

Abstract
BACKGROUND/AIMS
While the rise in non-alcoholic fatty liver disease (NAFLD) parallels the increase in obesity and diabetes, a significant increase in dietary fructose consumption in industrialized countries has also occurred. The increased consumption of high fructose corn syrup, primarily in the form of soft-drinks, is linked with complications of the insulin resistance syndrome. Furthermore, the hepatic metabolism of fructose favors de novo lipogenesis and ATP depletion. We hypothesize that increased fructose consumption contributes to the development of NAFLD.
METHODS
A dietary history and paired serum and liver tissue were obtained from patients with evidence of biopsy-proven NAFLD (n=49) without cirrhosis and controls (n=24) matched for gender, age (± 5 years), and body mass index (± 3 points).
RESULTS
Consumption of fructose in patients with NAFLD was nearly 2-3 fold higher than controls [365 kcal. vs 170 kcal (p<0.05)]. In patients with NAFLD (n=6), hepatic mRNA expression of fructokinase (KHK), an important enzyme for fructose metabolism, and fatty acid synthase, an important enzyme for lipogenesis were increased (p=0.04 and p=0.02 respectively). In an AML hepatocyte cell line, fructose resulted in dose-dependent increase in KHK protein and activity.
CONCLUSION
The pathogenic mechanism underlying the development of NAFLD may be associated with excessive dietary fructose consumption.
Keywords: hepatic steatosis, fructose, soft drinks, uric acid, fatty liver, non-alcoholic steatohepatitis
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD), a hepatic manifestation of the metabolic syndrome, is the most common explanation for liver aminotransferase elevation in obesity [1]. The prevalence of NAFLD exceeds the combined prevalence of chronic viral and alcohol-associated liver disease in the general United States population [2]. Population-based studies, such as NHANES III and the Dallas Heart Study [3, 4] confirm that obesity and type 2 diabetes are highly correlated with NAFLD. As the prevalence of obesity and diabetes increase, NAFLD has become the leading cause of chronic liver disease in developed countries.
The dramatic rise in prevalence of NAFLD and the metabolic syndrome strongly suggest a role for environmental factors in disease pathogenesis. Most attention has focused on increased energy (caloric) intake coupled with reduced physical activity as central mechanisms contributing to the obesity epidemic. However, an important, but not well-appreciated change in dietary habit has been the substantial increase in dietary fructose consumption acquired from sucrose and high fructose corn syrup (HFCS), a common sweetener used in the food industry [5]. For example, soft drinks and fruit drinks, which are a major source of HFCS or sugar, have increased from 3.9% of total energy intake in 1977 to 9.2% of total energy intake in 2001 [6]. Soft drink consumption has recently been linked with increased risk for weight gain [7, 8], type-2 diabetes [8, 9] and other features of the metabolic syndrome [10]. Soft drink consumption is associated with cardiometabolic risk factors and the metabolic syndrome in middle-aged adults [10, 11] and with the development of obesity in children [12].
Many studies suggest that the mechanism by which sugar or HFCS may induce metabolic syndrome is due to the fructose content [13, 14, 15, 16]. In animal models, diets high in fructose induce features of the metabolic syndrome including weight gain, insulin resistance, hypertriglyceridemia, and hypertension [17]. Similar effects are not observed with the administration of other simple sugars such as glucose [18]. Fructose (or sucrose) administration to humans also causes features of metabolic syndrome [13, 15, 16].
While it has not been emphasized, fructose may have a role in the pathogenesis of NAFLD. Fructose is lipogenic and stimulates triglyceride synthesis [19]. Splanchnic perfusion studies demonstrate that fructose produces higher rates of triglyceride secretion from the liver than equimolar amounts of glucose [20]. The long-term administration of fructose to rats results in hepatic macro- and microvesicular steatosis with a 198% increase in hepatic triglycerides and an 89% increase in hepatic cholesterol concentration [21]. Ducks fed high fructose diets also develop fatty liver [22]. Furthermore, the administration of a diet with 25% of total energy as sucrose (which contains 50% fructose) resulted in a rise in hepatic ALT and AST levels within 18 days [23]. This study, which was performed nearly 25 years ago, is all the more alarming as current sugar intake of Americans is in this same range [24]. Indeed, total fructose intake averages approximately 12% of total energy intake and may increase to 15% in some subgroups in the US population [15].
A potential mechanism by which fructose may cause liver injury also exists. The metabolism of fructose is distinct from glucose. Before converging with the glycolytic pathway, initial fructose metabolism involves phosphorylation of fructose to fructose-1-phosphate by fructokinase (ketohexokinase, KHK) using the substrate ATP. Unlike glucokinase, the phosphorylation of fructose by fructokinase is specific for fructose and not rate limited. The high activity of fructokinase in phosphorylating fructose to fructose-1-phosphate in the liver, could result in hepatic ATP depletion [15]. Indeed, fructose has been shown to cause ATP depletion in humans [25, 26, 27], and recovery from fructose-induced ATP depletion was found to be delayed in subjects with NALFD in studies that used phosphorus-1 magnetic resonance spectrosocopy to assess hepatic metabolism [27, 28]. In some regards, fructose-induced ATP depletion resembles hepatic ischemia [29]. In rats, fructose administration increases hepatic lipid peroxidation and activation of inflammatory pathways [30, 31]. We have also found that incubation of endothelial cells or renal tubular cells with postprandial concentrations of fructose reduces intracellular ATP and activates proinflammatory and pro-oxidative responses [Cirillo P, Sautin YY and Johnson RJ, unpublished]. Therefore, high fructose consumption may contribute to NAFLD pathogenesis because fructose-induced ATP depletion promotes hepatic necroinflammation.
Despite the evidence suggesting that fructose may be involved in the pathogenesis of NAFLD, no studies have specifically investigated this relationship. We conducted a case-control study to assess whether there is increased fructose intake in those with NAFLD and whether fructose correlates with features of the metabolic syndrome of insulin resistance. In addition, since fructose upregulates KHK activity in the liver and intestines of rats fed a high fructose diet [32, 33] we examined whether KHK is increased in the liver of subjects with and without NAFLD.
PATIENTS AND METHODS
Following approval by the Institutional Review Board of the University of Florida, we prospectively collected demographic, medical, and dietary information on patients with biopsy proven NAFLD (n=49) and no evidence of NAFLD (n=24) on liver biopsy. Each patient diagnosed with NAFLD underwent a comprehensive medical evaluation to exclude alternative causes for chronic liver disease. The presence of NAFLD was established by a comprehensive medical history, the presence of abnormal liver aminotransferases, negative serologies for alternative forms of chronic liver disease (i.e. viral, autoimmune or metabolic liver disease), hyperechoic liver by ultrasound imaging, and histologic features as defined by Brunt et al [34]. The control population was chosen on the basis of convenience sampling and was selected from a pool of healthy volunteers or patients who were evaluated in the Liver Clinics who did not have evidence of chronic liver disease following a comprehensive medical evaluation, appropriate serologic laboratory studies, imaging studies, or liver histology (if one had been performed) for the further evaluation of elevated liver enzymes. Each patient with a diagnosis of NAFLD was matched by age (± 5 years), gender, and body mass index (± 3 points) to a patient who lacked clinical, laboratory, radiologic, and/or histologic features of underlying NALFD. This study was designed as an exploratory observational pilot study.
NAFLD Cases
Stored liver biopsy tissue and clinical data were retrieved from our institutional IRB approved serum and tissue repository of patients with biopsy proven NAFLD (n=6). All patients included in this registry had clinical, radiologic and histologic confirmation of NALFD and no other form of chronic liver disease. The full histologic spectrum of NAFLD as defined by Brunt et al [34] was utilized for case-definition. The degree of alcohol consumption was estimated using specific volume estimates, type of alcohol consumed, and frequency of consumption. The presence of simple steatosis or steatohepatitis was considered to be non-alcoholic in origin if the patient consumed less than 20 grams of alcohol weekly. If the clinical and histologic criteria for NAFLD were not met, the patient was reclassified in the tissue and serum registry as non- NAFLD liver disease.
Non-NAFLD Controls
Stored liver tissue from non-fatty liver controls (n=6) were also retrieved from our institutional IRB approved repository of patients evaluated for concern of underlying liver disease but whose liver biopsy did not fulfill the Brunt histologic criteria for a diagnosis of NAFLD [34]. These subjects were selected on the basis of convenience sampling (patients who underwent a liver biopsy for the evaluation of underlying liver disease and who were deemed to have no identifiable cause for chronic liver disease). These subjects, matched by age (± 5 years), gender and body mass index (± 3 points) to a case with NAFLD, had no evidence of fatty liver by radiologic studies (either by abdominal ultrasound or computerized tomography scan), and absence of histologic features of NAFLD. Of the 6 control subjects, 5 patients had evidence of normal liver histology (1 live-donor liver transplant, 3 segmental resection of a benign hepatic lesion with surrounding normal liver parenchyma, and 1 liver biopsy performed for a non-specific elevation of aminotransferases), and 1 patient had non-specific histologic features of mild lobular disarray with no evidence of steatosis, inflammation or fibrosis.
Assessment of Dietary Fructose Consumption
Although HFSC is used extensively in many industrialized food products (carbonated beverages, artificially sweetened drinks, baked goods, candies, canned fruits, jams, jellies, and dairy products), we made a conservative estimate of daily dietary fructose consumption by asking our patients to self report the number of servings and frequency (days per week) of consumption of fructose containing beverages. Total energy consumption (kcal/day) from daily fructose intake was estimated based on reporting (frequency × amount) of kool-aid, fruit juices, and non-dietary soda per week. All dietary histories were obtained by the physician providing clinical care (MFA) and were performed within 1 month of the liver biopsy. Each patient was asked to recall their routine daily consumption fructose containing beverages over a 3 month period.
Plasma Biochemistry Analysis
Serum was from NAFLD (n=49) versus controls (n=24) was analyzed for variations in biochemical endpoints that may be associated with increased fructose consumption. Serum glucose, triglyceride, cholesterol and uric acid were measured with an autoanalyzer.
mRNA Isolation, Reverse Transcription, and Real-Time PCR of KHK, Fatty acid synthase (FAS), and xanthine dehydrogenase/oxidase (XO) from Human Liver Tissue
Liver tissue was analyzed for hepatic mRNA expression of fructokinase (KHK). Total RNA was isolated using the SV Total RNA Isolation kit (Promega, Madison, WI) according to the manufacturer’s protocol. The RNA was eluted with 50 ul of RNase-free water. All RNA was quantified by spectrophotometer, and the optical density 260/280 nm ratios were determined. Reverse transcription was performed in a one-step protocol using the iScript cDNA Synthesis Kit (Bio-Rad) according to the manufacturer’s protocols. Reactions were incubated at 25°C for 5 minutes, and 42°C for 30 minutes, 85°C for 5 min and cooled at 4°C in a Thermocycler (Eppendorf, Hamburg, Germany). Primers (Table 1) were designed by Genetool software (BioTools Inc., Edmonton, Canada) and oligonucleotides were synthesized by Sigma Genosys (Sigma- Genosys Ltd., Woodlands, TX). Real time PCR analyses were performed using the Opticon PCR machine (MJ Research, Waltham, MA). The SYBR Green master mix kit (BIO-RAD) was used for all reactions with real-time PCR. Briefly, PCR was performed as: 94°C for 2 minutes followed by 40 cycles of denaturation, annealing, and extension at 94°C for 15 seconds, 64°C for 30 seconds, 72°C for 45 seconds, respectively, and final extension at 72°C for 10 minutes. PCR reaction for each sample was done in duplicate for all of the products and for the glyceraldehyde-3-phospate dehydrogenase control. Ratios for each product/glyceraldehyde-3-phospate dehydrogenase mRNA were calculated for each sample and expressed as the mean ±SD.
Table 1.
Realtime PCR Primers Used in the Study
| Gene symbol | Gene description | Accession number | Forward primer sequence | Reverse primer sequence | Product size |
|---|---|---|---|---|---|
| KHK | Ketohexokinase (fructokinase) | NM_000221 | GGGGCTTGTATGGTCGTGTGAG | CCACCTGGCACCCGAATCTC | 229bp |
| FAS | fatty acid synthase | NM_004104 | CCCCCTCAGCCGCCATCTAC | GGGCCAGCGTCTTCCACACTAT | 136bp |
| XDH | Xanthine dehydrogenase | NM_000379 | ACCCCGTGTTCATGGCCAGTG | TCCGGGAGGCCTGCTTGAATG | 195bp |
Measurement of Enzymatic Activity of KHK in Mouse AML Hepatocyte Culture
Mouse AML-12 hepatocytes [ATCC] were cultured in the DMEM/F12 (1:1) medium containing 10% FBS, 40 ng/ml dexamethasone, 5 μg/ml insulin, 5 ng/ml sodium selenite, 5 μg/ml transferrin and 10 μg/ml gentamycin to 70-75% of confluence. For the stimulation with fructose, cells were transferred into glucose-free DMEM containing 10% dialyzed-FBS and 10 or 30 mM Glucose with or without 5 mM fructose and incubated for 72 h followed by collecting cell lysates for measuring KHK activity and immunoblotting.
To measure enzymatic activity of KHK, we used a coupled enzymatic assay based on existing methods [35, 36, 37]. Cells were harvested on ice in the buffer containing 25 mM HEPES (pH 7.1), 100 mM KCl, 1 mM DTT, 0.1 mM EDTA and homogenized by 100 strokes of the pestle. ADP, one of the products of the reaction was quantitatively converted to NAD+ and measured as a decrease of optical density at 340 nm in the reaction mixture containing 25 mM HEPES (pH 7.1), 6 mM MgCl2, 25 mM KCl, 10 mM NaF, 5 mM ATP, 5 mM D-fructose, 0.2 mM NADH, 1 mM phosphoenolpyruvate, 40 U/ml pyruvate kinase, 40 U/ml lactate dehydrogenase, and 50 mM N-acetyl-D-glucosamine (to inhibit hexokinase but retain KHK activity). Protein was measured using the bicinchoninic acid (BCA) protein assay (Pierce, Rockford IL).
For immunodetection of KHK, cells were lysed in the buffer containing 20 mM tris-HCl (pH 7.3), 1.5% nonyl glucoside, 0.6% CHAPS, 1 mM dithiothreitol, 1 mM EDTA, 1 mM EGTA, 10% glycerol, 2 mM sodium orthovanadate, 1 mM phenylmethylsulfonylfluoride, 40 μg/ml leupeptin, 5 μg/ml aprotinin, 1 μg/ml pepstatin. 20 μg protein of the total cell extract was resolved by SDS-PAGE and electroblotted onto PVDF membrane. Membranes were blocked in 10 mM Tris (pH 7.5), 100 mM NaCl, 0.1% Tween-20 containing 5% non-fat dry milk, followed by incubation with primary antibody against KHK (rabbit polyclonal; Abgent, SanDiego, CA). Membranes were washed three times and incubated with appropriate HRP-conjugated secondary antibody. The immunocomplexes were visualized by chemiluminescent detection with Phototope Western Detection System (LumiGLO®, Cell Signaling Technology, Beverly, MA). Then membranes were stripped and probed with monoclonal GAPDH antibody (Chemicon, Temecula, CA). The images were digitalized using the Alpha Ease FluorChem digital imaging system (Alpha Innotech Corp., San Leandro, CA). The optical density of the bands was quantified.
Statistical Analyses
All values presented are expressed as means ± SD and analyzed by one-way ANOVA or by unpaired Student’s t-test. Significance was defined as P < 0.05.
RESULTS
Consumption of non-dietary soft drinks and other sweetened beverages
Despite using the most conservative estimate of HFCS consumption possible, patients with biopsy proven NAFLD (n=49) had increased daily consumption of HFCS or sugar containing beverages when compared to their matched controls (n=24) [365 kcal/day versus 170 kcal/day, fructose intake, p<0.05]. Figure 1 depicts the difference in fructose energy consumption between groups. For comparison, the mean total energy intake from sweetened beverages in adults from the NHANES study performed in 1999-2001 was approximately 190 kcal/d [6]. This translates into approximately 80-100 kcal/day of fructose from sweetened beverages. Thus, the caloric intake from fructose reported in our study is significantly higher, both for controls and patients with NAFLD, than that reported in the NHANES study for 1999-2001.
Figure 1. HFCS consumption.
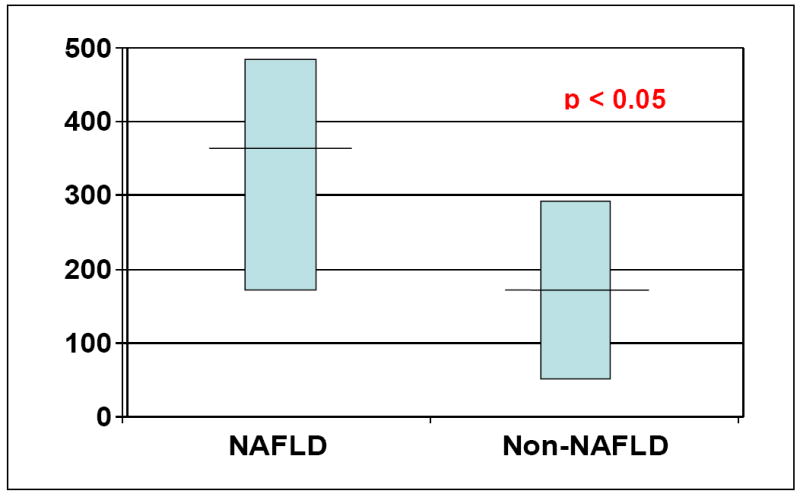
Energy consumption of fructose from sweetened beverages in patients with NAFLD was estimated as 356 kcal /day compared with 170 kcal /day in control patients with non -steatotic livers (p<0.05).
Plasma Biochemistry Profile
When compared to matched controls, patients with NAFLD had features of the metabolic syndrome (Table 2). Plasma cholesterol levels were higher in NAFLD groups compared to non-steatotic liver controls (p<0.05) whereas a trend was observed for serum triglycerides (p<0.05). No difference in fasting glucose levels was noted between groups. Interestingly, uric acid, which is commonly elevated in subjects with metabolic syndrome, was also significantly increased in the NAFLD group (p<0.05).
Table 2.
Metabolic Syndrome Biochemistry Profile
| Control (n=24) | NAFLD (n=49) | p-value | |
|---|---|---|---|
| Triglycerides mg/dL | 143 ± 81 | 258 ± 164 | < 0.05 |
| Cholesterol mg/dL | 168 ± 38 | 221 ± 73 | < 0.05 |
| Glucose mg/dL | 155 ± 79 | 138 ± 52 | 0.22 |
| Uric acid mg/dL | 4.8 ± 0.9 | 6.8 ± 1.6 | 0.03 |
Changes of Fructose-dependent Enzymes in Subjects with NAFLD
We extracted mRNA from liver biopsies from NAFLD patients (n=6) and non-fatty liver controls (n=6), and performed real time PCR for fructokinase (KHK), fatty acid synthase (FAS), and xanthine dehydrogenase (XDH). The mRNA expression of KHK, the first enzyme in fructose metabolism, and FAS (an important enzyme for lipogenesis) were both increased in NAFLD livers compared with non-steatotic liver control specimens (p<0.05). The mRNA expression of XDH tended to be higher in NAFLD subjects but did not reach statistical significance (p=0.16) [Figure 2].
Figure 2. Tissue Realtime PCR from Human Liver Tissue.
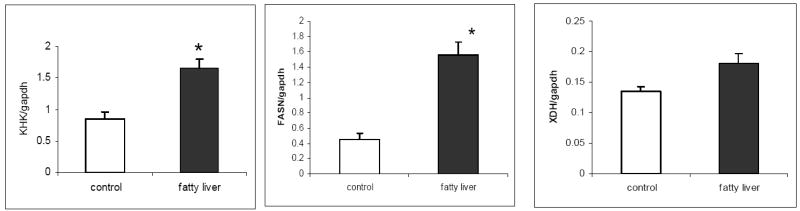
The mRNA expression of fructokinase (KHK), an important enzyme for fructose metabolism, and fatty acid synthase (FAS), an important enzyme for lipogenesis, are increased in NAFLD livers compared with non-steatotic liver control specimens (p<0.05). The mRNA expression of xanthine dehydrogenase (XDH) shows a trend for upregulation in NAFLD group but was not statistically significant (p=0.16).
Murine Liver Cells Show Increased KHK protein and activity to 5mM fructose
To determine if fructose can upregulate fructokinase activity in a murine hepatocyte cell line, AML12 hepatocytes were treated with different concentrations of fructose. Both KHK activity and expression were upregulated by the treatment with 5 mM fructose in the presence or absence of hyperglycemia; whereas hyperglycemia alone had no effect [Figure 3].
Figure 3. Effect of fructose on the enzymatic activity and expression of ketohexokinase (fructokinase, KHK) in the mouse hepatocytes AML-12.
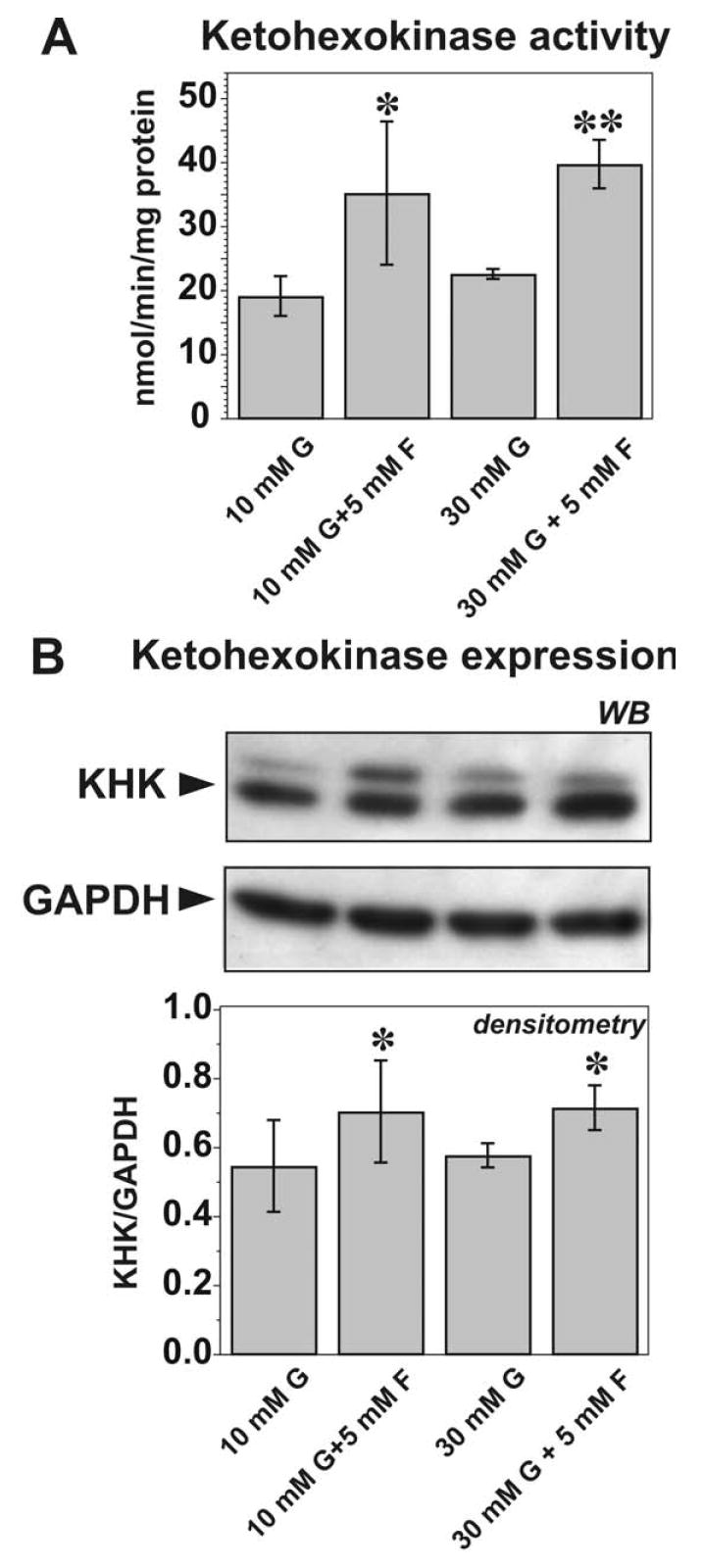
(A) Effect of fructose on the KHK activity. N=3 performed in triplicates. *, ** p<0.05, 0.01, respectively, in comparison to fructose-free control. (B) Effect of fructose on KHK expression. Representative blots from three independent experiments are shown. Densitometry is expressed as the ratio of the optical density of KHK band to GAPDH band detected after stripping the membrane.
DISCUSSION
The new millennium has witnessed a modern epidemic of obesity, metabolic syndrome, and diabetes. An increasingly recognized complication is non-alcoholic fatty liver disease (NAFLD), which can progress to cirrhosis over time in some individuals. In this study we investigated whether fructose could play a role in NAFLD, based on studies showing that fructose intake induces both features of metabolic syndrome and NAFLD in animals [17, 22] and correlates with the epidemic of metabolic syndrome [38]. Furthermore, administering high doses of sucrose (which contains 50% fructose) can also cause elevation of liver function tests in humans [13, 15, 16, 21, 23]. The Western diet, a “cafeteria diet” high in processed sugars and fat, has also been shown to cause deleterious effects with the development of hepatic steatosis in non-obese rats [39]. Thus, a strong rationale exists that suggests excessive fructose intake as a risk factor for developing NAFLD.
Our primary finding was that subjects with NAFLD have a significantly greater intake of sweetened beverages by history, representing two-fold greater than the mean intake in both controls and in population-based studies. The second finding was that the key initiating enzyme in fructose metabolism, KHK, was also increased two-fold in the liver biopsies of these patients compared to controls. The increase in KHK levels is consistent with the known effect of fructose to upregulate KHK in the liver of rats [32, 33]. Also consistent with this finding was our observation that fructose upregulates KHK mRNA, protein and activity in cultured hepatocytes.
There are several limitations to the study. First, it is limited by small sample size. Second, self-reporting of dietary intake is a relatively insensitive measure of dietary intake. We made every attempt to decrease reporting bias by obtaining the dietary history within one month of a liver biopsy; however, recall bias and reporting bias could exist. Despite these limitations, the study provides several different lines of evidence implicating a role for fructose in NAFLD.
A consequence of elevated KHK activity is that for the same dose of fructose, one might expect a greater or more prolonged degree of ATP depletion. One way this has been indirectly examined is to evaluate the effect of fructose on uric acid levels. Fructose, by causing ATP depletion, can rapidly generate uric acid which can be detected in the blood within minutes of ingestion [40]. Chronic fructose feeding can also lead to increased uric acid levels [41, 42] and intake of fructose correlates with uric acid levels in the population [43]. Furthermore, patients on a high fructose or sucrose diet show a greater uric acid response to a bolus of fructose [40, 41] consistent with upregulation of KHK activity. Finally, uric acid levels can predict the development of NAFLD [44]. It was thus of interest that uric acid levels were also higher in our patients than in the controls.
In conclusion, our studies raise the possibility that increased fructose intake may have a role in the pathogenesis of NAFLD. Furthermore, the upregulation of KHK by fructose may make subjects chronically drinking HFCS- or sugar-sweetened beverages more susceptible to the ATP-depleting effects of fructose. There is also increasing evidence that the rise in uric acid may also have a potential role in causing features of the metabolic syndrome [18], in part by the ability of uric acid to deplete endothelial nitric oxide levels [45] and by activating the adipocyte [32]. Further studies investigating fructose in NAFLD appear to be warranted.
Acknowledgments
Support for this study comes in part from NIH grants DK-52121, HL-68607, and DK-79352 and generous funds from Gatorade. MF Abdelmalek is supported by a NIH career development award, K23 DK062116.
Abbreviations
- NAFLD
non-alcoholic fatty liver disease
- KHK
hexokinase
- FAS
fatty acid synthase
- XDH
xanthine dehydrogenase
Footnotes
Financial Disclosures: RJ Johnson and Y Sautin are listed as inventors on patent applications by the University of Florida related to the role of fructose in hypertension and metabolic syndrome. Dr Johnson has also written a book on fructose for the lay public (Rodale Press, 2008).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Clark JM, Brancati FL, Diehl AM. Nonalcoholic fatty liver disease. Gastroenterology. 2002;122:1649–1657. doi: 10.1053/gast.2002.33573. [DOI] [PubMed] [Google Scholar]
- 2.Clark JM, Brancati FL, Diehl AM. The prevalence and etiology of elevated aminotransferase levels in the United States. Am J Gastroenterol. 2003;98:960–967. doi: 10.1111/j.1572-0241.2003.07486.x. [DOI] [PubMed] [Google Scholar]
- 3.Ioannou Gn, Boyko ES, Less SP. The prevalence and predictors of elevated serum aminotransferase activity in the Unites States in 1999-2002. Am J Gastroenterol. 2003;98:960–967. doi: 10.1111/j.1572-0241.2005.00341.x. [DOI] [PubMed] [Google Scholar]
- 4.Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, et al. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40:1387–1395. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- 5.Basciano H, Federico L, Adeli K. Fructose, insulin resistance, and metabolic dyslipidemia. Nutr Metab (Lond) 2005;21:5. doi: 10.1186/1743-7075-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nielsen SJ, Popkin BM. Changes in beverage intake between 1977 and 2001. Am J Prev Med. 2004;27:205–209. doi: 10.1016/j.amepre.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 7.Ludwig DS, Peterson KE, Gortmaker SL. Relation between consumption of sugar-sweetened drinks and childhood obesity: a prospective, observational analysis. Lancet. 2001;357:505–508. doi: 10.1016/S0140-6736(00)04041-1. [DOI] [PubMed] [Google Scholar]
- 8.Schulze MB, Manson JE, Lugwig DS, Colditz GA, Stamplfer MJ, Willett WC, et al. Sugar-sweetened beverages, weight gain, and incidence of type 2 diabetes in young and middle-aged women. JAMA. 2004;292:927–934. doi: 10.1001/jama.292.8.927. [DOI] [PubMed] [Google Scholar]
- 9.Gross LS, Ford ES, Liu S. Increased consumption of refined carbohydrates and the epidemic of type 2 diabetes in the United States: an ecological assessment. Am J Clin Nutr. 2004;79:774–779. doi: 10.1093/ajcn/79.5.774. [DOI] [PubMed] [Google Scholar]
- 10.Dhingra R, Sullivan L, Jacques PF, Wang TJ, Fox CS, Meigs JB, et al. Soft drink consumption and risk of developing cardiometabolic risk factors and the metabolic syndrome in middle-aged adults in the community. Circulation. 2007;116:480–488. doi: 10.1161/CIRCULATIONAHA.107.689935. [DOI] [PubMed] [Google Scholar]
- 11.Vartanian LR, Schwartz MB, Brownell KD. Effects of soft drink consumption on nutrition and health: a systematic review and meta-analysis. Am J Public Health. 2007;97:667–675. doi: 10.2105/AJPH.2005.083782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Welch JA, Cogswell ME, Rogers S, Rockett H, Mei Z, Grummer-Strawn LM. Overweight among low-income preschool children associated with the consumption of sweet drinks: Missouri, 1999-2002. Pediatrics. 2005;115:e223–e229. doi: 10.1542/peds.2004-1148. [DOI] [PubMed] [Google Scholar]
- 13.Segal MS, Gollub ES, Johnson RJ. Is the Fructose Index more relevant with regards to cardiovascular disease than the Glycemic Index? Eur J Nutr. 2007;46:406–417. doi: 10.1007/s00394-007-0680-9. [DOI] [PubMed] [Google Scholar]
- 14.Johnson RJ, Segal M, Sautin Y, Nakagawa T, Feig DI, Kang DH, et al. Potential Role of Sugar (Fructose) in the Epidemic of Hypertension, Obesity/Metabolic Syndrome, Diabetes, Kidney Disease, and Cardiovascular Disease. Am J Clin Nutr. 2007;86:899–906. doi: 10.1093/ajcn/86.4.899. [DOI] [PubMed] [Google Scholar]
- 15.Havel PJ. Dietary fructose: implications for dysregulation of energy homeostasis and lipid/carbohydrate metabolism. Nutr Rev. 2005;63:133–157. doi: 10.1301/nr.2005.may.133-157. [DOI] [PubMed] [Google Scholar]
- 16.Le KA, Tappy L. Metabolic effects of fructose. Curr Opin Clin Nutr Metab Care. 2006;9:469–475. doi: 10.1097/01.mco.0000232910.61612.4d. [DOI] [PubMed] [Google Scholar]
- 17.Mendeloff AI, Weichselbaum Role of the human liver in the assimilation of intravenously administered fructose. Metabolism. 1953;2:450–458. [PubMed] [Google Scholar]
- 18.Nakagawa T, Hu H, Zharikov S, Tuttle KR, Short RA, Glushakova O, et al. A causal role for uric acid in fructose-induced metabolic syndrome. Am J Physiol Renal Physiol. 2006;290:F625–F631. doi: 10.1152/ajprenal.00140.2005. [DOI] [PubMed] [Google Scholar]
- 19.Mayes PA. Intermediary metabolism of fructose. Am J Clin Nutr. 1993;58:754S–765S. doi: 10.1093/ajcn/58.5.754S. [DOI] [PubMed] [Google Scholar]
- 20.Wolfe BM, Ahuja SP, Marliss EB. Effects of intravenously administered fructose and glucose on splanchnic amino acid and carbohydrate metabolism in hypertriglyceridemic men. J Clin Invest. 1975;56:970–977. doi: 10.1172/JCI108177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ackerman Z, Oron-Herman M, Grozovski M, Rosenthal T, Pappo O, Link G, et al. Fructose-induced fatty liver disease: hepatic effects of blood pressure and plasma triglyceride reduction. Hypertension. 2005;45:1012–1018. doi: 10.1161/01.HYP.0000164570.20420.67. [DOI] [PubMed] [Google Scholar]
- 22.Davail S, Rideau N, Bernadet MD, André JM, Guy G, Hoo-Paris R. Effects of dietary fructose on liver steatosis in overfed mule ducks. Horm Metab Res. 2005;37:32–35. doi: 10.1055/s-2005-861029. [DOI] [PubMed] [Google Scholar]
- 23.Porikos KP, Van Itallie TB. Diet induced changes in serum transaminases and triglyceride levels in healthy men. Role of sucrose and excess calories. Am J Med. 1983;75:624–630. doi: 10.1016/0002-9343(83)90444-8. [DOI] [PubMed] [Google Scholar]
- 24.Howard BV, Wylie-Rosett J. Sugar and cardiovascular disease: A statement for healthcare professionals from the Committee on Nutrition of the Council on Nutrition, Physical Activity, and Metabolism of the American Heart Association. Circulation. 2002;106:523–527. doi: 10.1161/01.cir.0000019552.77778.04. [DOI] [PubMed] [Google Scholar]
- 25.Bode JC, Zelder O, Rumpelt HJ, Wittkamp U. Depletion of adenosine phosphates and metabolic effects of intravenous infusion of fructose or sorbitol in man and the rat. Eur J Clin Invest. 1973;3:436–441. doi: 10.1111/j.1365-2362.1973.tb02211.x. [DOI] [PubMed] [Google Scholar]
- 26.Hultman E, Nilsson H, Sahlin K. Adenine nucleotide content of human liver: normal values and fructose induced depletion. Scand J Clin Lab Invest. 1975;55:245–251. doi: 10.1080/00365517509095736. [DOI] [PubMed] [Google Scholar]
- 27.Oberhaensli RD, Galloway GJ, Taylor DJ, Bore PJ, Radda GK. Assessment of human liver metabolism by phosphorus-1 magnetic resonance spectrosocopy. Br J Radiol. 1986;59:695–699. doi: 10.1259/0007-1285-59-703-695. [DOI] [PubMed] [Google Scholar]
- 28.Cortez-Pinto H, Chatham J, Chacko VP, Arnold C, Rashid A, Diehl AM. Alterations in liver ATP homeostasis in human nonalcoholic steatohepatitis: a pilot study. JAMA. 1999;282:1659–1664. doi: 10.1001/jama.282.17.1659. [DOI] [PubMed] [Google Scholar]
- 29.Adachi F, Yu DT, Phillips MJ. An ultrastructural study of fructose-induced hepatic cell injury. Virchows Arch Abteilung B Zellpathol. 1972;10:200–209. doi: 10.1007/BF02899730. [DOI] [PubMed] [Google Scholar]
- 30.Nandhini AT, Balakrishnan SD, Anuradha CV. Response of liver antioxidant system to taurine in rats fed high fructose diet. Indian J Exp Biol. 2002;40:1016–1019. [PubMed] [Google Scholar]
- 31.Kelley GL, Allan G, Azhar S. High dietary fructose induces a hepatic stress response resulting in cholesterol and lipid dysregulation. Endocrinol. 2004;145:548–555. doi: 10.1210/en.2003-1167. [DOI] [PubMed] [Google Scholar]
- 32.Burant CF, Saxena M. Rapid reversible substrate regulation of fructose transporter expression in rat small intestine and kidney. Am J Physiol. 1994;267:G71–G79. doi: 10.1152/ajpgi.1994.267.1.G71. [DOI] [PubMed] [Google Scholar]
- 33.Korieh A, Crouzoulon G. Dietary regulation of fructose metabolism in the intestine and in the liver of the rat. Duration of the effects of a high fructose diet after the return to the standard diet. Arch Int Physiol Biochim Biophys. 1991;99:455–460. [PubMed] [Google Scholar]
- 34.Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol. 1999;94:2467–2474. doi: 10.1111/j.1572-0241.1999.01377.x. [DOI] [PubMed] [Google Scholar]
- 35.Asipu A, Hayward BE, O’Reilly J, Bonthron DT. Properties of normal and mutant recombinant human ketohexokinases and implications for the pathogenesis of essential fructosuria. Diabetes. 2003;52:2426–2432. doi: 10.2337/diabetes.52.9.2426. [DOI] [PubMed] [Google Scholar]
- 36.Davies R, Detheux M, Van Schaftingen E. Fructose 1-phosphate and the regulation of glucokinase activity in isolated hepatocytes. Eur J Biochem. 1990;192:283–289. doi: 10.1111/j.1432-1033.1990.tb19225.x. [DOI] [PubMed] [Google Scholar]
- 37.Weiser MM, Quill H. Estimation of fructokinase in crude tissue preparations. Anal Biochem. 1971;43:275–281. doi: 10.1016/0003-2697(71)90134-5. [DOI] [PubMed] [Google Scholar]
- 38.Bray GM, Nielson SJ, Popkin MB. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am J Clin Nutr. 2004;79:537–543. doi: 10.1093/ajcn/79.4.537. [DOI] [PubMed] [Google Scholar]
- 39.MacQueen HA, Sadler DA, Moore SA, Daya S, Brown JY, Shuker DEG, et al. Deleterious effects of a cafeteria diet on the livers of nonobese rats. Nutr Res. 2007;27:38–47. [Google Scholar]
- 40.Stirpe F, Della Corte E, Bonetti E, Abbondanza A, Abbati A, De Stefano F. Fructose-induced hyperuricaemia. Lancet. 1970;2:1310–1311. doi: 10.1016/s0140-6736(70)92269-5. [DOI] [PubMed] [Google Scholar]
- 41.Israel KD, Michaelis OE, Reiser S, Keeney M. Serum uric acid, inorganic phosphorus, and glutamic-oxalacetic transaminase and blood pressure in carbohydrate-sensitive adults consuming three different levels of sucrose. Ann Nutr Metab. 1983;27:425–435. doi: 10.1159/000176714. [DOI] [PubMed] [Google Scholar]
- 42.Reiser S, Powell AS, Scholfield DJ, Panda P, Ellwood KC, Canary JJ. Blood lipids, lipoproteins, apoproteins, and uric acid in men fed diets containing fructose or high-amylose cornstarch. Am J Clin Nutr. 1989;49:832–839. doi: 10.1093/ajcn/49.5.832. [DOI] [PubMed] [Google Scholar]
- 43.Gao X, Qi L, Qiao N, Choi HK, Curhan G, Tucker KL, et al. Intake of added sugar and sugar-sweetened drink and serum uric acid concentration in US men and women. Hypertension. 2007;50:306–312. doi: 10.1161/HYPERTENSIONAHA.107.091041. [DOI] [PubMed] [Google Scholar]
- 44.Sartorio A, Del Col A, Agosti F, Mazzilli G, Bellentani S, Tiribelli C, et al. Predictors of non-alcoholic fatty liver disease in obese children. Eur J Clin Nutr. 2007;61:877–883. doi: 10.1038/sj.ejcn.1602588. [DOI] [PubMed] [Google Scholar]
- 45.Khosla UM, Zharikov S, Finch JL, Nakagawa T, Roncal C, Mu W, et al. Hyperuricemia induces endothelial dysfunction. Kidney Int. 2005;67:1739–1742. doi: 10.1111/j.1523-1755.2005.00273.x. [DOI] [PubMed] [Google Scholar]