

Abstract
The coordinated expression of the different genes in an organism is essential to sustain functionality under the random external perturbations to which the organism might be subjected. To cope with such external variability, the global dynamics of the genetic network must possess two central properties. (a) It must be robust enough as to guarantee stability under a broad range of external conditions, and (b) it must be flexible enough to recognize and integrate specific external signals that may help the organism to change and adapt to different environments. This compromise between robustness and adaptability has been observed in dynamical systems operating at the brink of a phase transition between order and chaos. Such systems are termed critical. Thus, criticality, a precise, measurable, and well characterized property of dynamical systems, makes it possible for robustness and adaptability to coexist in living organisms. In this work we investigate the dynamical properties of the gene transcription networks reported for S. cerevisiae, E. coli, and B. subtilis, as well as the network of segment polarity genes of D. melanogaster, and the network of flower development of A. thaliana. We use hundreds of microarray experiments to infer the nature of the regulatory interactions among genes, and implement these data into the Boolean models of the genetic networks. Our results show that, to the best of the current experimental data available, the five networks under study indeed operate close to criticality. The generality of this result suggests that criticality at the genetic level might constitute a fundamental evolutionary mechanism that generates the great diversity of dynamically robust living forms that we observe around us.
Introduction
There is evidence that many complex dynamical systems found in nature are critical; namely, they operate close to a phase transition between two different dynamical regimes [1]. Avalanches [2], atmospheric phenomena [3], [4], financial markets [5], [6], earthquakes [7], [8], granular matter [9], and the brain [10]–[12], are typical examples. Critical systems exhibit remarkable properties which would be difficult to explain without the assumption of criticality. For instance, they can integrate, process and transfer information faster and more reliably than non critical systems [13]. Or they can detect and respond to external stimuli whose intensities span several orders of magnitude, like the brain [11]. These remarkable properties are mainly a consequence of the long-range correlations that emerge close to the critical point, producing collective behaviors and coordinated responses of the entire system. Thus, criticality confers on the system the ability to collectively respond and adapt to an often rapidly changing environment.
In the context of genetic regulatory networks (GRN), which are recognized as the main component in charge of cellular control [14], recent theoretical studies have shown that robustness and adaptability, two central properties of living organisms [15]–[20], exist simultaneously with the highest probability only in GRN operating at or close to criticality [21]. Thus, criticality is a property that can help us understand how the coordinate expression of the different genes in an organism is achieved under external perturbations, either to sustain cell functionality or to generate new phenotypes in order for the organisms to change and adapt to new environments [15]–[21]. Therefore, it is important to determine whether the GRN of real organisms are dynamically critical. Although some attempts have recently been made in order to answer this question [22]–[25], the definite answer has remained elusive for several decades. Here we present direct evidence that the GRN of five different organisms indeed exhibit critical dynamics. We do so by simulating in the computer the avalanche of perturbations in the gene expression profile of the genetic networks of these organisms. This allows us to compute the Derrida mapping M(x) for the five networks under consideration [26]. We will formally introduce the Derrida map in a further section. For the time being, it suffices to say that M(x) relates the size x(t) of the perturbation avalanche at time t, with the size x(t+1) of the avalanche at the next time step t+1. In other words: x(t+1) = M(x(t)). Therefore, M(x) contains all the information of the perturbation dynamics and can be used to directly measure the dynamical regime in which the network operates. Using this technique, we show that the dynamics of these avalanches are critical within numerical accuracy for the five different organisms studied.
However, in computing M(x) for the large networks of E. coli, S. cerevisiae and B. subtilis, we face the problem that the overwhelming majority of the regulatory functions (also termed regulatory phrases) that determine the combined effect of the regulators on their target genes are unknown. To circumvent this difficulty, we used random functions to model the dynamics of these networks. Although random, these functions were constructed in accordance with the fraction of positive regulatory phrases inferred from real gene expression profiles. Thus, the internal structure of the regulatory functions that we used for E. coli, S. cerevisiae and B. subtilis is statistically consistent with the one observed in microarray experiments.
Another important aspect that determines the dynamical regime in which the network operates (ordered, critical or chaotic) is the fraction of canalizing functions [27]–[31], which will be defined in a further section. Intuitively, these functions take into account the existence of dominant regulators such that, when present, override the effect of the other regulators. From the microarray experiments that we analyze it was not feasible to infer the fraction of canalizing functions present in the regulatory networks of E. coli, S. cerevisiae and B. subtilis. However, for these networks we varied in our simulations the fraction of canalizing functions around the statistically expected values. Interestingly, we observe a significant robustness of the critical dynamics under the addition or elimination of canalizing functions. This suggests that the critical behavior observed in the dynamics of the genetic networks of the organisms under study, is mainly produced by the network architecture rather than by the specific nature of the regulatory functions.
In the sections that follow we first present the Boolean network model that we use to implement the dynamics of the genetic networks, and the well known mean-field results that predict the existence of a phase transition from ordered to chaotic dynamics in this model. Then, we go beyond the mean-field theory by implementing the Boolean approach in the networks of real organisms, and show that in all the cases the Derrida map M(x) is consistent with critical dynamics. We then analyze how this map changes under the addition and removal of canalizing functions. In the last section we summarize and discuss our results.
Results
Boolean Models of Genetic Networks
Several models have been proposed to analyze the dynamics of GRN [32]–[35]. Although the details of the dynamics might change from one description (e.g. continuous) to another (e.g. discrete), we expect the general properties of the dynamics, such as criticality, to be model independent. In fact, recent work shows that continuous and discrete descriptions of GRN exhibit similar dynamical properties under very general conditions [36]. Here we use the Boolean approach [37]–[39], in which every gene is represented by a discrete variable g that can take two values: g = 1 if the gene is expressed and g = 0 otherwise. The genome is thus represented by a set of N binary variables, g1, g2, …, gN. The expression of each gene gn changes in time according to the equation
| (1) |
where 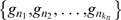 are the kn regulators of gn, and Fn is a Boolean function (also known as a logical rule), which is constructed according to the inhibitory or activatory nature of the regulators. The value acquired by the Boolean function for each configuration of the regulators is termed a regulatory phrase. For instance, if F(g
1, g
2) is a function of the two regulators g
1 and g
2 such that F(1,1) = 1, F(1,0) = 1, F(0,1) = 0, and F(0,0) = 0, then this function consists of the four regulatory phrases {1,1}→1, {1,0}→1, {0,1}→0, and {0,0}→0. We will refer to the regulatory phrases for which F = 1 as activatory, and those for which F = 0 as inhibitory. The fraction p of activatory phrases in the entire network, called the gene expression probability, is an important parameter that controls the dynamical regime in which the network operates (i.e. ordered, critical or chaotic). Recent work shows that the Boolean approach does capture the main aspects of the gene regulation dynamics, for it is able to reproduce gene expression patterns observed experimentally for several organisms [35], [40]–[43].
are the kn regulators of gn, and Fn is a Boolean function (also known as a logical rule), which is constructed according to the inhibitory or activatory nature of the regulators. The value acquired by the Boolean function for each configuration of the regulators is termed a regulatory phrase. For instance, if F(g
1, g
2) is a function of the two regulators g
1 and g
2 such that F(1,1) = 1, F(1,0) = 1, F(0,1) = 0, and F(0,0) = 0, then this function consists of the four regulatory phrases {1,1}→1, {1,0}→1, {0,1}→0, and {0,0}→0. We will refer to the regulatory phrases for which F = 1 as activatory, and those for which F = 0 as inhibitory. The fraction p of activatory phrases in the entire network, called the gene expression probability, is an important parameter that controls the dynamical regime in which the network operates (i.e. ordered, critical or chaotic). Recent work shows that the Boolean approach does capture the main aspects of the gene regulation dynamics, for it is able to reproduce gene expression patterns observed experimentally for several organisms [35], [40]–[43].
Phase Transition in the Boolean Network Model
In this section we present the mean-field theory results that show the existence of a dynamical phase transition from ordered to chaotic dynamics in the Boolean network model [26], [38], [44]. This allows us to introduce the tools that we use to characterize the dynamical regime in which the network operates. Although the phase transition was first obtained within the context of the mean-field approximation, we will show in the next section the remarkable result that the phase transition also occurs, almost identically, in networks with realistic topologies, for which the mean-field assumption does not necessarily apply. The phase transition is characterized by the temporal evolution of the Hamming distance x(t) between two different dynamical trajectories produced by two slightly different initial conditions. From a biological point of view, the Hamming distance x(t) is the average normalized size at time t of the avalanche of perturbations in the gene expression profile, produced by the perturbation (e.g. gene knockout or gene over expression) of a small fraction x(0) of genes at time t = 0. The temporal evolution of x(t) is given by a dynamical mapping x(t+1) = M(x(t)) which relates the size of the perturbation avalanche at two consecutive time steps [26], [38], [44]. Given an initial perturbation x(0) at time t = 0, successive iterations of this mapping will eventually converge to a stable fixed point 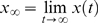 , which is the final size of the perturbation avalanche. The value x
∞ is the order parameter that characterizes the dynamical regime in which the network operates. Thus, if x
∞ = 0 (ordered regime), all the initial perturbations die out over time. On the contrary, if x
∞>0 (chaotic regime), the initial perturbation of even a small fraction of genes propagates across the entire system, finally altering the expression of a finite fraction x
∞ of genes in the genome. It turns out that M(x) is a continuous convex monotonically increasing function of x (in the mean field theory M(x) is a polynomial), with the property that M(0) = 0 and M(1)<1. Therefore, there is only one parameter that controls the phase transition, the so-called average network sensitivity S, which is given by S = [dM(x)/dx]x
= 0. If S<1 then the only fixed point is x
∞ = 0, whereas if S>1 then x
∞>0. The phase transition occurs at S = 1, for which the fixed point x
∞ = 0 is only marginally stable. In general, S depends on p and on the topology of the network. The dynamical mapping M(x) contains all the information about the dynamical regime in which the network operates. This is true even if M(x) cannot be obtained through a mean-field computation, which is the case for real networks. In the Supporting Information (Text S1) we provide a Java applet with the animation of the perturbation dynamics in networks with homogeneous random topology.
, which is the final size of the perturbation avalanche. The value x
∞ is the order parameter that characterizes the dynamical regime in which the network operates. Thus, if x
∞ = 0 (ordered regime), all the initial perturbations die out over time. On the contrary, if x
∞>0 (chaotic regime), the initial perturbation of even a small fraction of genes propagates across the entire system, finally altering the expression of a finite fraction x
∞ of genes in the genome. It turns out that M(x) is a continuous convex monotonically increasing function of x (in the mean field theory M(x) is a polynomial), with the property that M(0) = 0 and M(1)<1. Therefore, there is only one parameter that controls the phase transition, the so-called average network sensitivity S, which is given by S = [dM(x)/dx]x
= 0. If S<1 then the only fixed point is x
∞ = 0, whereas if S>1 then x
∞>0. The phase transition occurs at S = 1, for which the fixed point x
∞ = 0 is only marginally stable. In general, S depends on p and on the topology of the network. The dynamical mapping M(x) contains all the information about the dynamical regime in which the network operates. This is true even if M(x) cannot be obtained through a mean-field computation, which is the case for real networks. In the Supporting Information (Text S1) we provide a Java applet with the animation of the perturbation dynamics in networks with homogeneous random topology.
Beyond the mean-field theory: Existence of the Phase Transitions in Networks with Realistic Topologies
The mean-field theory that predicts the existence of the phase transition controlled by the average network sensitivity S, is based on the assumption that all the genes in the network are statistically independent and statistically equivalent [26], [44]. However, this assumption is certainly not true for real GRN, due to the existence of global regulators which correlate the expression of a large fraction of genes. Indeed, recent large-scale analysis [21] of the transcription regulatory networks of E. coli, S. cerevisiae, and B. subtilis indicate that these networks exhibit a Poisson-like input topology and a scale-free output topology (see Figure 1a). The output scale-free topology correlates the expression of a large fraction of the genes, and the assumption of statistical independence is not longer satisfied. Therefore, we do not expect the mean-field theory to be applicable for real networks because of their topological characteristics. Nonetheless, the phase transition predicted by the mean-field theory is identical to the one observed in randomly constructed Boolean networks with topologies statistically equivalent to the ones observed in real GRN. This is shown for the first time in Figure 1b. This result is quite remarkable, for we know that in many other systems the phase transition strongly depends on the network topology, and can even disappear for topologies that induce strong correlations between the elements (such as the scale-free topology, [45]).
Figure 1. Order-chaos phase transition in Boolean networks with realistic topologies.

(a) Graphic representation of the gene transcription network of E. coli K-12. For this network the probability for a given gene to have K regulators is P(K) = e −z zK/K! (Poisson input topology), whereas the probability to be a regulator to n other genes is P(n) = Cn −γ (scale-free output topology). (b) Phase transition diagram showing x ∞ as a function of S. The solid line in black is the theoretical result predicted by the mean-field theory. The dashed line in red was computed numerically for randomly constructed Boolean networks with N = 1000, Poisson input topology and scale-free output topology. The Boolean functions were randomly generated with a probability of gene expression p = 0.5. Remarkably, the phase transition predicted by the mean field theory is identical to the one obtained for random Boolean networks with topologies statistically equivalent to the real ones.
Critical Dynamics in Real Genetic Networks
To determine the dynamical regime in which the genetic network of a real organism operates, we have to compute the dynamical mapping M(x) directly from experimental data, without any mean-field assumptions. The actual form of M(x) depends on both the network topology and the particular set of Boolean functions. We computed numerically M(x) for five genetic networks: The network of flower morphogenesis in A. thaliana (15 genes; [41]); the network of segment polarity genes in D. melanogaster (60 genes; [42]); and the gene transcription networks of E. coli (1481 genes; [46]), S. cerevisiae (3459 genes; [47]), and B. subtillis (830 genes; [48]). In the first two cases the Boolean functions are already known. However, the overwhelming majority of regulatory phrases for the gene transcription networks of E. coli, S. cerevisiae and B. subtilis are still unknown. Due to this lack of information, to implement the Boolean dynamics on the GRN of these three organisms we used biased random Boolean functions generated with a gene expression probability p inferred from microarray experiments. (In the next section we show that the map M(x) does not change significantly for networks with a large fraction of canalizing functions.) Given the network topology, p can be estimated from microarray experiments by standard Bayessian parametric inference with two states (see Methods). Using 223 microarrays to sample the gene expression space in S. cerevisiae [49], 152 microarrays for E. coli [49]–[51], and 69 microarrays for B. subtillis [52], we inferred several regulatory phrases for each of these organisms. All the experiments used for this analysis are listed in the Supporting Information accompanying this article (Text S1). We only used regulatory phrases for which the average of the a posteriori probability distribution was greater than 70% (activatory phrase) or smaller than 30% (inhibitory phrase). The inference technique is explained below in the Materials and Methods section. With this technique, we obtained the following gene expression probabilities: p = 0.576±0.038 for E. coli (from 264 inferred phrases), p = 0.495±0.055 for S. cerevisiae (from 196 phrases), and p = 0.0531±0.035 for B. subtilis (from 307 phrases). We then constructed random Boolean functions with internal bias given by these probabilities.
We report in Figure 2 the Derrida curves (i.e. the graphs of M(x)) for the five genetic networks under consideration. It is apparent from this figure that in all five cases the Derrida curves arrive almost tangent to the identity close to the origin. The above is consistent with critical behavior at the genetic level. A polynomial regression analysis allows us to estimate the average network sensitivity S = [dM(x)/dx]x = 0 by computing the slope at the origin of the best-fit polynomial with a degree equal to the maximum number of regulators per gene in each case (this is the polynomial predicted by the mean-field theory). In all five cases the Regression Sum of Squares is below 10−4, and the average network sensitivities obtained in this way are: S = 1.028 for E. coli; S = 1.036 for S. cerevisiae; S = 0.826 for B. subtilis; S = 0.914 for D. melanogaster; and S = 1.127 for A. thaliana. Within numerical accuracy, these sensitivities show that the dynamics of these networks are very close to criticality. Note that this is particularly true for the two largest and most complete networks of E. coli and S. cerevisiae.
Figure 2. Critical dynamics in networks of real organisms.

Derrida curves exhibiting critical dynamical behavior in the genetic networks of five different organisms spanning four kingdoms of life: E. coli [46], S. cerevisiae [47], and B. subtilis [48], D. melanogaster [42] and A. thaliana [41]. Each point in the Derrida curve is the average over 20000 initial perturbations and the error bars indicate one standard deviation around this average. Additionally, the error bars in the curves of E. coli, B. subtilis and S. cerevisiae incorporate the uncertainty in the estimation of the p bias inferred from the microarray experiments. For comparison we show in the upper left corner three Derrida curves of randomly constructed networks with Poisson input topology operating in three dynamical regimes: Ordered (green), critical (black) and chaotic (blue). Note that criticality is characterized by the tangency of the Derrida curve to the identity close to x = 0.
Criticality and its robustness revealed by varying the degree of canalization
We have used random Boolean functions in our simulations because the overwhelming majority of the logical rules for E. coli, S. cerevisiae and B. subtilis, are still unknown. The caveat is that real biological functions are not random. It has been pointed out that canalizing functions are more realistic from a biological point of view [27], [28]. A formal definition of canalizing functions can be found in Ref. [31]. Here, it suffices to define a canalizing function as follows. Let F(g 1, g 2, …, gk) be a Boolean function of k arguments. We will say that F is canalizing on one of its arguments gi, if the value of F is determined by fixing the value of gi either to 0 or to 1. To illustrate this concept, Table 1 shows a function F(g 1, g 2, g 3) that depends on three arguments. In this example, F = 0 whenever g 2 = 1, regardless of the values of g 1 and g 3. Therefore, F is canalizing on g 2. The biological significance of canalizing functions is based on the existence of dominant regulators. Thus, in the example shown in Table 1, g 2 could represent a dominant repressor (like crp in E. coli) which, when present, blocks the transcription of the target gene regardless of the presence or absence of the activators.
Table 1. Example of canalizing function.
| g 1 | g 2 | g 3 | F(g 1, g 2, g 3) |
| 1 | 1 | 1 | 0 |
| 1 | 1 | 0 | 0 |
| 1 | 0 | 1 | 1 |
| 1 | 0 | 0 | 1 |
| 0 | 1 | 1 | 0 |
| 0 | 1 | 0 | 0 |
| 0 | 0 | 1 | 1 |
| 0 | 0 | 0 | 0 |
Example of a Boolean function of three arguments that is canalizing on one of them. Note that F = 0 whenever g 2 = 1, regardless of the values of the other two arguments.
It is known that the amount of canalizing functions in the system can change the dynamical regime in which the network operates [29], [30]. Therefore, it is important to determine if the dynamics of the large networks of E. coli, S. cerevisiae and B. subtilis are still critical when more realistic Boolean functions are used. However, from the microarray experiments that we analyzed it is impossible to know the fraction of canalizing functions present in these organisms, or if such functions have one, two or more canalizing inputs. For instance, according to the regulonDB, which gives the topology of the transcription regulatory network of E. coli [46], there are at least 817 genes with two or more regulators. These 817 genes are all regulated by a subset of 160 genes. Therefore, to determine whether or not the Boolean functions associated with these genes are canalizing, one would have to analyze microarray experiments probing at least 2160 different configurations for these genes. This is impossible for many reasons, but mainly because not all the 2160 configurations of the regulators are biologically attainable. (Consider for instance the configuration 00000….00 in which all the regulators are “off”, or the configuration 11111…11 in which all the regulators are “on”. Clearly, these two configurations are not attainable under any biological condition—without killing the organism.) Therefore, the 152 microarrays used to sample the gene expression space in E. coli represent a very tiny fraction of all the possible configurations necessary to determine the whole set of Boolean functions. However, these 152 experiments are absolutely relevant because they represent the observable and biologically attainable gene expression configurations of the organism. Thus, it might be irrelevant if the whole Boolean function of a given gene is canalizing because neither us nor the organism are sampling its whole set of (mathematically possible) configurations. For this reason, we do believe that the important quantity is the observed gene expression probability for the biologically attainable configurations.
Nonetheless, Random Boolean functions already contain canalizing functions. Table 2 gives the probability Pc(K) for a randomly generated Boolean function with K inputs to be canalizing on at least one of its inputs (data taken from [31]). As we can see from Table 2, the probability for a randomly generated Boolean function to be canalizing is high for K = 1, 2 and 3 (Boolean functions with K = 1 are canalizing, by definition). On the other hand, Table 3 shows the distribution PE(K) of the number of genes with K inputs in the genetic network of E. coli, according to the last update of the regulonDB [46].
Table 2. Fraction of canalizing functions.
| K | Pc(K) |
| 1 | 1 |
| 2 | 0.875 |
| 3 | 0.468 |
| 4 | 0.0536 |
| 5 | 3.0×10−4 |
| 6 | 5.5×10−9 |
| 7 | 1.6×10−18 |
| 8 | 6.7×10−38 |
| 9 | 3.1×10−76 |
| 10 | 2.9×10−153 |
Probability Pc(K) for a randomly generated Boolean function with K inputs to be canalizing. Data taken from [31].
Table 3. Distribution of regulators per gene in E. coli.
| K | PE(K) |
| 0 | 50 |
| 1 | 615 |
| 2 | 347 |
| 3 | 240 |
| 4 | 100 |
| 5 | 85 |
| 6 | 22 |
| 7 | 11 |
| 8 | 8 |
| 9 | 3 |
| 10 | 1 |
Distribution PE(K) of the number of genes with K regulators in the E. coli network according to the last update of the regulonDB [46].
From Tables 2 and 3 we obtain that, if the Boolean functions for E. coli are generated at random, just by chance about 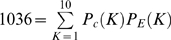 functions out of 1482 (70%) would be canalizing on at least one input. If we do not consider the 615 genes with only one regulator (because for such genes the Boolean function is trivially canalizing), and the 50 genes with no inputs, then there are 817 genes with two or more regulators. From the data listed in Tables 2 and 3 one obtains that, for the genes with K≥2, about
functions out of 1482 (70%) would be canalizing on at least one input. If we do not consider the 615 genes with only one regulator (because for such genes the Boolean function is trivially canalizing), and the 50 genes with no inputs, then there are 817 genes with two or more regulators. From the data listed in Tables 2 and 3 one obtains that, for the genes with K≥2, about 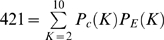 out of 817 of the randomly generated functions would be canalizing just by chance. It is important to note that these 421 canalizing functions mostly come from the genes with K = 2 and K = 3. Thus, combining the results presented in Tables 2 and 3 we obtain that the fraction fc of canalizing functions present just by chance for the genes with K≥2 is given by fc≈421/817≈0.51.
out of 817 of the randomly generated functions would be canalizing just by chance. It is important to note that these 421 canalizing functions mostly come from the genes with K = 2 and K = 3. Thus, combining the results presented in Tables 2 and 3 we obtain that the fraction fc of canalizing functions present just by chance for the genes with K≥2 is given by fc≈421/817≈0.51.
Therefore, in the simulations with random Boolean functions there is already a large fraction of canalizing functions. To further investigate the effect that canalizing functions might have on the dynamics of the genetic networks, we have added and removed canalizing functions to and from the ones already present just by chance. Our results indicate that the critical behavior observed in the genetic dynamics still exists even when the fraction of canalizing functions substantially deviates from the value expected for fully random functions.
Figure 3 shows the map M(x) for the E. coli network for several values of the fraction fc of genes with K≥2 regulated by canalizing functions. Figure 3a corresponds to the case in which there are more canalizing functions than the ones present just by chance, whereas Figure 3b shows the opposite case in which there are less canalizing functions. To compute fc we have ignored the genes with only one regulator, taking into account only the 817 genes with two or more regulators. We have already mentioned that in the E. coli network, fc = 0.51 for fully random Boolean functions. Additionally, from Table 2 we also see that for K = 2 and K = 3 the probability Pc(K) for a random Boolean function to be canalizing is relatively high (Pc(2) = 0.875 and Pc(3) = 0.468), whereas for K≥4 the probability is extremely low. Therefore, what we did to increase the value of fc was to add canalizing functions only to the genes with K≥4 in such a way as to preserve the overall value p = 0.576±0.038 of the gene expression probability observed in microarray experiments. There are 230 genes with K≥4, and we can consider that none of them are regulated by canalizing functions just by chance (the probability is very low). Therefore, to increase fc we forced a fraction q of these 230 genes to be regulated by canalizing functions. In Figure 3a we present the Derrida maps for q = 0.1, 0.3, 0.5, 0.7 and 0.9. When considering the other 421 genes already regulated by canalizing functions just by chance, these values of q correspond to fc = 0.544, 0.600, 0.656, 0.712, and 0.768, respectively. (We computed fc as fc = (421+230×q)/816). As we can see from Figure 3a, the map M(x) is practically the same even for fc = 0.656, namely, even when two thirds of the genes with two or more inputs are regulated by canalizing functions. Only for fc>0.7 significant deviations from criticality are observed (dotted-dashed curves in Figure 3).
Figure 3. Critical dynamics in E. coli using canalizing functions.

Derrida map M(x) for the E. coli network with different fractions fc of canalizing functions. To compute fc we took into consideration only the genes for which K≥2. For fully random Boolean functions fc = 0.51. (a) We increased the value of fc by adding canalizing functions to the 230 genes with K≥4. Note that M(x) remains critical even for fc≈0.66, but it starts to deviate from criticality towards the ordered region for fc>0.7 (dotted dashed curves). (b) We decreased the value of fc by removing canalizing functions from the 421 genes which originally are regulated by canalizing functions just by chance (most of them with K = 2 and K = 3). In this case the curve deviates from criticality towards the chaotic region only for fc<0.4. Each point in the curves is the average over 5000 initial conditions randomly chosen.
Analogously, to decrease the value of fc we removed a fraction q of the canalizing functions from the 421 genes which originally are regulated by canalizing functions just by chance. In other words, we forced a fraction q of these 421 genes to be regulated by non-canalizing functions. In this case, fc is given by fc = 421(1−q). Figure 3b shows the Derrida maps for q = 0.1, 0.3, 0.5, 0.7 and 0.9.
From Figure 3 it is clear that the Derrida map is less robust to the removal than to the addition of canalizing functions. However, in the region 0.4≤fc≤0.66 the Derrida map does not seem to change substantially.
To indicate the significance of the above results we present in Figure 4 the map M(x) for homogeneous random networks with different fractions fc of canalizing functions. In these networks each gene has exactly K = 2 regulators chosen randomly from anywhere in the system, and the bias p of the Boolean functions is p = 0.5. With fully random functions the network operates in the critical regime. Figure 4(a) corresponds to the addition and Figure 4(b) to the removal of canalizing functions. In both cases we used networks with N = 1481 (the same number of genes as in the E. coli network). In a homogeneous random network with N genes and connectivity K, just by chance there are Nc = N×Pc(K) genes regulated by canalizing functions, and Nnc = N×(1−Pc(K)) genes regulated by non-canalizing functions. By forcing a fraction q of these Nnc genes to be regulated by canalizing functions, the overall fraction fc of canalizing functions in the network increases according to fc = (Nc+q×Nnc)/N. Analogously, by removing a fraction q of the Nc canalizing functions present just by chance, the fraction fc of remaining canalizing functions is fc = Nc(1−q). In Figures 4(a) and 4(b) we present results for q = 0.1, 0.3, 0.5, 0.7 and 0.9, which produce the corresponding values of fc displayed in the Figure. As we can see from Figure 4, for homogeneous random networks the addition or removal of even a small fraction of canalizing functions, on top of the ones that are already present by chance, has a much bigger effect on the dynamics than for the E. coli network.
Figure 4. Dynamics of random networks using canalizing functions.

Derrida map M(x) for homogeneous random networks with N = 1481, K = 2, p = 0.5 and different fractions fc of canalizing functions. For fully random functions the networks operate in the critical regime and fc = 0.875. Panel (a) shows the results when more canalizing functions are added on top of the ones already present just by chance, whereas panel (b) corresponds to removing canalizing functions from the ones already present. Note that the addition or removal of even a small fraction of canalizing functions in homogeneous random networks has a considerably bigger impact on the dynamics than for the E. coli network. Each point in the curves is the average over 5000 initial conditions randomly chosen.
These results strongly suggest that the critical behavior observed in the dynamics of the genetic networks of real organisms, and its robustness to changes in the fraction of canalizing functions, are most probably due to the network architecture rather than to the logical rules that regulate the expression of the genes. If, for instance, there were a large fraction of genes with 5 inputs in the E. coli network, then the presence of canalizing functions would certainly affect the dynamical regime in which the network operates. But given that most of the genes have 1, 2 or 3 inputs, there is already a large fraction of canalizing functions just by chance. Adding more does not substantially affect the dynamical behavior.
Discussion
Our results show that, to the best of the current experimental data available, and within numerical accuracy, the Boolean dynamics of the GRN of five organisms from four different kingdoms are critical. We have used two small well documented networks for specific patterning processes in plants (A. thaliana) and animals (D. melanogaster), for which both the topology and the Boolean functions are known and correspond to thoroughly studied processes at the molecular level. We also tested larger GRN for unicellular organisms (E. coli, B. subtilis and S. cerevisiae) in which the logical rules are not known. Thus, in the absence of other knowledge, we used random regulatory phrases constructed with gene expression probabilities inferred from microarray experiments. How ever, the results are essentially the same when the fraction of canalizing functions with the same gene expression probabilities varies considerably. The fact that all these GRN, constructed with completely different approaches, for distinct organisms and of different sizes exhibit critical Boolean dynamics is of outmost interest and strongly suggest that this might be a generic characteristic with far reaching consequences. For there is evidence that criticality confers clear evolutionary advantages to living organisms, because it is only close to criticality that robustness and adaptability can coexist. It remains an open problem to determine how criticality has emerged throughout evolution, i.e. to devise biologically relevant models of network growth that generate critical dynamics. Such models must take into account not only the evolution of the network topology, but also the emergence of the regulatory phrases through which the genes interact. If this critical behavior is corroborated as more and better experimental data become available, and with more detailed dynamic models, criticality at the genetic level may become a fundamental evolutionary mechanism that renders the stability and diversity that we observe in living organisms.
Materials and Methods
Phase transition in networks with realistic topology
We computed the phase transition displayed in Fig. 2(b) by implementing the Boolean dynamics in networks with scale-free output topology and Poisson input topology. Such networks are easily generated in the computer by firs assigning to each gene gn its number of outputs ln, taken from a scale-free probability distribution Po(l) = Cl
−γ, where C is the normalization constant and γ is the scale-free exponent. Once every gene has been assigned with a number of outputs, the ln outputs of each gene gn are chosen randomly from anywhere in the network. By this process, the inputs of each gene are automatically set with a Poisson distribution 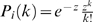 whose average z depends on the scale free exponent γ. There is a fraction Pi(0) = e
−z of genes which do not have inputs and hence remain frozen throughout the temporal evolution of the system. Those genes were not perturbed. Only genes with a nonzero number of inputs were perturbed. We used networks with N = 1000 genes since this is the order of magnitude of the gene transcription networks available in the databases. We run the dynamics for 1000 time steps, starting out from two initial conditions differing in 20 genes (2%). After these 1000 time steps we computed the Hamming distance between the resulting states. Each point in Fig. 2(b) is the average of this Hamming distance over 10000 different pairs of initial conditions.
whose average z depends on the scale free exponent γ. There is a fraction Pi(0) = e
−z of genes which do not have inputs and hence remain frozen throughout the temporal evolution of the system. Those genes were not perturbed. Only genes with a nonzero number of inputs were perturbed. We used networks with N = 1000 genes since this is the order of magnitude of the gene transcription networks available in the databases. We run the dynamics for 1000 time steps, starting out from two initial conditions differing in 20 genes (2%). After these 1000 time steps we computed the Hamming distance between the resulting states. Each point in Fig. 2(b) is the average of this Hamming distance over 10000 different pairs of initial conditions.
Once we know the regulators of every gene in the network, the Boolean functions are assigned as follows. A Boolean function of k inputs has 2k values, one for each of the 2k configurations of the k inputs. For each of these 2k configurations we generate a random number z uniformly distributed in the interval [0,1]. If z≤p we set the value of the Boolean function equal to 1 for the corresponding configuration of the inputs. If z>p, we set the value of the Boolean function equal to 0. We repeat this process for all the configurations of the Boolean function and for all the Boolean functions in the network. In this algorithm the parameter p is the gene expression probability inferred from microarray experiments.
Microarray Data
For E. coli all available microarray experiments in the Stanford Microarray Database (SMD, see http://genome-www5.stanford.edu) were incorporated in the inference algorithm, except by experiments that increased the number of false positives (see Assessing Inference Success below); 107 experiments were selected in total from SMD. Also, 45 microarray experiments were incorporated from Gosset et al, 2004 and Zhang et al, 2005. For B. subtilis all the available microarray experiments in the KEGG Expression Database (www.genome.jp/kegg/expression) were incorporated except experiments that increased the number of false positives; 69 experiments in total. For S. cerevisiae basically all the data form the three experimenters were retrieved from SMD. The name of the experimenter and the number of microarrays are as follows: Gasch, 138; DeRisi, 29; Spellman, 56. Only the cell cycle experiments from Spellman were rejected because they do not conform to the hypothesis of the Bayesian inference algorithm, which requires the absence of oscillating variables. See the Supporting Information (Text S1) for a complete relation of ID's of the incorporated experiments for the three organisms.
Data Normalization
The data were retrieved from SMD as Log Ratios (base 2). These data were already background corrected and mean normalized by SMD itself. Only features with no flags were selected. Data from (Gosset et al, 2004; Zhang et al, 2005) were background corrected and normalized by Affymetrix Microarray Suite 5.0. Log Ratios (base 2) were then calculated between the wild type experiments and the mutants with and without glucose. The data from the KEGG Expression Database were already normalized when retrieved. They were only background corrected by us; Log ratios (base 2) were obtained.
Inference Algorithm
Only Log Ratios greater than a certain threshold T 0 were considered (see Assessing Inference Success). With the filtered data we performed standard Bayesian parametric inference with variables of two states, inhibited (repressed) and induced (activated). The Equivalent Sample Size was set equal to 4. The induction or repression of a gene were established if the average of the a posteriori distribution was equal to or greater than 70% for induction, and equal to or lower than 30% for repression. A detailed example of the inference algorithm is presented below.
Assessing inference success
The nature of the regulation (activation or repression) of many of the established regulations in the networks of E. coli and B. subtilis is reported in the corresponding databases (Salgado et al, 2006; Makita et al, 2004). From this information it is possible to generate regulatory phrases only for those genes with one regulator. Thus we compared the one-regulator phrases already reported with our inferred one-regulator phrases. The inference success was established as the percentage of coincidences between our inferred phrases and the reported ones. The inference success increases as the threshold T 0 increases. However, the total number of phrases that can be inferred diminishes as T 0 increases. A good compromise between low percentage of false positives (high inference success) and a good statistics was achieved at T 0 = 1.50 for E. coli, and T 0 = 1.30 for B. subtilis. From these results we found that a good threshold for S. cerevisiae, for which the nature of the regulations is not reported, is T 0 = 1.50. Note that a threshold of 1.5 in Log2 Ratios is equivalent to almost a three fold change in expression intensities.
Example of inference
We illustrate the inference technique to obtain the value of the parameter p from microarray experiments with a specific example. Suppose that gene A is regulated by genes B and C, and that we want to determine the regulatory phrases, i.e. how A changes its expression due to the joint combinatorial changes of B and C. In order to do so, we need a set of microarray experiments in different conditions that have been already normalized and their background noise subtracted. We use the data shown in Table 4 for the gene expression level of three genes (log ratios base 2), obtained from two color spotted microarrays. In this table, “Condi” refers to one microarray experiment in the ith condition (not necessarily all the conditions have to be different). For instance, Cond1 may be the wild type condition, whereas Cond2 may be a condition in which a global regulator has been knocked out. In general, the level of expression of a given gene is different from one condition to another. Namely, there may be over expressions or under expressions across the different conditions. To filter out only the biggest changes in the level of expression of the genes we use a threshold T 0, and indicate the positive changes (those greater than the threshold) with an arrow pointing upwards. Analogously, we indicate the negative changes (those lower than the threshold) with an arrow pointing downwards. When no change is detected we use the symbol (—). Using the data shown in Table 4 and setting the threshold value to T 0 = 1.5, we get the discrete representation shown in Table 5.
Table 4. Gene expression data.
| Cond1 | Cond2 | Cond3 | Cond4 | Cond5 | Cond6 | Cond7 | |
| A | 2.34 | 1.56 | 2.05 | 2.56 | 0.65 | −3.45 | −2.55 |
| B | 1.76 | 1.95 | 2.86 | 2.67 | −1.89 | −2.06 | −1.79 |
| C | 3.36 | 1.45 | 1.35 | 1.97 | −1.78 | −1.67 | −1.99 |
Typical example of the gene expression data obtained from microarray experiments. Each number represents the change (increase or decrease) of the gene expression level of the corresponding gene in Condition i (Condi), relative to its level of expression in a given reference condition. This change is measure as a log ratio in base 2. Thus, a positive number corresponds to an increase in the level of expression, whereas a negative number represents a decrease.
Table 5. Discretization of the gene expression change.
| Cond1 | Cond2 | Cond3 | Cond4 | Cond5 | Cond6 | Cond7 | |
| A | ↑ | ↑ | ↑ | ↑ | — | ↓ | ↓ |
| B | ↑ | ↑ | ↑ | ↑ | ↓ | ↓ | ↓ |
| C | ↑ | — | — | ↑ | ↓ | ↓ | ↓ |
If the change of the level of expression reported in Table 4 is larger than a given threshold T 0, we write an arrow pointing upwards (↑), whereas if the change of the level of expression is smaller than −T 0 then we write an arrow pointing downwards (↓). The symbol (—) indicates that no significant change was detected. In this example we used a threshold T 0 = 1.5 to discretize the numbers given in Table 4.
By counting how many times A changes for the different combinations of B and C given in Table 5, we obtain the data shown in Table 6. Some table entries are equal to zero, indicating that there are no data for that particular combination of B and C. Note that we have considered all the cases with three symbols (↓,—,↑), and two regulators (B and C). Now we reduce Table 6 by considering only the entries where a change can be detected (i.e., we eliminate all entries with “—”). This gives the results displayed in Table 7. For reasons that will be clear in a moment, it is necessary to add one unit of a priori evidence to every entry of Table 7. After this unit has been added, we obtain Table 8. Observe from this last table that, for the first combinatorial change of B and C (first row), there are three evidences where the level of expression of A decreases and one in which it increases. If these were all the evidences, we could say that from 100% of the cases (3+1), in 75% of them the expression level of A increases and in 25% of them it decreases. Thus, the a priori evidence that we have added has the effect of changing the importance the a posteriori evidence. There are several ways of distributing the a priori evidence; in this work we have used an Equivalent Sample Size S = 4, which means that the 4 units of a priori evidence are distributed equally among all the possibilities.
Table 6. Occurrence count of evidences.
| B | C | A | ||
| ↓ | — | ↑ | ||
| ↓ | ↓ | 2 | 1 | 0 |
| — | ↓ | 0 | 0 | 0 |
| ↑ | ↓ | 0 | 0 | 0 |
| ↓ | — | 1 | 0 | 0 |
| — | — | 0 | 0 | 0 |
| ↑ | — | 0 | 1 | 2 |
| ↓ | ↑ | 0 | 0 | 0 |
| — | ↑ | 0 | 0 | 0 |
| ↑ | ↑ | 0 | 0 | 2 |
Counting of how many times A changes positively (↑), negatively (↓), or it does not change (—) for every combined instance of changes of B and C, according to the results displayed in Table 5.
Table 7. A priori and a posteriori evidence I.
| B | C | A | |
| ↓ | ↑ | ||
| ↓ | ↓ | 2 | 0 |
| ↑ | ↓ | 0 | 0 |
| ↓ | ↑ | 0 | 0 |
| ↑ | ↑ | 0 | 2 |
After removing all the instances in Table 6 where there is no change, we end up with the data shown in this table. Note that there are some configurations of B and C for which there is no evidence for a corresponding change in A.
Table 8. A priori and a posteriori evidence II.
| B | C | A | |
| ↓ | ↑ | ||
| ↓ | ↓ | 3 | 1 |
| ↑ | ↓ | 1 | 1 |
| ↓ | ↑ | 1 | 1 |
| ↑ | ↑ | 1 | 3 |
To every instance of A shown in the previous table, we add an a priori evidence of 1, which results in the data displayed here. This has the effect of changing the importance of the a posteriori evidence.
Finally, given the a posteriori evidence, in order to decide whether the expression level of A increases or decreases we use a second threshold T 1. If the percentage of a posteriori evidence is greater or equal than T 1, then a regulatory phrase has been established. Using a threshold T 1 = 75% we obtain the results shown in Table 9. We have used the symbol “—” to state that the evidence does not support a decision. To infer the regulatory phrases from the microarray experiments analyzed in this work we used the value T 1 = 75%.
Table 9. Inferred table of regulation.
| B | C | A |
| ↓ | ↓ | ↓ |
| ↑ | ↓ | — |
| ↓ | ↑ | — |
| ↑ | ↑ | ↑ |
With a threshold T 1 = 75% for the a posteriori evidence in Table 8, we inferred the most probable effect of the combined changes of B and C on the expression of A. With the information available, only two regulatory phrases could be inferred in this example.
Once a set of phrases has been successfully established, we determine the probability of gene expression as the quotient of activatory phrases between the total of inferred phrases. In the case shown in Table 9, two phrases have been successfully established, one activatory and the other inhibitory. Thus, the estimated probability of gene expression in this example would be p = ½.
To validate our methodology for inferring regulatory phrases we use the nature of the regulation already reported in the data bases for the case of simple regulations, namely, when only one regulator determines the expression of the regulated gene (gene A is only regulated by gene B). For example, in E. coli the gene alaW is regulated (positively) only by fis. In the Regulon Data Base this regulation is represented as:
The plus sign at the end indicates that alaW is positively retulated by fis. The table with the two phrases follows immediately:
fis alaW
↓ ↓
↑ ↑
Note that this information in the RegulonDB cannot be used to validate our methodology for genes with more than one regulator. This is because the regulation is combinatorial. To validate our methodology we compared only the inferred phrases for simple regulations with the ones reported in the RegulonDB or the DBTBS. Figure 5 shows the inference success (the fraction of regulatory phrases form simple regulations that matched the phrases obtained from the curated databases) as a function of the threshold T 0. In the same graphs we have plotted the total number of inferred phrases (including the phrases from simple regulations). As we can see, the inference becomes better as the threshold T 0 increases. However, the total number of inferred phrases decreases with the threshold. In order to have a good statistics (more than 250 inferred phrases) and a high inference success (around 90%), in this work we have chosen T 0 = 1.5 in E. coli and T 0 = 1.3 in B. subtilis.
Figure 5. Inference success.

This figure shows the compromise between the inference success (solid line), and the total number of inferred phrases (dashed line) as functions of the threshold T 0 for (a) E. coli and (b) B. subtilis. Note that increasing the inference success decreases the number of inferred phrases. A compromise has to be established by adequately choosing T 0. We have chosen T 0 = 1.5 for E. coli and T 0 = 1.3 for B. subtilis.
Supporting Information
Java Applet of the dynamics and list of ID numbers for the microarray experiments used in this work.
(0.05 MB DOC)
Acknowledgments
We thank the reviewers of this work for their useful comments and suggestions.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work was partially supported by the National Institute of Health grants GM070600 (I.S. and S.A.K.), GM072855 (I.S.), and P50 GM076547 (I.S.), CONACyT grants P47836-F (M.A.), CO1.41848/A-1, CO1.0538/A-1 and CO1.0435.B-1 (E.R.A.B.), and PAPIIT-UNAM grants IN112407-3 (M.A.) and IN230002 (E.R.A.B.). E.B. and A.C. acknowledge CONACyT for Ph.D. scholarships.
References
- 1.Hohenberg PC, Halperin BI. Theory of dynamic critical phenomena. Rev Mod Phys. 1977;49:435–479. [Google Scholar]
- 2.Turcotte DL. Self-organized criticality. Rep Prog Phys. 1999;62:1377–1429. [Google Scholar]
- 3.Peters O, Neelin JD. Critical phenomena in atmospheric precipitation. Nat Phys. 2006;2:393–396. [Google Scholar]
- 4.Arakawa A. Scaling tropical rain. Nat Phys. 2006;2:373–374. [Google Scholar]
- 5.Lux T, Marchesi M. Scaling and criticality in a stochastic multi-agent model of a financial market. Nature. 1999;397:498–500. [Google Scholar]
- 6.Mantegna RN, Stanley HE. Scaling behavior in the dynamics of an economic index. Nature. 1995;376:46–49. [Google Scholar]
- 7.Heimpel M. Critical behavior and the evolution of fault strength during earthquake cycles. Nature. 1997;388:865–868. [Google Scholar]
- 8.Scholz CH. Earthquakes and friction laws. Nature. 1998;391:37–42. [Google Scholar]
- 9.Ostojic S, Somfai E, Nienhuis B. Scale invariance and universality of force networks in static granular matter. Nature. 2006;439:828–830. doi: 10.1038/nature04549. [DOI] [PubMed] [Google Scholar]
- 10.Chialvo DR. Are our senses critical? Nat Phys. 2006;2:301–302. [Google Scholar]
- 11.Kinouchi O, Copelli M. Optimal dynamical range of excitable networks at criticality. Nat Phys. 2006;2:348–352. [Google Scholar]
- 12.Werner G. Metastability, criticality and phase transitions in brain and its models. BioSystems. 2007 doi: 10.1016/j.biosystems.2006.12.001. doi:10.1016/j.biosystems.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 13.Sethna JP, Dahmen K A, Myers CR. Crackling noise. Nature. 2001;410:242–250. doi: 10.1038/35065675. [DOI] [PubMed] [Google Scholar]
- 14.Kioussis D. Kissing chromosomes. Nature. 2005;435:579–480. doi: 10.1038/435579a. [DOI] [PubMed] [Google Scholar]
- 15.Levine M, Tjian R. Transcription regulation and animal diversity. Nature. 2003;424:147–151. doi: 10.1038/nature01763. [DOI] [PubMed] [Google Scholar]
- 16.Kitano H. Biological robustness. Nat Rev Genet. 2004;5:826–837. doi: 10.1038/nrg1471. [DOI] [PubMed] [Google Scholar]
- 17.de Visser J, Hermisson J, Wagner GP, Meyers LA, Bagheri-Chaichian H, et al. Perspective: evolution and detection of genetic robustness. Evolution. 2003;57:1959–1972. doi: 10.1111/j.0014-3820.2003.tb00377.x. [DOI] [PubMed] [Google Scholar]
- 18.Kirshner M, Gerhart J. Evolvability. Proc Natl Acad Sci U S A. 1998;95:8420–8427. doi: 10.1073/pnas.95.15.8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nehaniv C. Evolvability. BioSystems. 2003;69:77–81. doi: 10.1016/s0303-2647(02)00130-2. [DOI] [PubMed] [Google Scholar]
- 20.Wagner A. Robustness and evolvability in living systems. Oxford University Press; 2007. [Google Scholar]
- 21.Aldana M, Balleza E, Kauffman S, Resendis O. Robustness and evolvability in gene regulatory networks. J Theor Biol. 2007;245:433–448. doi: 10.1016/j.jtbi.2006.10.027. [DOI] [PubMed] [Google Scholar]
- 22.Serra R, Villani M, Semeria A. Genetic network models and statistical properties of gene expression data in knock-out experiments. J Theor Biol. 2004;227:149–157. doi: 10.1016/j.jtbi.2003.10.018. [DOI] [PubMed] [Google Scholar]
- 23.Serra R, Villani M, Graudenzi A, Kauffman S. Why a simple model of genetic regulatory networks describes the distribution of avalanches in gene expression data. J Theor Biol. 2007;246:449–460. doi: 10.1016/j.jtbi.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 24.Shmulevich I, Kauffman S, Aldana M. Eukariotic cells are dynamically oredred or critical but not chaotic. Proc Natl Acad Sci U S A. 2005;102:13439–13444. doi: 10.1073/pnas.0506771102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nykter M, Price ND, Aldana M, Ramsey SA, Kauffman SA, Hood LE, Yli-Harja O, Shmulevich I. Gene expression dynamics in the macrophage exhibit criticality. Proc. Natl. Acad. Sci. U.S.A. 2008;105(6):1897–1900. doi: 10.1073/pnas.0711525105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Derrida B, Stauffer D. Phase transitions in two dimensional Kauffman cellular automata. Europhys Lett. 1986;2:739–745. [Google Scholar]
- 27.Harris S, Sawhill B, Wuensche A, Kauffman S. A model of transcriptional regulatory networks based on biases observed in the regulatory rules. Complexity. 2002;7:23–40. [Google Scholar]
- 28.Shmulevich I, Lähdesmäki H, Dougherty ER, Astola J, Zhang W. The role of certain Post classes in Boolean network models of genetic networks. Proc. Natl. Acad. Sci. U.S.A. 2003;100 (19):10734–10739. doi: 10.1073/pnas.1534782100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nikolajewa S, Friedel M, Wilhelm T. Boolean networks with biologically relevant rules show order behavior. BioSystems. 2007;90:40–47. doi: 10.1016/j.biosystems.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 30.Karlsson F, Hörnquist M. Order and chaos in Boolean gene networks depends on the mean fraction of canalizing functions. Physica A. 2007;384:747–575. [Google Scholar]
- 31.Just W, Shmulevich I, Konvalina J. The number and probability of canalizing functions. Physica D. 2004;197:211–221. [Google Scholar]
- 32.Bower JM, Bolouri H, editors. Computational Modeling of Genetic and Biochemical Networks. The MIT Press; 2001. [Google Scholar]
- 33.Smolen P, Baxter DA, Byrne JH. Modeling transcriptional control in gene networks - methods, recent results, and future directions. Bull Math Biol. 2000;62:247–292. doi: 10.1006/bulm.1999.0155. [DOI] [PubMed] [Google Scholar]
- 34.Hasty J, McMillen D, Isaacs F, Collins JJ. Computational studies of gene regulatory networks: In Numero molecular biology. Nat Rev Genet. 2001;2:268–279. doi: 10.1038/35066056. [DOI] [PubMed] [Google Scholar]
- 35.Bornholdt S. Less is more in modeling large genetic networks. Science. 2005;310:449–451. doi: 10.1126/science.1119959. [DOI] [PubMed] [Google Scholar]
- 36.Chaves M, Sontag ED, Albert R. Methods of robustness analysis for Boolean models of gene control networks. IIE Proc-Syst Biol. 2006;153:154–167. doi: 10.1049/ip-syb:20050079. [DOI] [PubMed] [Google Scholar]
- 37.Kauffman S. Metabolic stability and epigenesis in randomly constructed genetic nets. J Theor Biol. 1969;22:437–467. doi: 10.1016/0022-5193(69)90015-0. [DOI] [PubMed] [Google Scholar]
- 38.Aldana M, Coppersmith S, Kadanoff LP. Boolean dynamics with random couplings. In: Kaplan E, Marsden JE, Sreenivasan KR, editors. Perspectives and Problems in Nonlinear Science. Springer NY; 2003. pp. 23–89. [Google Scholar]
- 39.Kauffman S. At Home in the Universe. Oxford University Press; 1995. [Google Scholar]
- 40.Mendoza L, Alvarez-Buylla ER. Genetic regulation of root hair development in Arabidopsis thaliana: a network model. J Theor Biol. 2000;204:311–326. doi: 10.1006/jtbi.2000.2014. [DOI] [PubMed] [Google Scholar]
- 41.Espinosa-Soto C, Padilla-Longoria P, Alvarez-Buylla ER. A gene regulatory network model for cell-fate determination during Arabidopsis thaliana flower development that is robust and recovers experimental gene expression profiles. The Plant Cell. 2004;16:2923–2939. doi: 10.1105/tpc.104.021725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Albert R, Othmer HG. The topology of the regulatory interactions predicts the expression pattern of the segment polarity genes in Drosophila melanogaster. J Theor Biol. 2002;223:1–18. doi: 10.1016/s0022-5193(03)00035-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang S, Ingber DE. Shape-dependent control of cell growth, differentiation, and apoptosis: Switching between attractors in cell regulatory networks. Exp Cell Res. 2000;262:91–103. doi: 10.1006/excr.2000.5044. [DOI] [PubMed] [Google Scholar]
- 44.Aldana M. Boolean dynamics of networks with scale-free topology. Physica D. 2003;185:45–66. [Google Scholar]
- 45.Pastor-Satorras R, Vespignani V. Epidemis spreading in scale-free networks. Phys Rev Lett. 2001;86:3200–3203. doi: 10.1103/PhysRevLett.86.3200. [DOI] [PubMed] [Google Scholar]
- 46.Salgado H, Gamma-Castro S, Martínez-Antonio A, Diaz-Peredo E, Sanchez-Solano F, et al. RegulonDB (version 5.0): Escherichia coli K-12 transcriptional regulatory network, operon organization, and growth conditions. Nucleic Acids Res. 2006;34 (Database issue):D394–397. doi: 10.1093/nar/gkj156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luscombe NM, Babu MM, Yu H, Snyder M, Teichmann SA, et al. Genomic analysis of regulatory network dynamics reveals large topological changes. Nature. 2004;431:308–312. doi: 10.1038/nature02782. [DOI] [PubMed] [Google Scholar]
- 48.Makita Y, Nakao M, Ogasawara N, Nakai K. DBTBS: database of transcriptional regulation in Bacillus subtilis and its contribution to comparative genomics. Nucleic Acids Res. 2004;32:D75–77. doi: 10.1093/nar/gkh074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stanford Microarray Database ( http://genome-www5.stanford.edu)
- 50.Gosset G, Zhang Z, Nayyar S, Cuevas WA, Saier MH., Jr Transcriptome analysis of crp-dependent catabolite control of gene expression in Escherichia coli. J Bacteriology. 2004;186:3516–3524. doi: 10.1128/JB.186.11.3516-3524.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Z, Gosset G, Barabote R, González CG, Cuevas WA, et al. Functional interactions between the carbon and iron utilization regulators, crp and fur, in Escherichia coli. J Bacteriology. 2005;187:980–990. doi: 10.1128/JB.187.3.980-990.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. KEGG Expression Database ( www.genome.jp/kegg/expression)
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Java Applet of the dynamics and list of ID numbers for the microarray experiments used in this work.
(0.05 MB DOC)