level of apoptosis than HPV-negative SCCs (41). Hence, our findings suggest that S4TdR/UVA might be particularly effective in treating SCCs independently of their HPV status. In contrast, basal cell carcinomas—which are generally slow growing—would be expected to be less responsive to S4TdR/UVA.
Despite high proliferation rates and a potential sensitivity to DNA-damaging drugs, the effectiveness of therapeutic treatments can be compromised by down-regulation or inactivation of death pathways in tumor cells. One example is the resistance to DNA-damaging therapeutics that often accompanies p53 inactivation. p53 mutations are very common among NMSCs (24, 25). One significant finding of our study is that the S4TdR/UVA sensitivity of the p53 wild-type HT1080 and p53-null RTS3b cells are similar. Although S4TdR/UVA treatment induced p53 stabilization and downstream components of the p53 DNA damage response, its toxicity seems to be largely independent of p53 status.
HT1080 and RTS3b cell lines are not isogenic, however, and any extrapolation of findings derived from comparisons between them should be made with caution. The behavior of HT1080 clones expressing E6 proteins derived from different oncogenic HPVs provides a more appropriate comparison of the effects of p53 inactivation. These experiments provide additional evidence that S4TdR/UVA cytotoxicity is largely independent of p53 status. Neither HPV18 E6, from an anogenital HPV type known to target p53 for destruction (11), nor HPV5, associated with NMSC in patients with epidermodysplasia verruciformis, which does not promote p53 degradation (16), significantly affected S4TdR/UVA-induced cell death.
Our findings raise the question of the nature of the DNA lesions introduced by S4TdR/UVA and the DNA damage response that they provoke. Based on chemical evidence and the relative sensitivity of xeroderma pigmentosum cell lines to S4TdR/UVA, we have suggested that S4TdR/UVA produces thietane lesions (42) that resemble the (6-4)pyrimidine-pyrimidone adducts produced by UVC (and UVB) radiation, and that these contribute to cytotoxicity (26). The differential effect of HPV E6 status on UVB and S4TdR/UVA-mediated apoptosis suggests that alternative DNA lesion(s) might be the key to S4TdR/UVA toxicity. In agreement with this possibility, DNA damage produced by S4TdR/UVA is not recognized by antibodies that recognize UVB-induced (6-4)pyrimidine-pyrimidone photoproducts, and S4TdR/UVA treatment induces a burst of reactive oxygen species in the cell nucleus.5 Despite this uncertainty, the finding that S4TdR/UVA cytotoxicity is independent of HPV E6 status suggests that this may provide a useful approach to the treatment of skin disorders in which there is HPV involvement.
Acknowledgments
We are grateful to Drs. Gary Warnes and Derek Davies from the Cancer Research UK FACS laboratory, for their help with the flow cytometry analysis.
Grant support: Translational fellowship (no. TR4020) from Cancer Research UK (O. Reelfs, A. Storey, and P. Karran).
Footnotes
Supplementary material for this article is available at Molecular Cancer Therapeutics Online (http://mct.aacrjournals.org/).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Reelfs O, Montaner B. unpublished data.
References
- 1.Alam M, Ratner D. Cutaneous squamous cell carcinoma. N Eng J Med. 2001;344:975–83. doi: 10.1056/NEJM200103293441306. [DOI] [PubMed] [Google Scholar]
- 2.Black HS, deGruijl FR, Forbes PD, et al. Photocarcinogenesis: an overview. J Photochem Photobiol B. 1997;40:29–47. doi: 10.1016/s1011-1344(97)00021-3. [DOI] [PubMed] [Google Scholar]
- 3.Harris RB, Alberts DS. Strategies for skin cancer prevention. Int J Dermatol. 2004;43:243–51. doi: 10.1111/j.1365-4632.2004.01966.x. [DOI] [PubMed] [Google Scholar]
- 4.Friedberg EC, Walker GC, Siede W. DNA repair and mutagenesis. Washington (DC): ASM Press; 1995. [Google Scholar]
- 5.Kraemer KH, Lee MM, Scotto J. Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch Dermatol. 1987;123:241–50. doi: 10.1001/archderm.123.2.241. [DOI] [PubMed] [Google Scholar]
- 6.Akgul B, Cooke JC, Storey A. HPV-associated skin disease. J Pathol. 2006;208:165–75. doi: 10.1002/path.1893. [DOI] [PubMed] [Google Scholar]
- 7.Lu S, Syrjanen K, Havu VK, Syrjanen S. Expression of PCNA is associated with the presence of HPV DNA in skin warts. Arch Dermatol Res. 1996;289:35–9. doi: 10.1007/s004030050149. [DOI] [PubMed] [Google Scholar]
- 8.McCance DJ, Kopan R, Fuchs E, Laimins LA. Human papillomavirus type 16 alters human epithelial cell differentiation in vitro. Proc Natl Acad Sci U S A. 1988;85:7169–73. doi: 10.1073/pnas.85.19.7169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herber R, Liem A, Pitot H, Lambert PF. Squamous epithelial hyperplasia and carcinoma in mice transgenic for the human papillomavirus type 16 E7 oncogene. J Virol. 1996;70:1873–81. doi: 10.1128/jvi.70.3.1873-1881.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pfister H. Chapter 8: Human papillomavirus and skin cancer. J Natl Cancer Inst Monogr. 2003;31:52–6. doi: 10.1093/oxfordjournals.jncimonographs.a003483. [DOI] [PubMed] [Google Scholar]
- 11.Zur Hausen H. Papillomaviruses causing cancer: evasion from host-cell control in early events in carcinogenesis. J Natl Cancer Inst. 2000;92:690–8. doi: 10.1093/jnci/92.9.690. [DOI] [PubMed] [Google Scholar]
- 12.Harwood CA, McGregor JM, Proby CM, Breuer J. Human papillomavirus and the development of non-melanoma skin cancer. J Clin Pathol. 1999;52:249–53. doi: 10.1136/jcp.52.4.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orth G. Human papillomaviruses and the skin: more to be learned. J Invest Dermatol. 2004;123:XI–XIII. doi: 10.1111/j.0022-202X.2004.23243.x. [DOI] [PubMed] [Google Scholar]
- 14.Jablonska S, Orth G. Epidermodysplasia verruciformis. Clin Dermatol. 1985;3:83–96. doi: 10.1016/0738-081x(85)90052-5. [DOI] [PubMed] [Google Scholar]
- 15.Surentheran T, Harwood CA, Spink PJ, et al. Detection and typing of human papillomaviruses in mucosal and cutaneous biopsies from immunosuppressed and immunocompetent patients and patients with epidermodysplasia verruciformis: a unified diagnostic approach. J Clin Pathol. 1998;51:606–10. doi: 10.1136/jcp.51.8.606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jackson S, Storey A. E6 proteins from diverse cutaneous HPV types inhibit apoptosis in response to UV damage. Oncogene. 2000;19:592–8. doi: 10.1038/sj.onc.1203339. [DOI] [PubMed] [Google Scholar]
- 17.Giampieri S, Garcia-Escudero R, Green J, Storey A. Human papillomavirus type 77 E6 protein selectively inhibits P53-dependent transcription of proapoptotic genes following UV-B irradiation. Oncogene. 2004;23:5864–70. doi: 10.1038/sj.onc.1207711. [DOI] [PubMed] [Google Scholar]
- 18.Giampieri S, Storey A. Repair of UV-induced thymine dimers is compromised in cells expressing the E6 protein from human papillomaviruses types 5 and 18. Br J Cancer. 2004;90:2203–9. doi: 10.1038/sj.bjc.6601829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jackson S, Harwood C, Thomas M, Banks L, Storey A. Role of BAK in UV-induced apoptosis in skin cancer and abrogation by HPV E6 proteins. Genes Dev. 2000;14:3065–73. doi: 10.1101/gad.182100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gasparro FP. The role of PUVA in the treatment of psoriasis. Photobiology issues related to skin cancer incidence. Am J Clin Dermatol. 2000;1:337–48. doi: 10.2165/00128071-200001060-00002. [DOI] [PubMed] [Google Scholar]
- 21.Lever LR, Farr PM. Skin cancers or premalignant lesions occur in half of high-dose PUVA patients. Br J Dermatol. 1994;131:215–9. doi: 10.1111/j.1365-2133.1994.tb08494.x. [DOI] [PubMed] [Google Scholar]
- 22.van Praag MC, Bavinck JN, Bergman W, et al. PUVA keratosis. A clinical and histopathologic entity associated with an increased risk of nonmelanoma skin cancer. J Am Acad Dermatol. 1993;28:412–7. [PubMed] [Google Scholar]
- 23.Santamaria AB, Davis DW, Nghiem DX, et al. P53 and Fas ligand are required for psoralen and UVA-induced apoptosis in mouse epidermal cells. Cell Death Differ. 2002;9:549–60. doi: 10.1038/sj.cdd.4401007. [DOI] [PubMed] [Google Scholar]
- 24.Brash DE, Rudolph JA, Simon JA, et al. A role for sunlight in skin cancer: UV induced P53 mutations in squamous cell carcinoma. Proc Natl Acad Sci U S A. 1991;88:10124–8. doi: 10.1073/pnas.88.22.10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ziegler A, Leffell DJ, Kunala S, et al. Mutation hotspots due to sunlight in the P53 gene of nonmelanoma skin cancers. Proc Natl Acad Sci U S A. 1993;90:4216–20. doi: 10.1073/pnas.90.9.4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Massey A, Xu YZ, Karran P. Photoactivation of DNA thiobases as a potential novel therapeutic option. Curr Biol. 2001;11:1142–6. doi: 10.1016/s0960-9822(01)00272-x. [DOI] [PubMed] [Google Scholar]
- 27.Rasheed S, Nelson-Rees WA, Toth EM, Arnstein P, Gardner MB. Characterization of a newly derived human sarcoma cell line (HT-1080) Cancer. 1974;33:1027–33. doi: 10.1002/1097-0142(197404)33:4<1027::aid-cncr2820330419>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 28.Rapp B, Pawellek A, Kraetzer F, et al. Cell-type-specific separate regulation of the E6 and E7 promoters of human papillomavirus type 6a by the viral transcription factor E2. J Virol. 1997;71:6956–66. doi: 10.1128/jvi.71.9.6956-6966.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rheinwald JG, Green H. Serial cultivation of strains of human epidermal keratinocytes: the formation of keratinizing colonies from single cells. Cell. 1975;6:331–44. doi: 10.1016/s0092-8674(75)80001-8. [DOI] [PubMed] [Google Scholar]
- 30.Xu YZ, Zheng Q, Swann PF. Simple synthesis of 4-thiothymidine, 4-thiouridine and 6-thio-2′-deoxyguanosine. Tetrahedron Lett. 1991;32:2817–20. [Google Scholar]
- 31.Griffiths GJ, Dubrez L, Morgan CP, et al. Cell damage-induced conformational changes of the pro-apoptotic protein BAK in vivo precede the onset of apoptosis. J Cell Biol. 1999;144:903–14. doi: 10.1083/jcb.144.5.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Otto AI, Riou L, Marionnet C, Mori T, Sarasin A, Magnaldo T. Differential behaviours toward ultraviolet A and B radiation of fibroblasts and keratinocytes from normal and DNA-repair-deficient patients. Cancer Res. 1999;59:1212–8. [PubMed] [Google Scholar]
- 33.Gelinas C, White E. BH3-only proteins in control: specificity regulates MCL-1 and BAK-mediated apoptosis. Genes Dev. 2005;19:1263–8. doi: 10.1101/gad.1326205. [DOI] [PubMed] [Google Scholar]
- 34.Wei MC, Lindsten T, Mootha VK, et al. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 2000;14:2060–71. [PMC free article] [PubMed] [Google Scholar]
- 35.Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of P53 alleviates inhibition by MDM2. Cell. 1997;91:325–34. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- 36.She QB, Chen N, Dong Z. ERKs and p38 kinase phosphorylate P53 protein at serine 15 in response to UV radiation. J Biol Chem. 2000;275:20444–9. doi: 10.1074/jbc.M001020200. [DOI] [PubMed] [Google Scholar]
- 37.Huibregtse JM, Scheffner M, Howley PM. A cellular protein mediates association of P53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J. 1991;10:4129–35. doi: 10.1002/j.1460-2075.1991.tb04990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Howell SB, Herbst K, Boss GR, Frei E. Thymidine requirement for the rescue of patients treated with high-dose methotrexate. Cancer Res. 1980;40:1824–9. [PubMed] [Google Scholar]
- 39.Jackman AL, Taylor GA, Calvert AH, Harrap KR. Modulation of anti-metabolite effects. Effects of thymidine on the efficacy of the quinazoline-based thymidylate synthetase inhibitor, CB3717. Biochem Pharmacol. 1984;33:3269–75. doi: 10.1016/0006-2952(84)90089-3. [DOI] [PubMed] [Google Scholar]
- 40.Al-Sader MH, Doyle E, Kay EW, et al. Proliferation indexes-a comparison between cutaneous basal and squamous cell carcinomas. J Clin Pathol. 1996;49:549–51. doi: 10.1136/jcp.49.7.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jackson S, Ghali L, Harwood C, Storey A. Reduced apoptotic levels in squamous but not basal cell carcinomas correlates with detection of cutaneous human papillomavirus. Br J Cancer. 2002;87:319–23. doi: 10.1038/sj.bjc.6600431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Connolly BA, Newman PC. Synthesis and properties of oligonucleotides containing 4-thiothymine, 5-methyl-2-pyrimidinone-1-β-D(2′-deoxyriboside) and 2-thiothymine. Nucleic Acids Res. 1989;17:4957–74. doi: 10.1093/nar/17.13.4957. [DOI] [PMC free article] [PubMed] [Google Scholar]